1.Department of Physics and Information Engineering, Cangzhou Normal University, Cangzhou 061001, China 2.State Key Laboratory of Nonlinear Mechanics (LNM), Institute of Mechanics, Chinese Academy of Science, Beijing 100190, China 3.School of Mathematics and Physics, University of Science and Technology Beijing, Beijing 100083, China
Fund Project:Project supported by the National Key Research and Development Program of China (Grant No. 2016YFB0700500), the Key Research and Development Program of Hebei Province, China (Grant No. 18211233), the Key Sciencific Studies Program of the Higher Education Institute of Hebei Province, China (Grant No. ZD2018301), and the Natural Science Foundation of Cangzhou, China (Grant No. 177000001)
Received Date:29 March 2019
Accepted Date:26 July 2019
Available Online:01 November 2019
Published Online:05 November 2019
Abstract:Graphene is a two-dimensional (2D) crystal of carbon atoms packed in a honeycomb lattice. Because of this unique structure, it shows a number of intriguing properties. Interface between neighboring 2D layers or between 2D overlayers and substrate surfaces provides confined space for chemical process. The interlayer spacing between bilayer graphenes of van der Waals material is expected to modify the properties of atoms and molecules confined at the atomic interfaces. In this paper, the carbon monoxide (CO) and oxygen (O) in bilayer graphene are studied by density functional theory (DFT). The quantitative relationship between the interlayer spacing of bilayer graphene (d) and the reaction energy barrier ($ {{E_{\rm{a}}}} $) is obtained. Five values of d between 4.7 ? and 5.9 ? are used. The calculated results show that the total energy of the initial state, the transition state, the final state system and the reaction barrier are sensitive to the variation of the interlayer distance: the reaction barrier increases gradually with interlayer distance decreasing. The calculated energy barrier is 1.13 eV when the interlayer distance is 4.7 ?, while the energy barrier is 0.39 eV when the interlayer distance is 5.9 ?. It is also found that adsorption energy between O and graphene at the top site and the bridge site increase gradually with interlayer distance decreasing. Therefore, the atomic-level regulation of the reaction barrier can be achieved by changing the interlayer spacing of bilayer graphene. The charge density difference shows that when the distance between two layers of graphene is small, there is an obvious charge accumulation between C atoms in transition state O—C=O and C atoms in the upper or lower layer of graphene. This results in sp orbital hybridization, which leads the interaction between two C atoms to be enhanced. It is difficult to form a weak O—C bond of transition state O—C=O with O atoms adsorbed on graphene because of a binding force which exists in the z-axis direction. The DFT calculation of CO oxidation reaction barrier can be reduced by adjusting the spacing of bilayer graphene, which provides a theoretical support for the application of graphene and the preparation of new carbon-based intercalated composites. Keywords:grapheme/ confined reaction/ density functional theory/ reaction energy barrier
表1五个不同间距情况下的$E_{{\rm{ads}}{\text{-}}{\rm{O}}}^{{\rm{Top}}}({\rm{IS}})$, $E_{{\rm{ads}}{\text{-}}{\rm{O}}}^{{\rm{Bridge}}}({\rm{IS}})$, ${E_{\rm{a}}}$和$\Delta H$ Table1.Adsorption energy at top site and bridge site, the reaction energy barrier and reaction heat at five different interlayer distances.
图3给出了不同层间距时, 各状态对应的结构图. 图 3 各状态对应结构的侧视图 (a)初态; (b)过渡态; (c)末态 Figure3. Side view of local configurations at various states along the reaction pathway: (a) Initial state; (b) transition state; (c) final state.
表2五个不同间距时初态(IS)、过渡态(TS)、末态(FS)的CO中C—O键长, O与CO分子间距以及CO2 (O=C=O)中O—C与C—O键长(分别对应dC—O(CO), dCO—O, dO—C(CO2), dC—O(CO2), 单位为?) Table2.The C—O bond lengths of the initial, transition and final states of CO at five different distances, the molecular distances between O and CO, and the O—C and C—O bond lengths in CO2 (O=C=O). They correspond to dC—O(CO), dCO—O, dO—C(CO2), dC—O(CO2) with the units of ?.
我们研究了从初态IS到末态FS不同层间距离下一氧化碳氧化反应路径, 具体结果如图4所示. 图 4 不同层间距下, 从初态到末态一氧化碳氧化反应路径 (a) d = 4.7 ?; (b) d = 5.0 ?; (c) d = 5.3 ?; (d) d = 5.6 ?; (e) d = 5.9 ? Figure4. Reaction pathway of the oxidation of CO and O from the initial state to final state under different interlayer distance: (a) d = 4.7 ?, (b) d = 5.0 ?, (c) d = 5.3 ?, (d) d = 5.6 ?, (e) d = 5.9 ?.
