全文HTML
--> --> -->在Al-Cu-Li三元体系里, 铝中Li的加入降低了合金的密度, 增加了合金的刚度, 而与Cu的结合促进了析出强化, 从而获得高强度的沉淀析出物. 在众多的析出物中, T1相(Al6Cu4Li3)是主要的强化析出物, 它是常见并且深入研究的中间相之一. 然而, T1相的存在可能增加合金的腐蚀敏感性, 以往的研究表明, T1相通常作为合金基体的阳极, 导致阳极的溶解和腐蚀[11-13]. Zhang等[14]发现2A97-T6合金中的T1相是优先溶解相, 导致合金发生晶内腐蚀. Buchheit等[15]研究AA2090发现T1相的浓度较高时, 亚晶界对局部腐蚀尤为敏感. 实验研究表明, T1相对Al-Cu-Li合金的腐蚀起着关键作用, 研究T1相的腐蚀机理具有重要的意义.
研究Al-Cu-Li合金体系腐蚀机理, 其前提是获得合理可信的T1相晶体结构. 事实上, 已有大量的实验和理论工作构建和预测T1相的晶体结构, 但仍然存在较大的争议. 准确晶体结构的缺乏, 也导致对T1相的腐蚀行为研究不够深入. 最近, Kim等[3]基于密度泛函理论(density functional theory, DFT)的第一原理计算和HAADF-STEM实验图像, 提出一种新的T1相合理结构, 使得原子尺度上的T1相的研究成为可能.
腐蚀的本质是一个电化学过程, 材料表面发生阳极反应, 金属离子溶解到溶液中. 材料表面的电子功函数描述材料表面电子逃逸难度, 是表征材料耐腐蚀性强弱的重要参数[16]. 为了进一步揭示T1相的腐蚀机理, 本文从电子层面入手, 利用密度泛函理论结合表面能、电子功函数研究不同终结面的表面稳定性, 针对T1相的不同面, 讨论应力对其影响, 同时计算Ag, Mg和Zn在Al/T1界面替位能, 以此讨论合金元素对T1相析出的影响.
2
2.1.表面能的计算
T1相晶格平面通常有不同的终结面, 它们也具有不同的表面能. 为了计算表面能(Esurf), 结合表面能计算的常规方法[19,20], 遵循文献[20]提出的计算方法, 其中考虑到了截断能(Ecle)和弛豫能(Erel). 截断能的计算公式为


2
2.2.表面电子功函数的计算
功函数是材料的固有表面特性, 描述了电子从材料发射的势垒[21], 大小等于固体表面电子克服能量势垒脱离表面逃到真空所需要的能量. 电子功函数是固体表面的宏观物理量, 它的数值随晶面的不同而不同, 能够很好地反应晶面变化. 不同材料具有不同的电子功函数, 即使是同一种材料, 不同终结面, 电子功函数也不尽相同[16]. 材料表面的电子功函数越大, 表明电子越不易从材料表面逸出, 则材料的耐蚀性越好; 反之, 材料表面的电子功函数越小, 表明电子越易从材料表面逸出, 则材料的耐蚀性越差, 即功函数较高的材料表面更加稳定[22]. 因此, 可以用电子功函数的大小表示材料耐蚀性的强弱.文献[23]将模型的电子功函数(Φ)定义为真空能级(Evacumm)与费米能级(EFermi)之差, 具体的计算为
2
2.3.替位能的计算
掺杂替位能可以很好地表征常见掺杂元素对T1相形成和材料耐蚀性的影响, 因此, 构建了Al/T1相界面, 计算不同掺杂元素、不同位置的替位能. 根据文献[24], 替位能计算公式为
先对T1相模型进行几何优化, 而后再沿(001), (010)和(100)面进行切面, 采用切面的方向和方法不同, 共得到10个不同的终结面模型, 如图1所示, (001), (010)和(100)面分别有4种、2种、4种截取方式. 每个终结面的原子类型分别为A (Al, Cu, Li), B (Cu), C (Al, Cu, Li), D (Al), E (Al, Cu, Li), F (Al, Cu, Li), G (Al, Cu), H (Al, Li), I (Al, Cu), J (Li). 每个终结面的原子类型模型如图2所示. 为了讨论它们与Al基体的本征电势差, 需要了解不同终结面的电子功函数情况.
