全文HTML
--> --> -->有机-无机杂化卤化物钙钛矿材料(ABX3; A = CH3NH3, HC(NH2)2; B = Pb, Sn; X = Cl, Br, I)拥有许多优异的光电性质, 如高的光吸收系数、高的载流子迁移率、长的平衡载流子寿命、低的激子结合能以及大的极化子等, 使其成为新型太阳能电池领域研究的热点[7-10]. 2009年至今, 钙钛矿基太阳能电池的光电转化效率由最初的不足4%提升到了超过23%[11,12]. 但是, 由于这类钙钛矿材料的A位为有机阳离子, 表现出对热、光、电子束等极差的稳定性, 并且包含有毒的铅元素, 阻碍其商业化的大规模应用[10,13]. 目前人们投入大量的科研力量, 在理论和实验上开展了大量工作优化、设计新型钙钛矿, 致力于解决上述问题. 其中一个最简单且可行的策略是: 替换A位、B位或X位上的离子[11, 14-18]. 例如, 利用Cl或Br替换I离子已经被实验证实可以提高材料的稳定性[19]. 采用无机阳离子(Cs+, Rb+等)取代A位有机阳离子的方法也已经有相关的报道[10,14].
全无机钙钛矿CsPbX3 (X = Cl, Br, I)可以通过低成本的溶液旋涂法合成, 具有与有机-无机杂化钙钛矿类似的光电性质, 同时其稳定性得到明显提升[10,20,21]. 但是, 材料中仍然含有有毒的铅. 因此, 寻找适合的无毒或少毒的二价阳离子替换Pb2+是该材料体系研究的一个重要方向. 到目前为止, Sn, Ge, Ba, Mg, Ca, Sr, Mn和Eu等元素已经被应用于B位替换[11,15-17]. 其中, Sn2+, Ge2+和Pb2+同属于第Ⅳ主族, 具有类似的物理化学性质, 是替换Pb2+最有希望的候选者[22,23]. 但是, Sn2+和Ge2+在空气中极易被氧化, 阻碍了其大规模商业化应用[22,23]. Mn2+, Sr2+和Eu2+等二价阳离子在无机钙钛矿中的混溶度较小(< 5%), 且具有较小的带隙调控区间[16,24,25]. 2016年, Edvinsson研究组[26]通过第一性原理计算系统研究了Ca, Sr和Ba三种碱土金属元素替换CH3NH3PbI3中的Pb元素对体系电子结构的影响, 发现较高的导带边位置、宽的带隙以及电子和空穴有效质量的显著差异使其不适合作为光吸收材料, 但是可以用于太阳能电池器件中的载流子选择层材料. 2019年3月, 研究者通过理论和实验双重手段证实, 利用Mg2+替换CsPbBr3中的Pb2+可以提高光致发光量子产率和场致发光效率, 并降低CsPbBr3的卤素空位缺陷[27]. 近几年, 大量的实验和理论文献报道了Ba2+替换CH3NH3PbI3中的Pb2+可以提高稳定性以及调控光电特性[11,15,26,28]. 但是, 对于Ba掺杂全无机CsPbX3钙钛矿的研究尚未见报道.
本文结合无序合金结构搜索方法和第一性原理计算对Ba掺杂CsPbX3 (X = Cl, Br, I)钙钛矿体系的稳定性和光电特性进行了系统地研究. 理论计算结果表明, 高的Ba掺杂浓度下, 可以形成无序固溶体合金CsPb1–xBaxX3 (X = Cl, Br, I); 材料所具有的较大带隙使其在短波段(如紫外或近紫外光)发光二极管或辐射探测器等领域具有潜在的应用价值[29,30].
计算使用基于密度泛函理论的第一性原理计算方法的VASP (Vienna ab-initio simulation package)仿真包[35]. 采用广义梯度近似的Perdew-Burke-Ernzerhof (PBE)交换关联泛函进行结构优化和总能计算[36,37]. 在整个计算中, 平面波基矢的截断能设置为300 eV; 合金相CsPb1–xBaxX3 (X = Cl, Br, I; x = 0.25, 0.5, 0.75)的k点网格密度设为


| 晶格常数/? 理论[实验] | 带隙/eV | |||||||
| a | b | c | PBE | HSE | HSE + SOC | 实验 | ||
| CsPbCl3 | 7.993 [7.902] | 11.365 [11.248] | 7.953 [7.899] | 2.42 | 3.19 | 2.08 | 2.91[14] | |
| CsPbBr3 | 8.388 [8.252] | 11.978 [11.753] | 8.353 [8.203] | 1.99 | 2.67 | 1.57 | 2.27[14] | |
| CsPbI3 | 9.021 [8.845] | 12.768 [12.524] | 8.760 [8.612] | 1.74 | 2.32 | 1.23 | 1.75[20] | |
表1CsPbX3 (X = Cl, Br, I)的晶格常数[14,20]和带隙的理论计算值与实验值的对比
Table1.Experimental lattice parameters and band gaps in comparison with the computational (this work) results for CsPbX3 (X = Cl, Br, I).
