1.College of Materials Science and Engineering, Nanjing Tech University, Nanjing 211816, China 2.Jiangsu Collaborative Innovation Center for Advanced Inorganic Function Composites, Nanjing Tech University, Nanjing 211816, China
Fund Project:Project supported by the National Natural Science Foundation of China (Grant Nos. 11604047, 51672127), the Natural Science Foundation of Jiangsu Province, China (Grant No. BK20160694), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), China
Received Date:29 April 2019
Accepted Date:17 July 2019
Available Online:01 October 2019
Published Online:05 October 2019
Abstract:Since the discovery of graphene, graphene-based gas sensors have been widely studied, but the inherent zero band gap of graphene limits the response sensitivity of gas sensors. Transition metal dichalcogenides (TMDs) are ideal materials for designing nanoscaled highly-sensitive gas sensors due to their moderate band gaps, large surface-to-volume ratios and high carrier mobilities. Tungsten ditelluride (WTe2), as an important member of TMDs family, has outstanding advantages such as high specific surface area, excellent selectivity, and fast response. The WTe2 has quite a high carrier mobility and thus can provide a great response speed for gas sensor compared with graphene, which motivates us to further explore WTe2 as a promising sensing material. Recent studies have reported that monolayered and multilayered WTe2 films have been successfully synthesized, and the precise control of the number of atomic layers of monolayered WTe2 has been achieved. In this work, by density functional theory calculation, we examine the most stable adsorption configuration, adsorption energy, charge transfer, electrical and magnetic properties for each of the gas molecules (CO, CO2, NH3, NO and NO2) adsorbed on WTe2 monolayer. The results show that all the adsorptions of these gas molecules are physical adsorptions, and the adsorption energy of nitrogen-based gas is smaller than that of carbon-based gas, indicating that WTe2 is more sensitive to the adsorption of N-based gas molecules. The adsorption of NH3 behaves as a charge donor with electron obtained from WTe2 monolayer. The adsorption of CO, CO2, NO, and NO2 are charge acceptors, which accept charges from the WTe2 monolayer. Moreover, compared with the adsorption of CO, CO2 and NH3 gas molecules, the adsorption of NO and NO2 gas molecules introduce impurity states near the Fermi level, which are mainly contributed by the N p orbital and O p orbital. In addition, the adsorption of NO and NO2 induce magnetic moments of 0.99 μB and 0.80 μB, respectively. The results obtained in this work not only conduce to further understanding the charge transfer mechanism of gas molecules adsorbed on WTe2 monolayer, but also indicate the promising prospects of developing WTe2-based ultra-sensitivity gas sensing nanodevices. Keywords:transition metal dichalcogenides/ density functional theory/ gas sensors
其中${E_{{\rm{total}}}}$是分子吸附在WTe2表面体系的总能量, ${{E_{{\rm{WT}}{{\rm{e}}_2}}}}$为单层WTe2的能量, ${{E_{{\rm{gas}}}}}$为气体分子的能量. 根据这一定义, Ea为负值时表明吸附过程为放热反应, 其中Ea的绝对数值越大表明吸附强度越强. 计算结果如图2所示, CO气体分子吸附在WTe2表面的吸附强度最低, 其吸附能的大小为–0.117 eV, 而NO2气体分子吸附在WTe2表面的吸附强度最高(–0.366 eV). CO2, NO和NH3气体分子吸附在WTe2表面的吸附能大小分别为–0.175, –0.238, –0.225 eV. 结果表明, N基气体分子在WTe2表面的吸附能明显小于CO和CO2在WTe2表面的吸附能, 这与图2中的吸附距离有着很好的关联性, 一般来说, 气体分子在WTe2表面的吸附距离越小, 其对应的吸附能也越小. 这说明WTe2对N基气体分子的吸附更为敏感, 与基于二硫化钼和石墨烯的气敏传感器的研究结果相似[36,42]. 同时, 与以往石墨烯的研究结果相比(CO, NH3, NO和NO2气体分子在石墨烯表面的吸附能分别为0.014, 0.031, 0.029, 0.067 eV)[42], WTe2表面气体分子的吸附能绝对值远大于石墨烯表面气体分子的吸附能, 这表明WTe2的气体吸附性能明显优于石墨烯. 图 2 CO, CO2, NH3, NO, NO2气体分子与单层WTe2之间的吸附距离和吸附能 Figure2. Adsorption distance and adsorption energy for CO, CO2, NH3, NO, and NO2 on WTe2 monolayer.