 图 1 T1相(001), (100), (010) 3个晶面的切面方向
图 1 T1相(001), (100), (010) 3个晶面的切面方向Figure1. Surface selection of (001), (100), (010) phase.
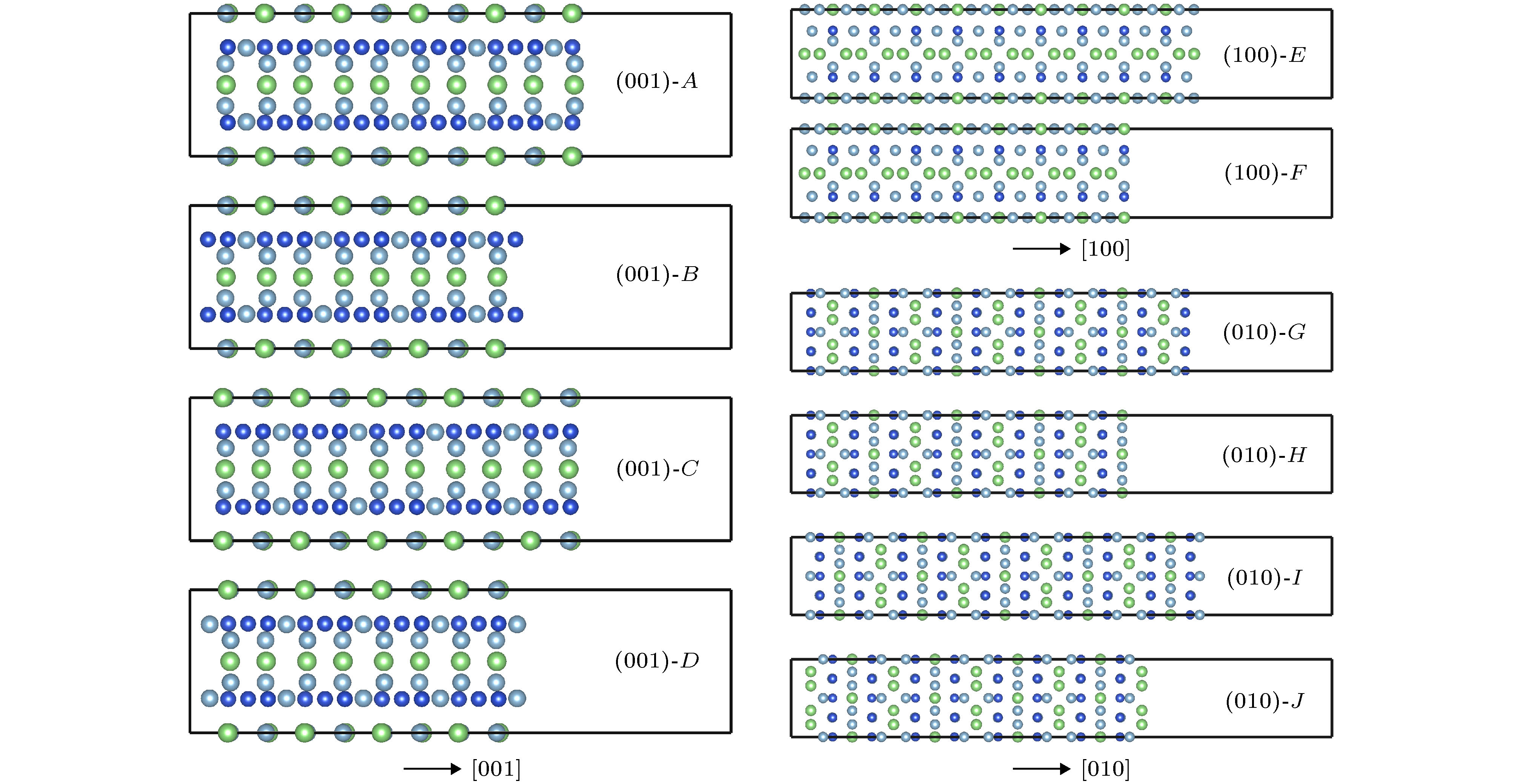 图 2 不同面的原子构型
图 2 不同面的原子构型Figure2. The configurations of different surfaces.