自旋轨道耦合(SOC)相互作用作为一种相对论效应, 是原子轨道角动量和电子自旋之间的耦合作用. 在大多数质量较轻的元素中, 自旋轨道耦合作用较弱, 一般不予考虑. 但是, 对于含有重元素(如Bi, Pb, Sb, Hg, Te, Se, Ir, Os等)的化合物体系, 自旋轨道耦合作用较为显著. 在此类化合物的电子结构计算中, 需要把SOC考虑进来[32,39,40]. 一般来说, 对于SOC作用显著的体系, 考虑SOC之后会使得体系的带隙值明显降低[41]. 此外, 普遍认为采用非局域的HSE06[42]交换关联泛函可以得到比PBE更为精确的带隙值. 这是因为PBE[43]方法通常会低估带隙值. 为了获得更为可靠的带隙值, 我们测试了PBE, HSE06和HSE06 + SOC三种方法. 由表1可以发现, HSE06方法高估了带隙值, HSE06 + SOC方法又大大低估了带隙值, 而PBE的结果反而与实验值符合较好. 考虑到HSE06和SOC存在补偿作用, 以及PBE方法较好的准确性和计算效率, 本文在预测带隙大小及变化规律上采用了PBE方法作为交换关联泛函. 同时, 考虑到SQS结构为超胞结构, 计算得到的能带结构存在折叠现象. 我们采用了BandUP软件[44,45]将折叠的能带结构展开到对应的原胞上, 便于对比合金化前后的能带结构变化.
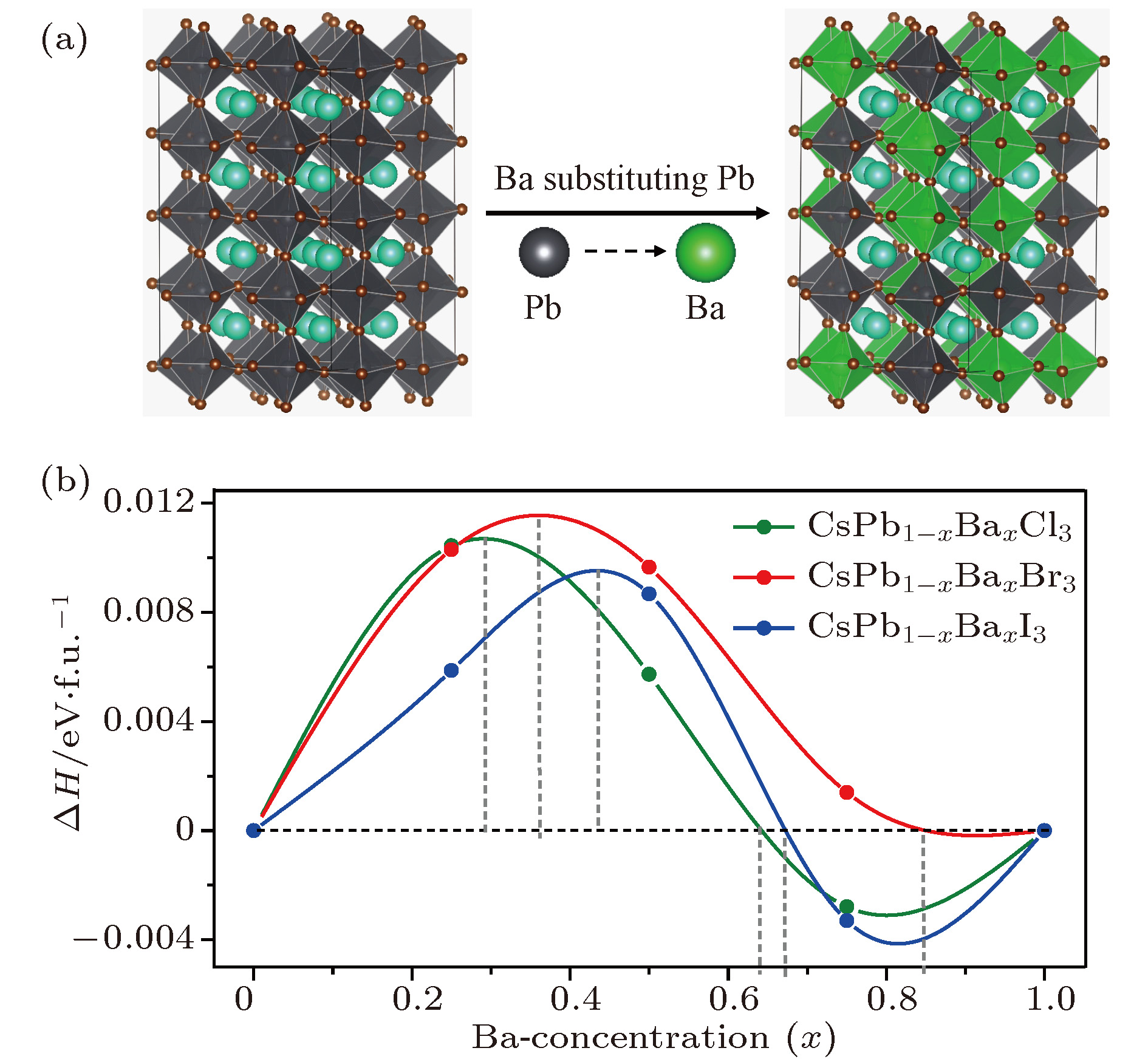 图 1 CsPb1-xBaxX3 (X = Cl, Br, I; x = 0, 0.25, 0.5, 0.75, 1)合金钙钛矿体系的(a)晶体结构示意图, (b)计算得到的形成能
图 1 CsPb1-xBaxX3 (X = Cl, Br, I; x = 0, 0.25, 0.5, 0.75, 1)合金钙钛矿体系的(a)晶体结构示意图, (b)计算得到的形成能Figure1. (a) Schematic diagram of crystal structure, (b) density functional theory-calculated formation energies of the alloyed perovskite CsPb1-xBaxX3 (X = Cl, Br, I; x = 0, 0.25, 0.5, 0.75, 1).