为了进一步分析双层石墨烯不同层间距对一氧化碳的反应能垒的影响, 探究双层石墨烯与一氧化碳、氧的电子转移, 分别计算了不同层间距对应过渡态与双层石墨烯间的电荷差分密度, 结果如图5所示. 图 5 CO与O反应, 在不同间距的双层石墨烯层间过渡态的电荷差分密度图, 其中等值面数值分别为 (a) 0.00014 e (a.u.)3; (b) 0.00020 e (a.u.)3; (c) 0.00025 e (a.u.)3; (d) 0.00035 e (a.u.)3; (e) 0.00048 e (a.u.)3 Figure5. Electron density different of the transition states between the bilayer grapheme and the isosurface is (a) 0.00014 e (a.u.)3, (b) 0.00020 e (a.u.)3, (c) 0.00025 e (a.u.)3, (d) 0.00035 e (a.u.)3, (e) 0.00048 e (a.u.)3, respectively.

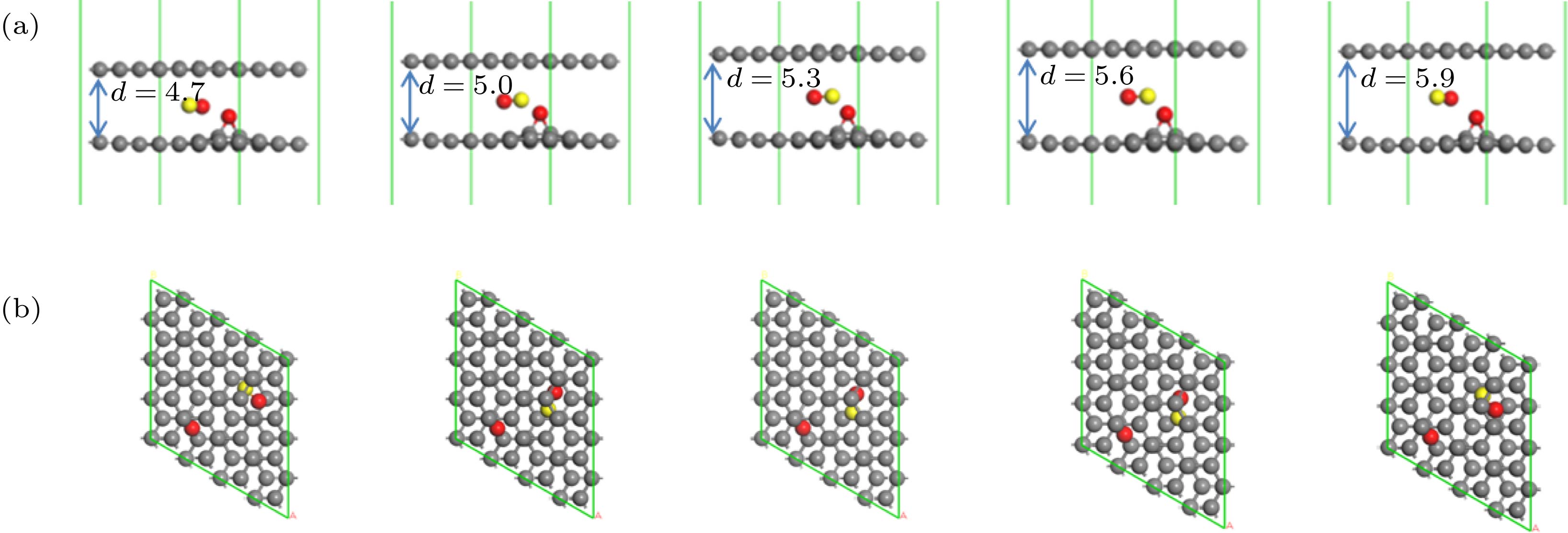 图 1 初始结构优化模型 (a)侧面图; (b)俯视图
图 1 初始结构优化模型 (a)侧面图; (b)俯视图


















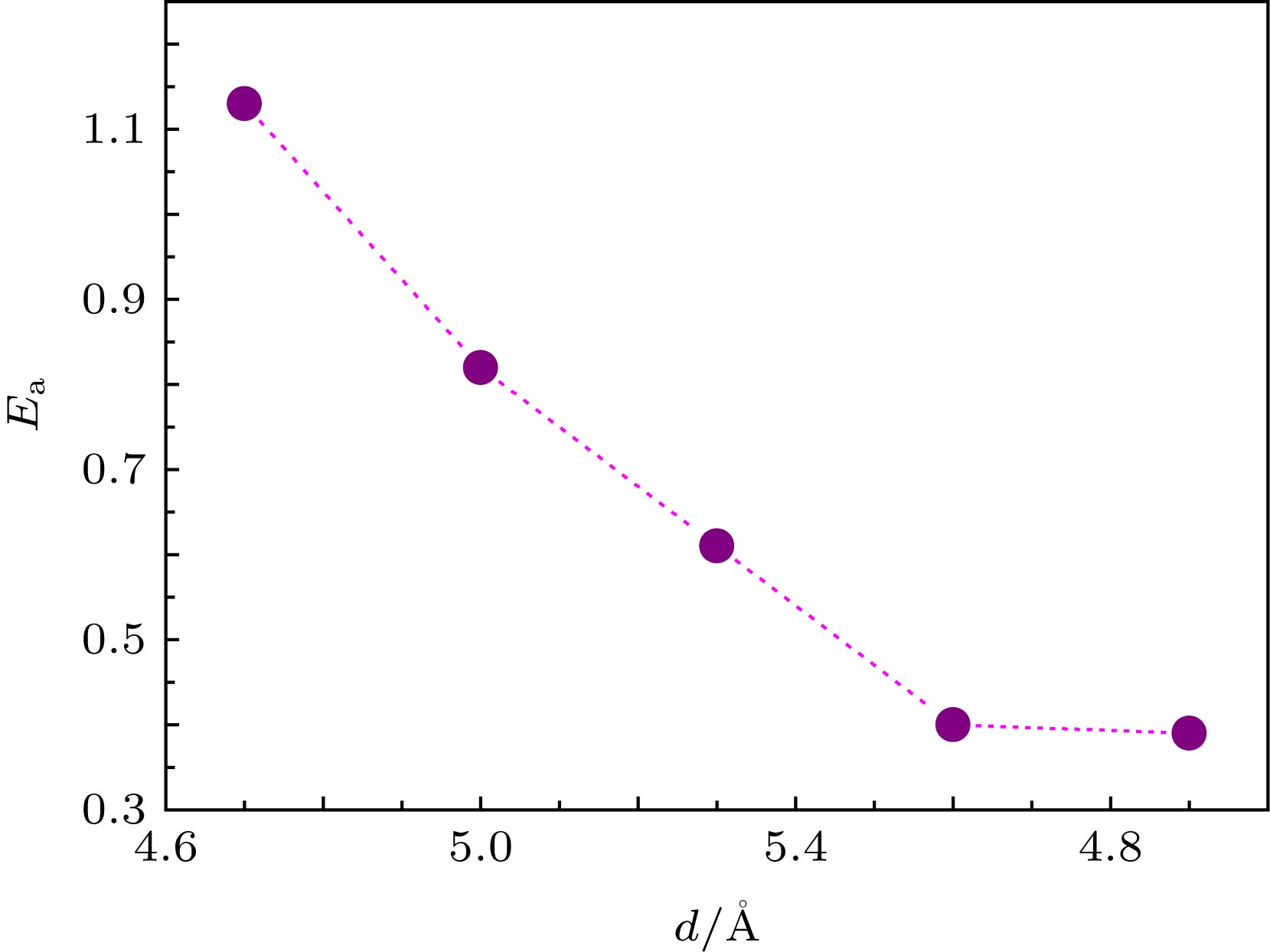 图 2 反应能垒
图 2 反应能垒

 图 3 各状态对应结构的侧视图 (a)初态; (b)过渡态; (c)末态
图 3 各状态对应结构的侧视图 (a)初态; (b)过渡态; (c)末态 图 4 不同层间距下, 从初态到末态一氧化碳氧化反应路径 (a) d = 4.7 ?; (b) d = 5.0 ?; (c) d = 5.3 ?; (d) d = 5.6 ?; (e) d = 5.9 ?
图 4 不同层间距下, 从初态到末态一氧化碳氧化反应路径 (a) d = 4.7 ?; (b) d = 5.0 ?; (c) d = 5.3 ?; (d) d = 5.6 ?; (e) d = 5.9 ?