为了深入了解电荷密度的重新分布, 以及吸附气体分子与WTe2之间的相互作用, 计算了差分电荷密度(如图3所示), 差分电荷密度定义为 图 3 (a) CO, (b) CO2, (c) NH3, (d) NO和 (e) NO2气体分子与单层WTe2之间的差分电荷密度. 等值面取6.0 × 10–4e/?3, 电子积累(损耗)分别用黄色(蓝色)表示, 同时标注了电荷转移的方向(用箭头表示)和电荷转移量 Figure3. The charge difference between WTe2 monolayer and gas molecules for (a) CO, (b) CO2, (c) NH3, (d) NO and (e) NO2. The isosurface is taken as 6.0 × 10–4e/?3. The electron accumulation (depletion) is indicated by yellow (blue) color. The direction (indicated by an arrow) and value of the charge transfer are shown.
其中${\rho _{{\rm{gas}}/{\rm{WT}}{{\rm{e}}_2}}}$, ${\rho _{{\rm{gas}}}}$和${\rho _{{\rm{WT}}{{\rm{e}}_2}}}$分别代表气体分子吸附在WTe2表面体系、孤立的气体分子和本征单层WTe2的电荷密度. 同时采用Bader分析方法计算了CO, CO2, NH3, NO, NO2气体分子与WTe2之间的电荷转移量, 进一步评估WTe2表面吸附气体分子后的电荷转移. 计算结果表明(如图3(a)和图3(b)所示), 当CO和CO2吸附在WTe2表面时, 从WTe2转移到CO和CO2气体分子上的电荷量较小(分别为0.027 e和0.036 e), 这说明WTe2与C基气体分子之间的相互作用较弱. 然而, 对于N基气体分子在WTe2表面的吸附, 可以观察到NH3, NO和NO2气体分子和WTe2之间的电荷转移量明显增加(分别为0.013 e, 0.107 e和0.376 e, 如图3(c)—图3(e)所示), 这表明N基气体分子与WTe2之间的相互作用较强, 其中NO2气体分子吸附在WTe2表面后的电荷转移量最高. 图3所示的电荷转移量与图2所示的吸附能(吸附强度)显示出明显的关联性, 即电荷转移量越大, 相应的吸附体系的吸附强度越强(吸附能越小), 这与MoS2气体传感器的研究结果相似[36]. 因此, 从电荷转移的角度, 可以进一步理解气体分子吸附在单层WTe2表面的作用机理. 