形成能的计算公式如下:
图2为CsPb1–xBaxI3 (x = 0, 0.25, 0.5, 0.75, 1)合金钙钛矿体系的能带结构, 均显示出直接带隙. 随着Ba掺杂浓度增加, 最高占据能级处的带边变得越来越平坦, 导致空穴有效质量显著增大; 从0%, 25%, 50%到75%, 最低非占据能级处的带边也变得越来越平坦, 电子有效质量也在逐步增大, 而CsBaI3的电子有效质量略有减小. 因为Ba掺杂之后[BX6]八面体之间的相互连接的方式发生改变, 由于量子限域效应导致B位原子之间的轨道波函数在空间和能量上的重叠程度减小, 致使能带色散变小. 这些平坦的带边态导致较大的电子/空穴有效质量. 而在纯的CsPbX3和CsBaX3中, 电子维度的三维连续性不会被打破, 所以有效质量较小. 而价带顶和导带底随着Ba掺杂浓度的变化先从Γ点移到S点, 然后又回到Γ点, 是由于结构的对称性改变导致的.
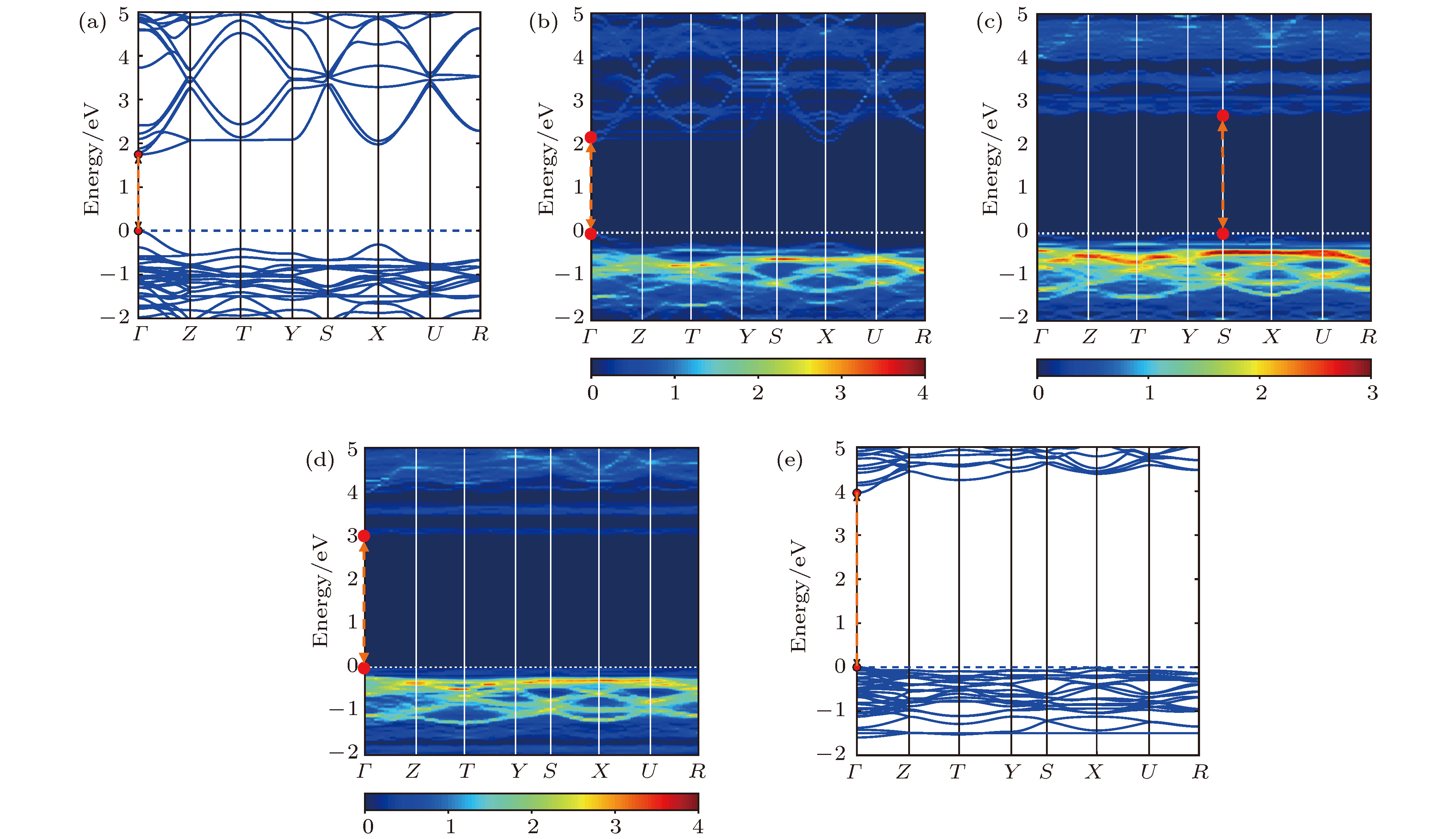 图 2 计算得到的CsPb1–xBaxI3合金钙钛矿体系的能带结构 (a) x = 0%; (b) x = 25%; (c) x = 50%; (d) x = 75%; (e) x = 100%; 其中, 图(b)?(d)是通过能带展开技术得到的, 彩色刻度尺代表指定波矢下穿过能量区间的原胞能带数目
图 2 计算得到的CsPb1–xBaxI3合金钙钛矿体系的能带结构 (a) x = 0%; (b) x = 25%; (c) x = 50%; (d) x = 75%; (e) x = 100%; 其中, 图(b)?(d)是通过能带展开技术得到的, 彩色刻度尺代表指定波矢下穿过能量区间的原胞能带数目Figure2. Calculated band structures of the alloyed perovskite CsPb1–xBaxI3, x = (a) 0%, (b) 25%, (c) 50%, (d) 75%, (e) 100%; panels (b)?