Fund Project:Project supported by the Fundamental Research Fund for the Central Universities, China (Grant No. XDJK2018B034)
Received Date:19 April 2019
Accepted Date:01 July 2019
Available Online:01 October 2019
Published Online:20 October 2019
Abstract:The electronic structures and absorption spectra of LiTaO3 (LT) crystal and Fe:Mg:LiTaO3 crystal with different Mg concentrations are investigated by first-principles through using the density functional theory. The supercell crystal structures are established with 60 atoms with four models: the near-stoichiometric pure LiTaO3 crystal (LT); the iron doped LiTaO3 crystal (Fe:LT), with the charge compensation model expressed as FeLi2+-2VLi–; the iron and magnesium co-doped LiTaO3 crystal (Fe:Mg:LT), with the charge compensation model taken as FeLi2+-MgLi+-3VLi–; the other iron and magnesium co-doped LiTaO3 crystal (Fe:Mg(E):LT), with Mg ion concentration near threshold value (slightly less than 6 mol%) and taking the charge compensation model as 2MgLi+-FeTa2–. The geometry optimization results show that the total energy values of all models can achieve certain stable values, which means that the models used in this paper are very close to the actual crystal structures. In the electronic structures, the extrinsic defect energy levels in the forbidden band of Fe:LT crystal are mainly contributed from the Fe 3d orbital, and the band gap of Fe:LT about 3.05 eV is narrower than that of LT, the band gap of Fe:Mg:LT and Fe:Mg(E):LT sample are 2.72 eV and 2.45 eV respectively. The results show that the orbit of Fe 3d, Ta 5d and O 2p are superposed with each other, forming covalent bonds, which results in conduction band and valence band shifting toward low energy in iron doped LiTaO3 crystal. The Fe 3d orbit is split into Eg orbit and T2g orbit under the influence of the crystal field. There are two absorption peaks at 417 nm (2.97 eV) and 745 nm (1.66 eV) in the Fe:LiTaO3 crystal. The first one is attributed to the transfer of the T2g orbital electron to conduction band. The last one can be taken as the result of Eg electron transfer of Fe2+ in crystal. The intensities and positions of these peaks vary with the concentration of Mg ion. Specially, with the concentration of Mg ion attaining the threshold value, the peak at 745 nm disappears, and the other peak moves slightly to 457 nm (2.71 eV). With the Mg ion concentration at the threshold value, the Fe ions can occupy Ta positions. This occupying condition makes the Eg orbital energy change greatly compared with the scenario in the FeLi condition, and it affects hardly the T2g orbital energy. So, if the absorption nearby 745 nm waveband can be taken as the useful process in holographic storage application, it is better to take smaller concentration of Mg ions (less than threshold value). On the other hand, nearby 457 nm waveband, concentration of Mg ions can be chosen as a large value. Keywords:LiTaO3 crystal/ Fe and Mg co-doped/ electronic structure/ absorption spectrum/ first-principles
对模型几何优化所得到各体系总能量变化如图2所示, 横坐标表示迭代次数, 纵坐标为相应的体系能量. 从图2可以看出, 各体系的总能量随迭代增加而不断减小, 最终趋于稳定, 表明所建立的模型与晶体的真实结构相符合. 表2为LT晶体晶格常数的几何优化值与实验值, 两者的相对误差仅在2%左右, 优化后体系的结构变化微小, 表明采用的理论模型和计算方法是合理可靠的. 图 2 各体系几何优化总能量 Figure2. Total energy of geometry optimization for every system.
Lattice parameter
a/nm
b/nm
c/nm
V/nm3
Experimental value
1.0308
0.5154
1.3863
637.83 × 10–3
Optimization result
1.0521
0.5260
1.4127
677.19 × 10–3
表2LT 晶格常数的几何优化值与实验值 Table2.Geometry optimization result and experiment values of LT crystal.