此外, 从图3中可以进一步看出, 对于大多数气体分子(CO, CO2, NO, NO2)在WTe2表面的吸附, 电荷均从WTe2表面转移到气体分子上, 表现为得电子体, 而NH3气体分子吸附后, 电荷从NH3气体分子转移到WTe2表面, 表现为给电子体. 这种现象类似于气体分子吸附在石墨烯和MoS2表面的情况[36,42]. 对于n型WTe2来说, 导带中已经存在一些电子. 当CO, CO2, NO, NO2气体分子吸附在WTe2表面, 电子从WTe2转移到气体分子上, 会导致WTe2的电阻增大, 因而电流减小. 综上, 气体分子吸附在WTe2表面后的电荷转移引起WTe2体系电阻的变化, 同时电荷转移量反映出WTe2对不同气体分子的吸附性能, 这进一步解释了WTe2吸附N基气体分子具有较低的吸附能. 接下来研究气体吸附对单层WTe2电学性能的影响. 首先计算本征单层WTe2的能带结构, 其带隙为1.05 eV, 这与以往的研究结果一致[38-41]. 能带结构的计算结果如图4所示. 当CO和CO2气体分子吸附在WTe2表面时, WTe2的价带和导带没有明显的变化(如图4(a)—图4 (c)所示), 表明CO和CO2气体分子与WTe2之间的相互作用较弱, 这与它们高的吸附能(低的吸附强度)相对应. 并且由图5(a)和图5(b)的分态密度可知, 能带结构图中的杂质能带主要由O原子和C原子的p轨道组成. 