3.1.表面能及其表面电子功函数
对T1相截面可以得到10种不同的终结面, 通过计算各个晶面的表面能、电子功函数来探讨材料表面稳定性问题. 各个表面的表面能及电子功函数计算结果如表1所示.| 面 | 终结面 | 表面能/J·m–2 | 功函数/eV |
| A | Al-Cu-Li | 1.24 | 3.64 |
| B | Cu | 1.10 | 3.91 |
| C | Al-Cu-Li | 1.20 | 3.70 |
| D | Al | 1.28 | 4.27 |
| E | Al-Cu-Li | 1.02 | 3.89 |
| F | Al-Cu-Li | 1.07 | 4.35 |
| G | Al-Cu | 0.84 | 4.29 |
| H | Al-Li | 0.86 | 4.53 |
| I | Al-Cu | 0.59 | 4.12 |
| J | Li | 0.83 | 3.40 |
表1T1相10个终结面的表面能和电子功函数
Table1.The surface energies and electron work functions of ten surfaces.
T1相的表面能在0.59—1.28 J/m2之间, 全Al组成的D终结面表面能最大, 而Al-Cu组成终结面的I面表面能最小, 金属键材料表面能的高低通常由表面原子密度以及原子弛豫重构情况决定. 对于I面, 表面原子经过弛豫之后, 由于应力的释放, 表面原子位置发生重构, 致使其表面能降低. 为了具体描述其原子结构, 给出了I面弛豫前后的结果, 如图3(a)所示. 可以看出, 弛豫后表层的Al原子向次外层偏移(平均偏移1.02 ?), 导致新形成表面的原子密度高达0.23 atom/?2. 由于其表面原子密度的增大, 原子间库仑相互作用力增强, 使其结构更加稳定, 进而得到更低的表面能.
 图 3 (a)面I, (b)面F, (c)面H 3个终结面弛豫前后原子构型
图 3 (a)面I, (b)面F, (c)面H 3个终结面弛豫前后原子构型Figure3. Atomic configurations of (a) surface I, (b) surface F and (c) surface H before and after relaxation.
表1也给出了电子功函数的结果, 不同于表面能, 结果显示功函数主要由表面原子种类决定. 由表可知, T1相的电子功函数的值在3.40—4.53 eV之间. 可以看出, 当终结面中含有Li原子, 其功函数相对来说较小. 但也有一些特例, 如F, H面, 尽管含有Li原子, 但其功函数相对较高, 分别为4.35 eV和4.53 eV. F面虽然为Al-Cu-Li面, 但是其表面只含有一个Li原子, 所占比例较小, 其表面功函数主要由Al, Cu原子所决定, 如图3(b)所示. 而H面虽为Al-Li终结面, 但经过弛豫后Li原子下沉到表层以下, 导致最外层原子主要为Al原子, 如图3(c)所示. 此外, 对于纯Li终结的J面来说, 全Li组成导致了最低的功函数.
以上结果可以看出, 决定材料耐蚀性的功函数, 主要与材料表面原子组成相关. 而电负性常用于描述元素原子对电子的约束能力, Cu, Al和Li的电负性分别为1.90, 1.61和0.98. T1相中的3种元素中, Li的电负性最小, 对电子的束缚能力最弱, 电子更容易从表面逸出, 导致材料的电子功函数下降. 为了进一步说明, 当终结面含Li时对电子功函数的影响, 我们给出含Li表面, 即A, C, E和J面的电荷密度分布图. 由图4可以看出Al, Cu原子周围有大量电子, 而Li周围基本无电子, Al, Cu原子比Li原子具备更强的电子束缚能力. 这一结果基本符合原子电负性的规律.