图4为各体系的态密度图以及Fe:Mg:LT的分态密度图, 图中显示价带主要由O 2p轨道组成, 导带主要由Ta 5d轨道组成, O 2p和Ta 5d轨道在价带有很多的交叠, 形成较强的共价键. Fe:LT晶体形成的杂质能级主要由Fe的3d轨道组成, 并与O的2p轨道有一定的相互作用. 双掺样品在–42 eV附近的态密度峰非常窄, 是由Mg的2p轨道贡献, 局域性很强, 说明Mg原子在晶体中以离子键为主, 几乎没有与其他原子形成共价键. 当掺Mg 浓度达到阈值时, 晶体的态密度峰整体向低能方向移动2 eV, 即掺杂降低了各原子的轨道能量. 这与能带结构中导带和价带的移动是一致的. 图 4 晶体态密度图 (a) LT及不同掺Mg浓度的Fe:Mg:LT晶体态密度图; (b) Fe:Mg:LT晶体分态密度图 Figure4. Density of states of crystals: (a) Density of states of LT and Fe:Mg:LT crystals; (b) partial density of states of Fe:Mg:LT crystals.
禁带附近分态密度如图5所示. 图中显示了在晶体场的影响下, Fe的3d轨道分裂为能量较高的Eg轨道和能量较低的T2g轨道[23]. 