而当NH3气体分子吸附在WTe2表面后, 从图4(d)可知, 在费米能级下方的–3和–8 eV处形成了杂质能带, 结合分态密度图(图5(c))可知, 这些杂质能带主要由N原子的p轨道所贡献. 虽然CO, CO2和NH3这三种气体分子吸附在WTe2表面后, 在能带结构中引入了杂质能带, 但未改变费米能级附近的能带结构, 带隙仍为1.05 eV, 这表明CO, CO2和NH3气体分子在WTe2表面的吸附没有影响WTe2的电子结构. 图 4 (a) 本征WTe2的能带结构图; (b) CO, (c) CO2和 (d) NH3吸附在单层WTe2表面的能带结构图; (e), (f) NO和 (g), (h) NO2吸附在单层WTe2表面的能带结构图. 其中蓝线和红线分别表示自旋向上和自旋向下的能带结构, 橄榄色的点线表示吸附气体分子的投影能带结构 Figure4. (a) The band structure of pristine WTe2; band structure of (b) CO, (c) CO2 and (d) NH3 adsorbed on WTe2 monolayer; band structure of (e), (f) NO and (g), (h) NO2 adsorbed on WTe2 monolayer, the blue and red lines represent the band structure of spin-up and spin-down, respectively. The olive dots represent the projected band structure of the adsorbed gas molecules.
图 5 (a) CO, (b) CO2, (c) NH3, (d) NO和 (e) NO2分别吸附在单层WTe2上的分态密度图; (f) NO和 (g) NO2吸附在WTe2表面的自旋密度分布图 Figure5. The density state of (a) CO, (b) CO2, (c) NH3, (d) NO and (e) NO2 adsorbed on WTe2 monolayer, respectively. The spin density distribution of (f) NO and (g) NO2 adsorbed on WTe2 monolayer
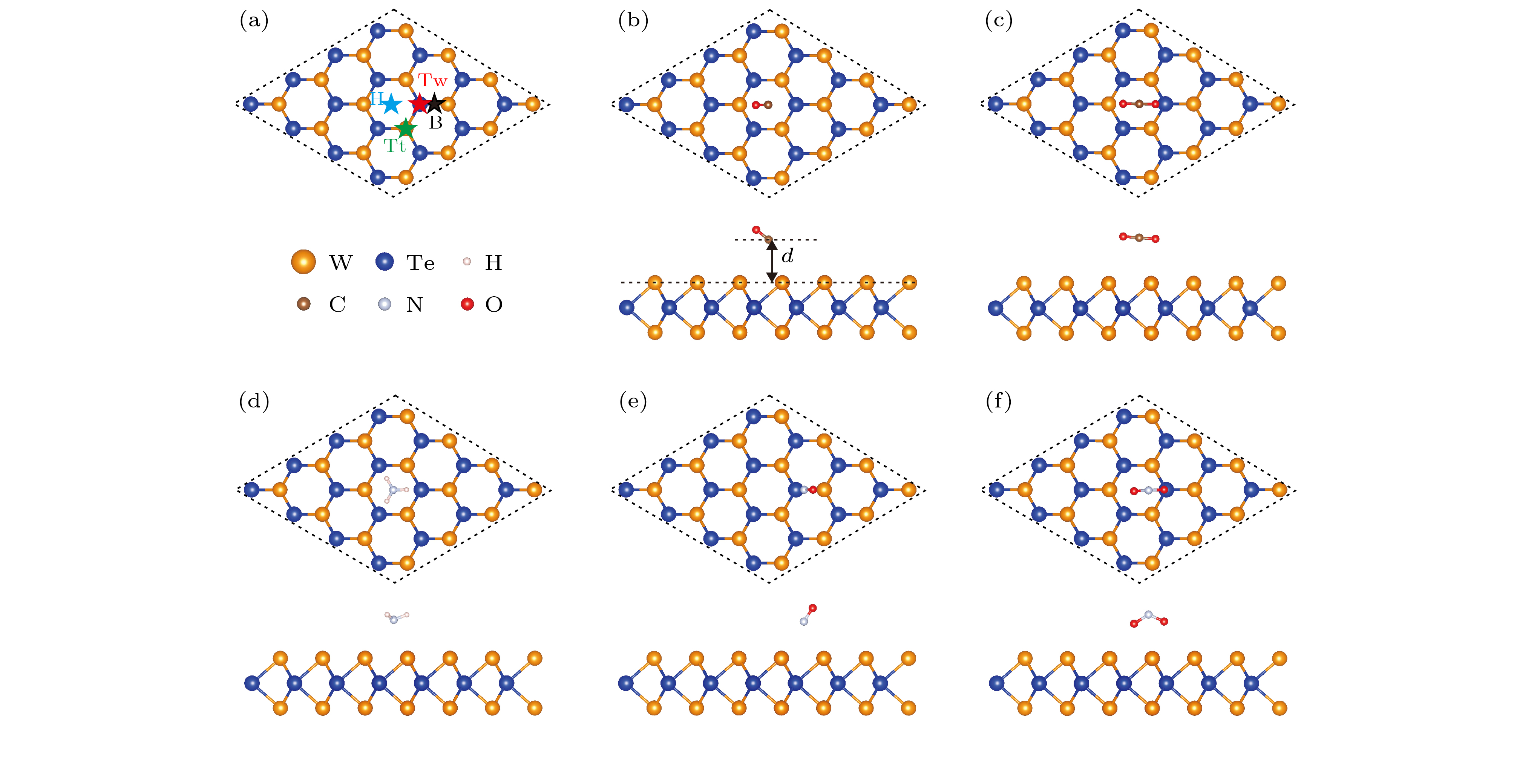 图 1 (a) 四个不同的吸附位点示意图; (b) CO, (c) CO2, (d) NH3, (e) NO和(f) NO2吸附在单层WTe2表面最稳定构型的俯视图和侧视图; 吸附距离d的定义如 (b) 所示