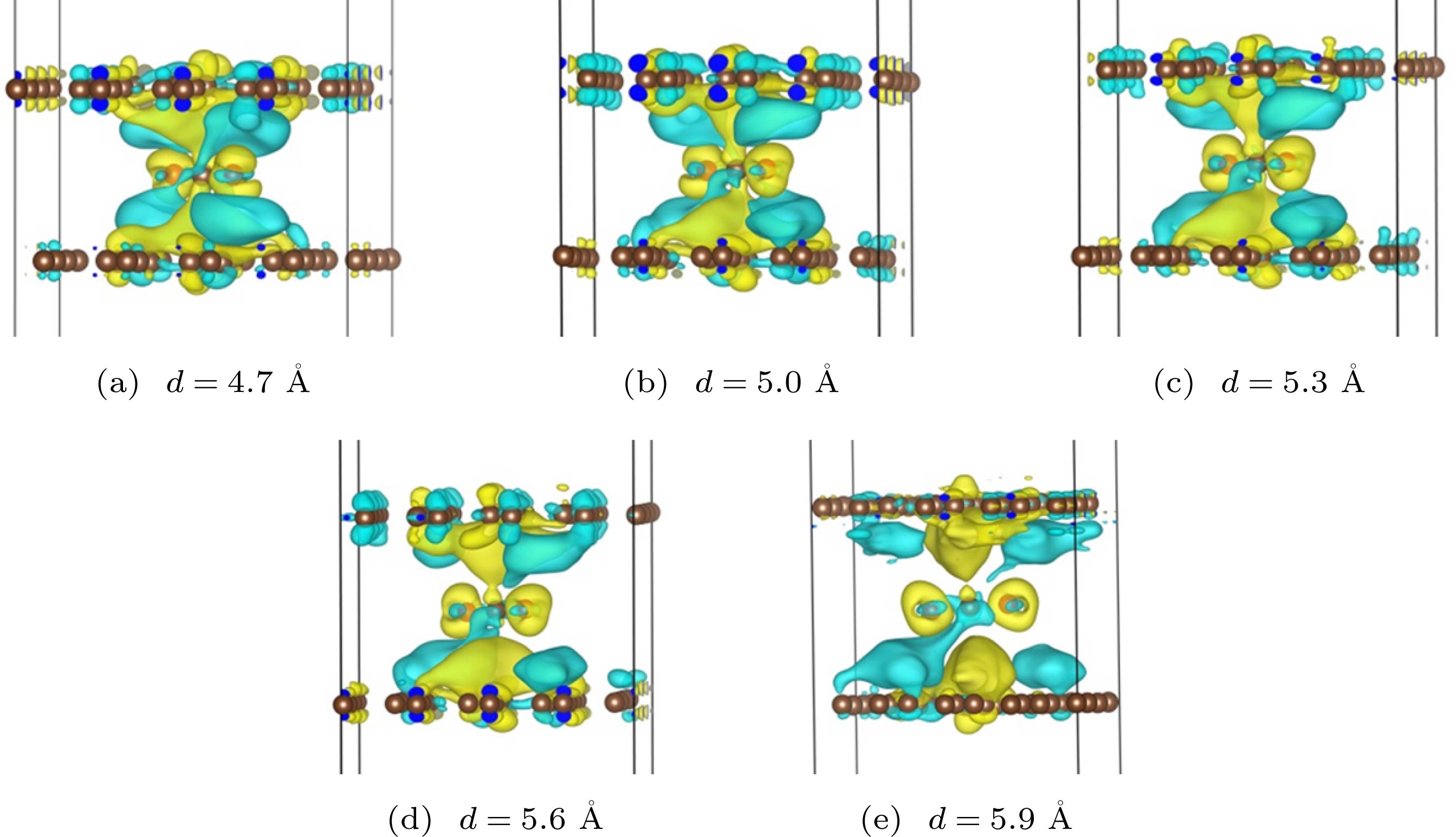 图 5 CO与O反应, 在不同间距的双层石墨烯层间过渡态的电荷差分密度图, 其中等值面数值分别为 (a) 0.00014 e (a.u.)3; (b) 0.00020 e (a.u.)3; (c) 0.00025 e (a.u.)3; (d) 0.00035 e (a.u.)3; (e) 0.00048 e (a.u.)3
图 5 CO与O反应, 在不同间距的双层石墨烯层间过渡态的电荷差分密度图, 其中等值面数值分别为 (a) 0.00014 e (a.u.)3; (b) 0.00020 e (a.u.)3; (c) 0.00025 e (a.u.)3; (d) 0.00035 e (a.u.)3; (e) 0.00048 e (a.u.)3