 图 4 (a)面A, (b)面C, (c)面E以及(d)面J的电子密度分布
图 4 (a)面A, (b)面C, (c)面E以及(d)面J的电子密度分布Figure4. Electron density distribution of (a) surface A, (b) surface C, (c) surface E, and (d) surface J.
2
3.2.应力对表面能和功函数的影响
作为结构材料的铝合金通常服役于应力条件, 而应力状态下的腐蚀也有别于无应力条件下. 有研究表明, 材料在应力条件下强度和刚度降低, 材料的腐蚀性能与无应力时有所差别[25-28]. 因此研究应力状态下T1相的耐蚀性, 对于探索应力腐蚀机理也具有一定意义.根据应变方向, 以1.0%为应变步长, 计算了T1相10个终结面, 在X和Y方向上应变从–2.0%到2.0%时(共计25个状态)的表面能和电子功函数. 为了方便比较, 这里着重讨论应变条件下, T1相表面能和电子功函数波动情况. 表2为在25个状态下, 各个面的表面能和电子功函数最高值与最低值之差.
| 面 | 表面能变化量/J·m–2 | 功函数变化量/eV |
| A | 0.41 | 0.14 |
| B | 0.59 | 0.09 |
| C | 0.25 | 0.09 |
| D | 0.16 | 0.12 |
| E | 0.21 | 0.14 |
| F | 0.31 | 0.07 |
| G | 0.34 | 0.08 |
| H | 0.37 | 0.05 |
| I | 0.37 | 0.15 |
| J | 0.18 | 0.04 |
表2应变条件下表面能和功函数的波动量
Table2.Fluctuation of surface energy and work function with strain.
由表2可以看出施加应变后, 每个面的表面能和功函数变化值是大小不一, 这主要是因为每个终结面的原子种类不同, 应变对表面的影响也不同. 在表面能方面, 应变的影响在0.19—0.59 eV, 其中B面的表面能变化最大. 由图5可知, 在无应变情况下, 以及在X和Y方向上应变情况相反时, 表面能基本不变; 而应变情况相同时会导致表面能上升, 且张应变导致的表面能上升更明显. 这与纯金属的研究结果略有不同, 纯金属在应力的作用下, 表面能的变化一般是呈现对称的, 但B面的表面能变化却不对称, 这主要是因为T1相属于三元合金相, 而非纯金属块体相. 压和张应变会导致表面的重构, 进而对表面能产生不一样的影响.
 图 5 面B应变条件下表面能的变化
图 5 面B应变条件下表面能的变化Figure5. Surface energy of surface B with strain.
相对于表面能的变化, 功函数变化较小, 不超过0.15 eV. 图6为I面的电子功函数随应变变化的等高线图. 拉应变的施加会致使I面功函数增大, 进而导致表面耐腐蚀性能的提升; 而压应变却使功函数变小, 材料的耐蚀性能减弱. 这与以往的大多数研究纯金属[29]结论相反, 以往结论通常认为拉应变使得原子之间距离增大, 电荷密度减小, 进而使得电子功函数变小. 对于I面而言, 拉应变使得表层的Al原子进一步与次外层融合, 如图3(a)所示, 反而增加了原子密度和电子密度, 获得较高的电子功函数, 类似的结果也出现在其他金属相中[30].
 图 6 面I应变条件下电子功函数的变化
图 6 面I应变条件下电子功函数的变化Figure6. Work function of surface I with strain state.