从图5(a)可以看出, Ta 5d轨道和O 2p轨道完全交叠, 轨道之间发生了明显的杂化, 形成较强的共价键, 导致价带和导带同时向低能方向移动, 能带间隙较LT明显减小(见图3). 如图5(b)所示, Mg以低浓度掺入 Fe:LT时, Mg对禁带附近的态密度并没有贡献, 却使得带隙较单掺Fe时减小(见图3(c)). 这是由于Mg2+的极化能力强于Li+, Mg2+占锂位后与其周围的氧原子形成共价键, 导致O2–的极化度升高, 其电子云变形增强, 使得带隙变窄. Mg的浓度达到阈值时, Mg占锂位, Fe占钽位, 虽然Fe3+相比Ta5+对O2–的极化能力弱些, 使O2-的电子云变形减弱, 但该模型中锂位Mg离子的个数是前一模型的一倍, 仍使得图3(d)中带隙比图3(c)减小. 图 5 晶体禁带附近分态密度 (a) Fe:LT; (b) Fe:Mg:LT; (c) Fe:Mg(E):LT Figure5. Partial density of states near the forbidden band: (a) Fe:LT; (b) Fe:Mg:LT (c) Fe:Mg(E):LT.
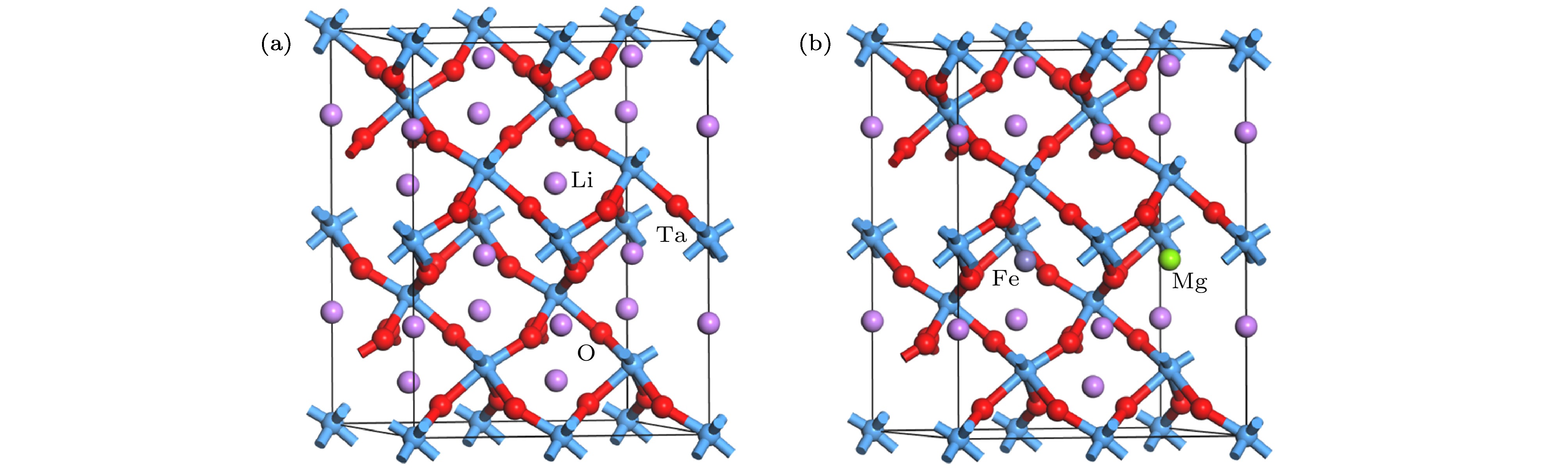 图 1 晶体结构模型 (a) LT; (b) Fe:Mg:LT
图 1 晶体结构模型 (a) LT; (b) Fe:Mg:LT