图 1 (a) 四个不同的吸附位点示意图; (b) CO, (c) CO2, (d) NH3, (e) NO和(f) NO2吸附在单层WTe2表面最稳定构型的俯视图和侧视图; 吸附距离d的定义如 (b) 所示


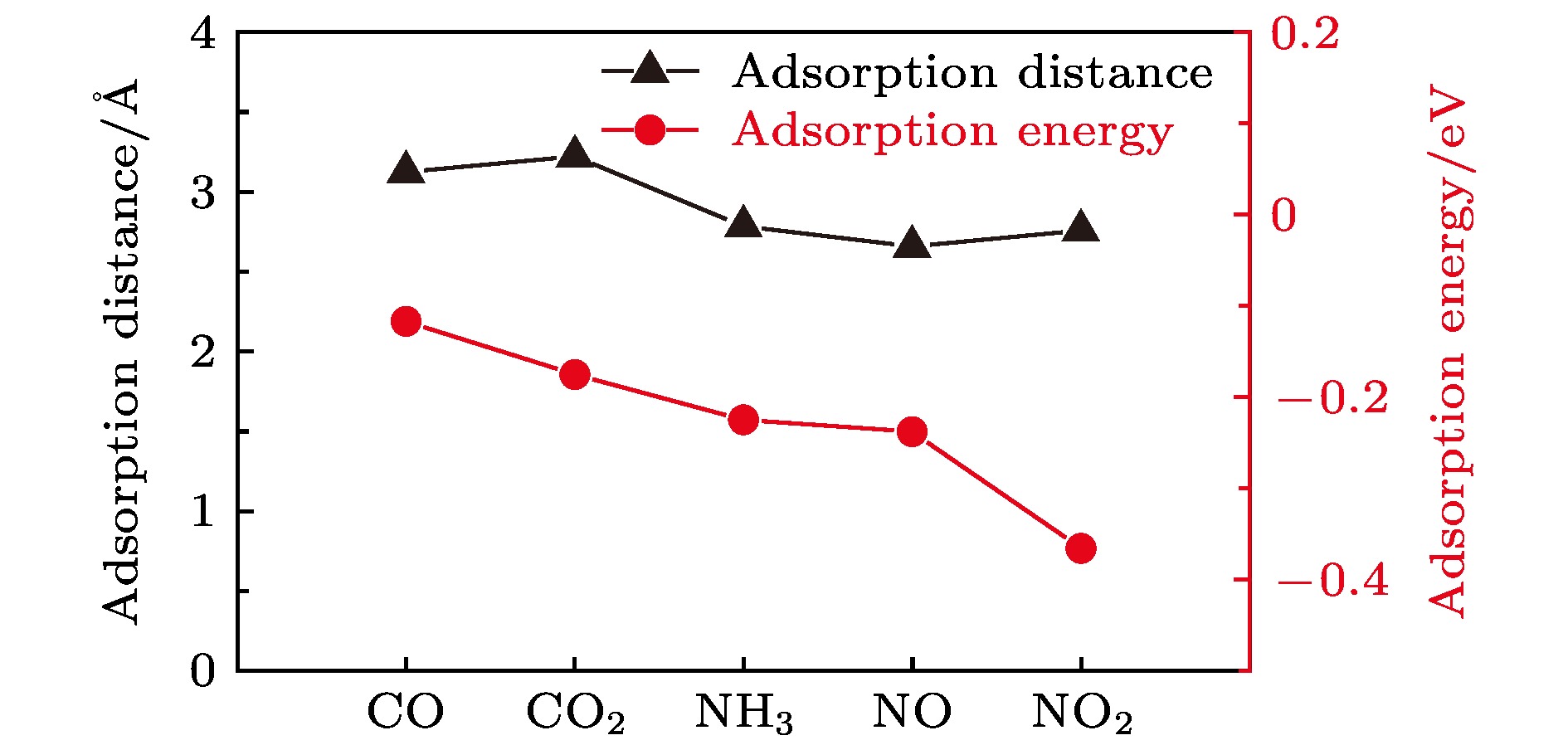 图 2 CO, CO2, NH3, NO, NO2气体分子与单层WTe2之间的吸附距离和吸附能
图 2 CO, CO2, NH3, NO, NO2气体分子与单层WTe2之间的吸附距离和吸附能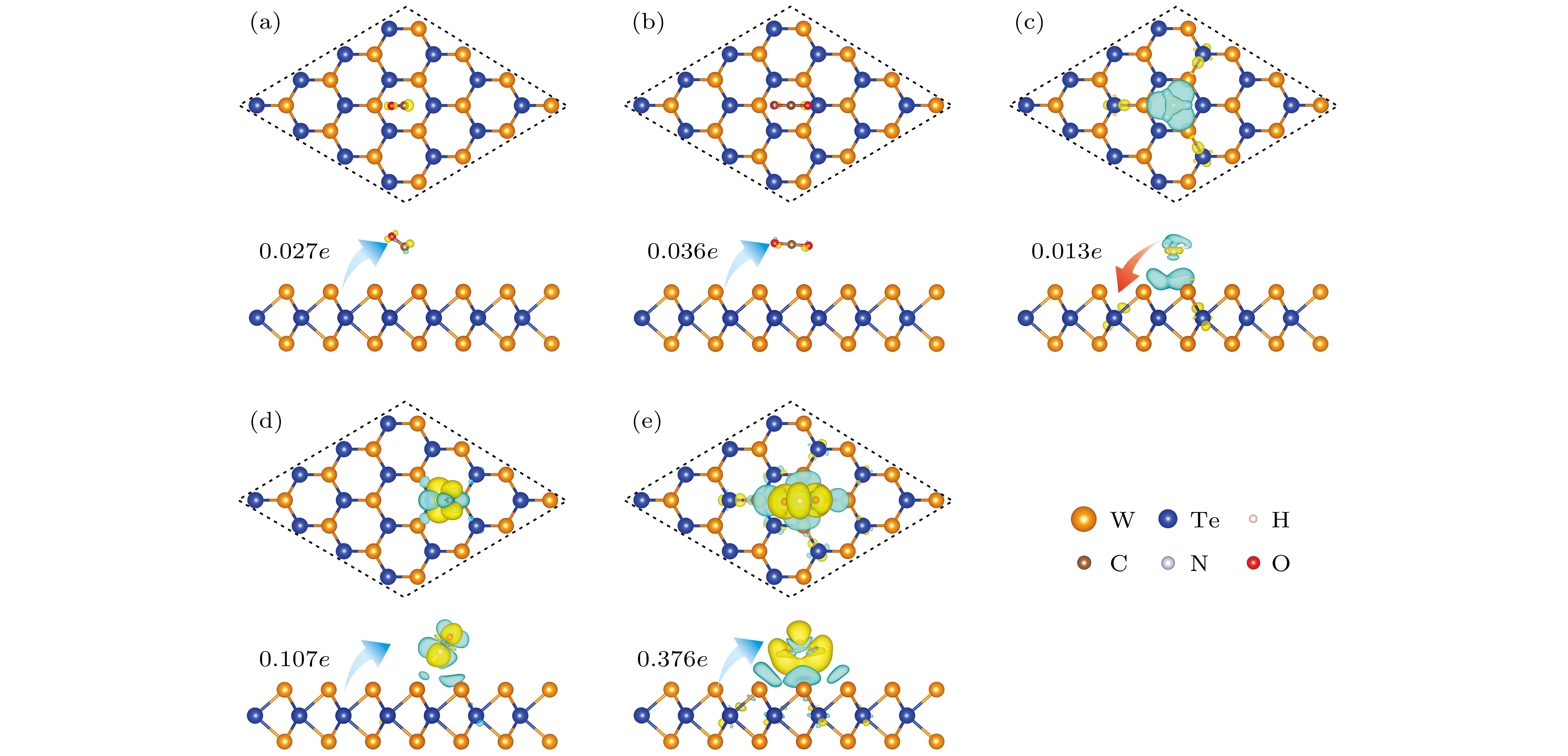 图 3 (a) CO, (b) CO2, (c) NH3, (d) NO和 (e) NO2气体分子与单层WTe2之间的差分电荷密度. 等值面取6.0 × 10–4 e/?3, 电子积累(损耗)分别用黄色(蓝色)表示, 同时标注了电荷转移的方向(用箭头表示)和电荷转移量
图 3 (a) CO, (b) CO2, (c) NH3, (d) NO和 (e) NO2气体分子与单层WTe2之间的差分电荷密度. 等值面取6.0 × 10–4 e/?3, 电子积累(损耗)分别用黄色(蓝色)表示, 同时标注了电荷转移的方向(用箭头表示)和电荷转移量