2
3.3.掺杂对T1相形成的影响
T1相通常是在热处理阶段析出的, 为了促进其析出, 会添加Ag, Zn和Mg等合金元素. 而合金元素作用机理及其在Al/T1界面行为仍然有待研究. 根据已有的模型[3], 我们构建Al(111)/T1(010)界面模型, 如图7所示, 模型包含11层Al原子和1个单元T1相, 其中中间3层Al原子固定. 基于这模型, 采用文献[31]所用的替位能的计算方法, 计算Ag, Zn和Mg 3种原子在界面15处位置的替位能变化.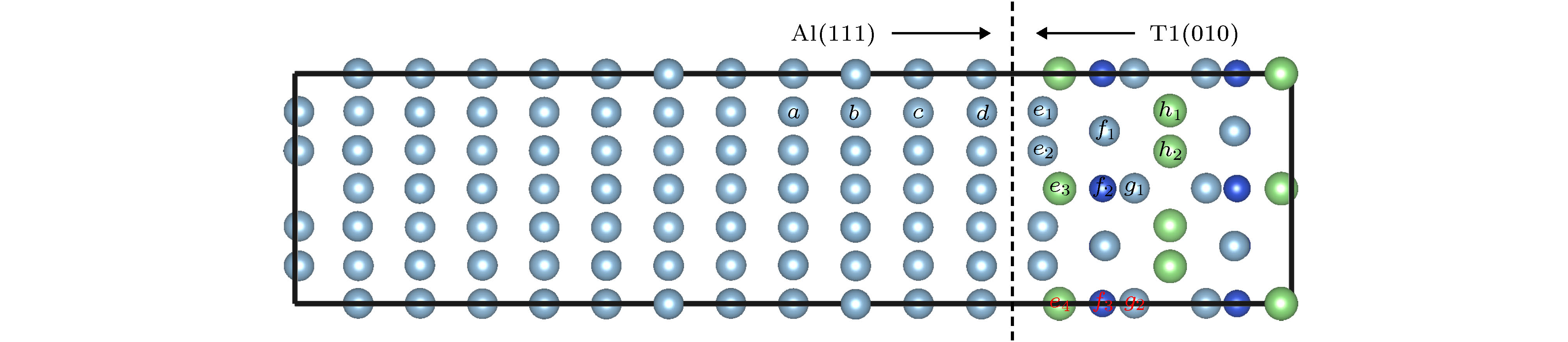 图 7 界面模型及其原子替换位置
图 7 界面模型及其原子替换位置Figure7. Interface model and atomic substitution positions.
图8是Ag, Zn和Mg原子替换15个特征位置后替位能的结果. 在Al基体中, 随之不断靠近界面, 替位能不断降低; 但越过界面, 在T1相中各种合金原子替位能重新变大, 且为正值, 表明Ag, Zn和Mg原子不易固溶至T1相中. 以上结果说明合金元素原子更倾向于在Al/T1相界面处偏聚, 这也与实验结果一致[32]. 比较3种原子, Ag原子具有更低的替位能, 且在e1和e2位置已经为负值, 可以看出, 大多数情况下, Ag的替位能最小, 这主要与其原子半径有关, Zn, Al, Ag和Mg 4种原子的半径分别为134, 143, 144, 160 pm. 在合金或中间相中, 原子间通过金属键结合, 原子半径是替位能的主导因素, Ag与Al具有最接近的原子半径, 替位导致的晶格畸变最小, 也因此获得最低的替位能. 因此, 当Ag富集在界面处, 能有效降低Al/T1相的界面应力, 从而促进T1相的析出, 这也符合实验观察的现象[33,34]. 此外, 由图8可以发现, 当Ag, Zn和Mg原子替位Li原子时, 无论是界面还是T1相内, 通常可以获得较低的替位能, 这一结果与Li原子本身特性有关. Li是碱金属, 对电子束缚能力弱, 被替位后, 由于替位原子的电负性大于Li, 电子重新被吸引过来, 进而降低了能量. 同时结合第2节的计算结果可知, 当Ag, Zn和Mg原子替位表面的Li后, 由于具有高于Li的电负性, 会使得电子功函数提高, 进而提高T1相的耐蚀能力.
 图 8 Mg, Zn和Ag在Al(111)/T1(010)界面的替位能
图 8 Mg, Zn和Ag在Al(111)/T1(010)界面的替位能Figure8. The substitution energies of Mg, Zn and Ag in Al(111)/T1(010) interface.