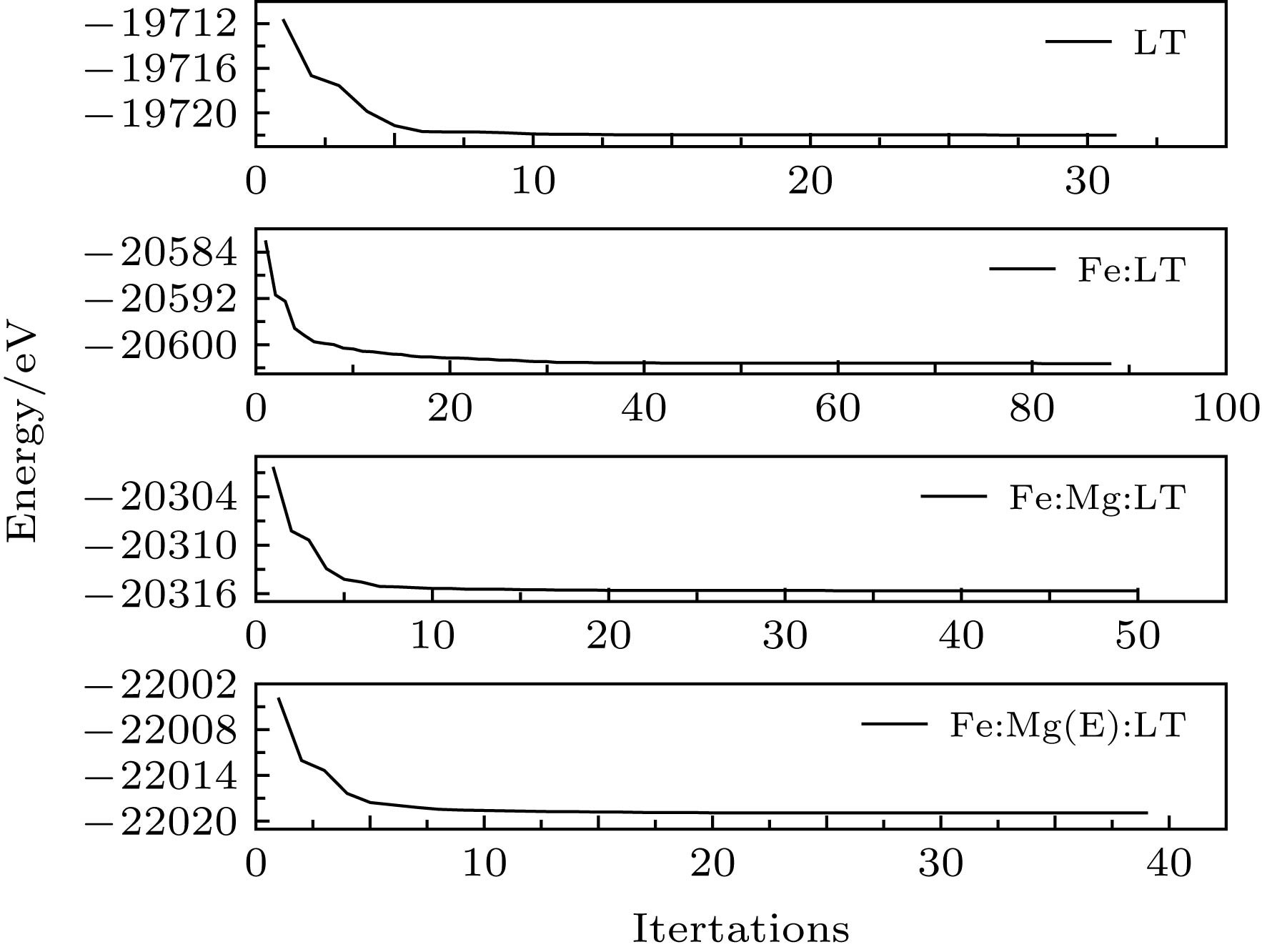 图 2 各体系几何优化总能量
图 2 各体系几何优化总能量 图 3 能带结构图 (a) LT; (b) Fe:LT; (c) Fe:Mg:LT; (d) Fe:Mg(E):LT
图 3 能带结构图 (a) LT; (b) Fe:LT; (c) Fe:Mg:LT; (d) Fe:Mg(E):LT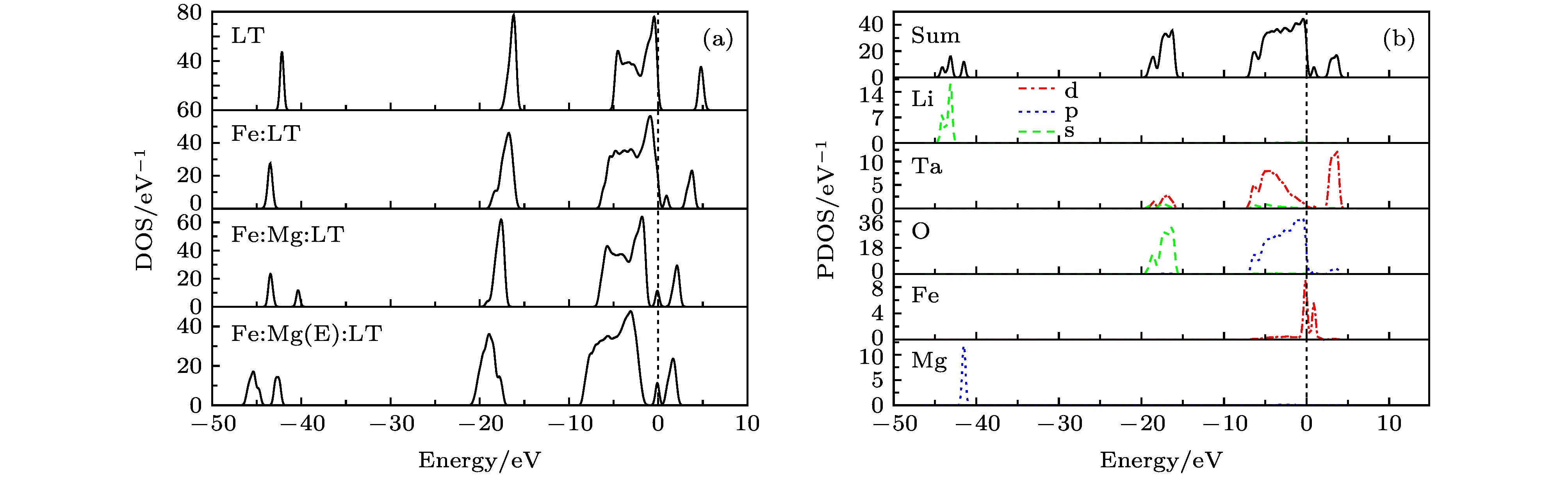 图 4 晶体态密度图 (a) LT及不同掺Mg浓度的Fe:Mg:LT晶体态密度图; (b) Fe:Mg:LT晶体分态密度图
图 4 晶体态密度图 (a) LT及不同掺Mg浓度的Fe:Mg:LT晶体态密度图; (b) Fe:Mg:LT晶体分态密度图 图 5 晶体禁带附近分态密度 (a) Fe:LT; (b) Fe:Mg:LT; (c) Fe:Mg(E):LT
图 5 晶体禁带附近分态密度 (a) Fe:LT; (b) Fe:Mg:LT; (c) Fe:Mg(E):LT











 图 6 LT及不同掺Mg浓度的Fe:Mg:LT晶体吸收光谱, 插图(a), (b): Fe:Mg:LT晶体吸收谱实验值[13]
图 6 LT及不同掺Mg浓度的Fe:Mg:LT晶体吸收光谱, 插图(a), (b): Fe:Mg:LT晶体吸收谱实验值[13]






