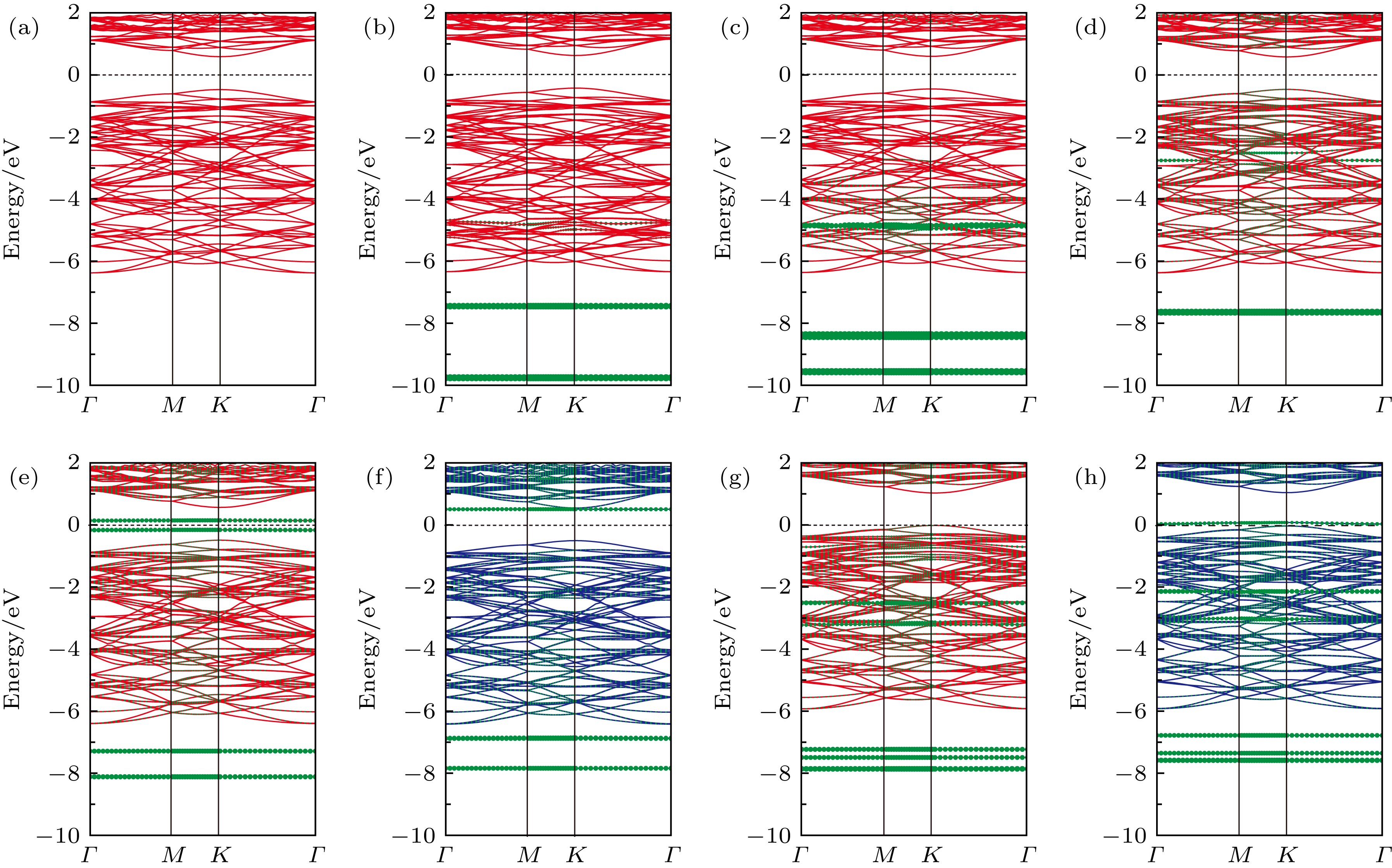 图 4 (a) 本征WTe2的能带结构图; (b) CO, (c) CO2和 (d) NH3吸附在单层WTe2表面的能带结构图; (e), (f) NO和 (g), (h) NO2吸附在单层WTe2表面的能带结构图. 其中蓝线和红线分别表示自旋向上和自旋向下的能带结构, 橄榄色的点线表示吸附气体分子的投影能带结构
图 4 (a) 本征WTe2的能带结构图; (b) CO, (c) CO2和 (d) NH3吸附在单层WTe2表面的能带结构图; (e), (f) NO和 (g), (h) NO2吸附在单层WTe2表面的能带结构图. 其中蓝线和红线分别表示自旋向上和自旋向下的能带结构, 橄榄色的点线表示吸附气体分子的投影能带结构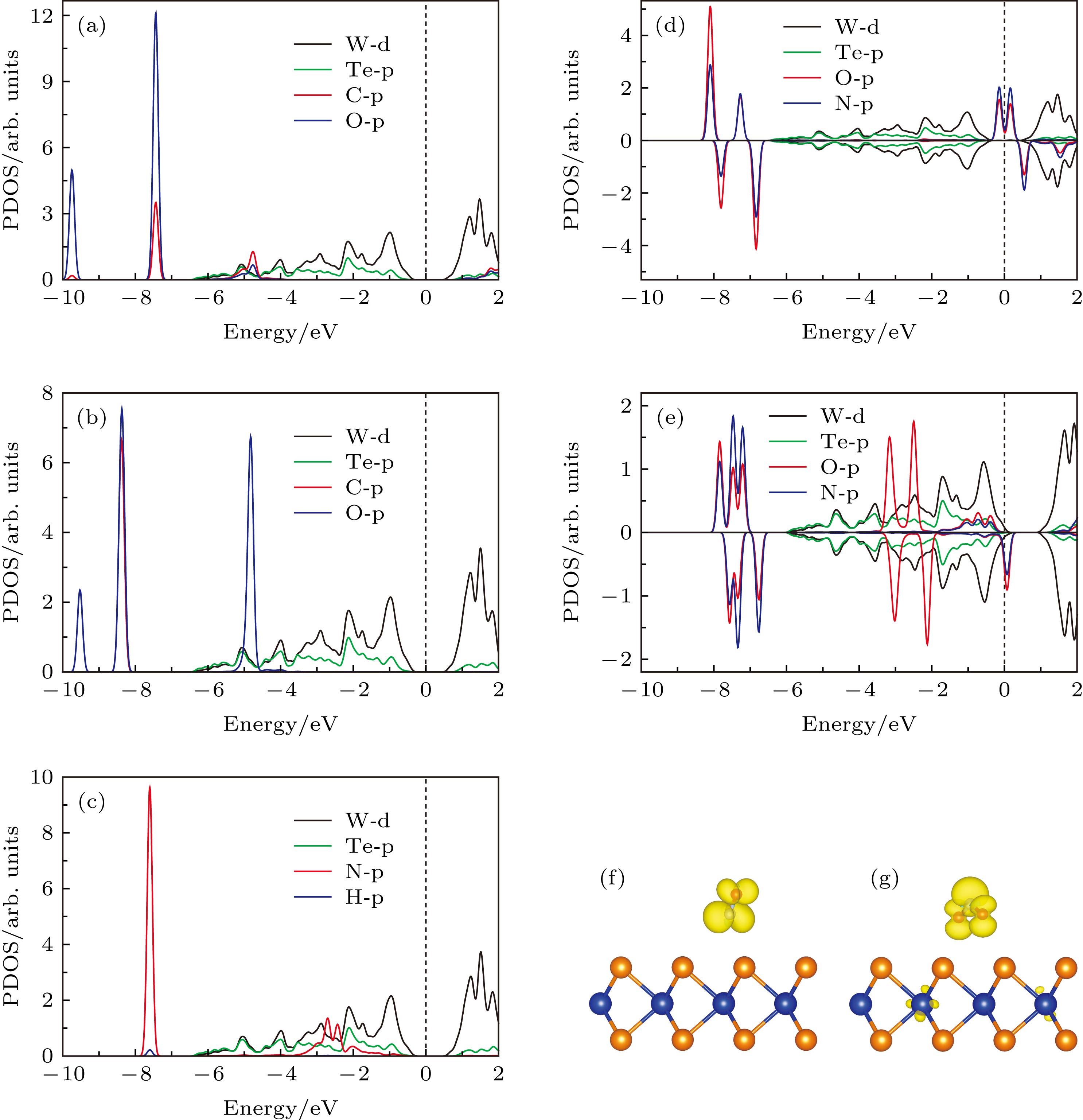 图 5 (a) CO, (b) CO2, (c) NH3, (d) NO和 (e) NO2分别吸附在单层WTe2上的分态密度图; (f) NO和 (g) NO2吸附在WTe2表面的自旋密度分布图
图 5 (a) CO, (b) CO2, (c) NH3, (d) NO和 (e) NO2分别吸附在单层WTe2上的分态密度图; (f) NO和 (g) NO2吸附在WTe2表面的自旋密度分布图