(d) are obtained by band unfolding technique. The color scale represents the number of the primitive cell bands crossing the energy interval at a given primitive wave vector.
CsPb1–xBaxX3 (X = Cl, Br, I)合金钙钛矿体系的带隙大小随Ba掺杂浓度的变化规律见图3. 结果显示, 随着Ba掺杂浓度增大, 带隙单调递增, 并存在很宽的变化范围(CsPb1–xBaxCl3, 2.42—5.12 eV; CsPb1–xBaxBr3, 1.99—4.44 eV; CsPb1–xBaxI3, 1.74—3.96 eV). 这是因为Ba和Pb的电负性存在显著差异. Ba—X为离子键, 成键时阳离子外层的电子会完全转移到卤素阴离子上, 成键/反键态相对于它们成键前的原子能级有显著的下/上移, 它们形成的离子晶体相比于Pb-X形成的共价晶体有明显高得多的带隙. 随着Ba掺杂浓度增加, 体系中B—X的成键逐渐从共价型向离子型过渡. 由于体系带隙来自于B—X的成键环境, 随着Ba浓度的增大, 带隙也会逐步过渡到其离子晶体对应的带隙值上. 此外, 同一浓度下, Eg(CsPb1–xBaxCl3) > Eg(CsPb1–xBaxBr3) > Eg(CsPb1–xBaxI3). 其原因是: 从卤素的最外层p轨道的原子能级来看, Cl的最低(Cl-3p: –0.3204 eV), 其次是Br (Br-4p: –0.2953 eV), I具有最高的能级(I-5p: –0.2679 eV). 因此, 当Ba (Ba-6s: –0.1190 eV)与这些卤素形成离子键时, Ba—Cl的成键态-反键态的能级差最大, Ba—I对应的能级差最小. 如此宽的带隙调节范围有利于CsPb1–xBaxX3 (X = Cl, Br, I)合金钙钛矿体系在多种领域的应用, 如太阳能电池、发光二极管、辐射探测器等[14].
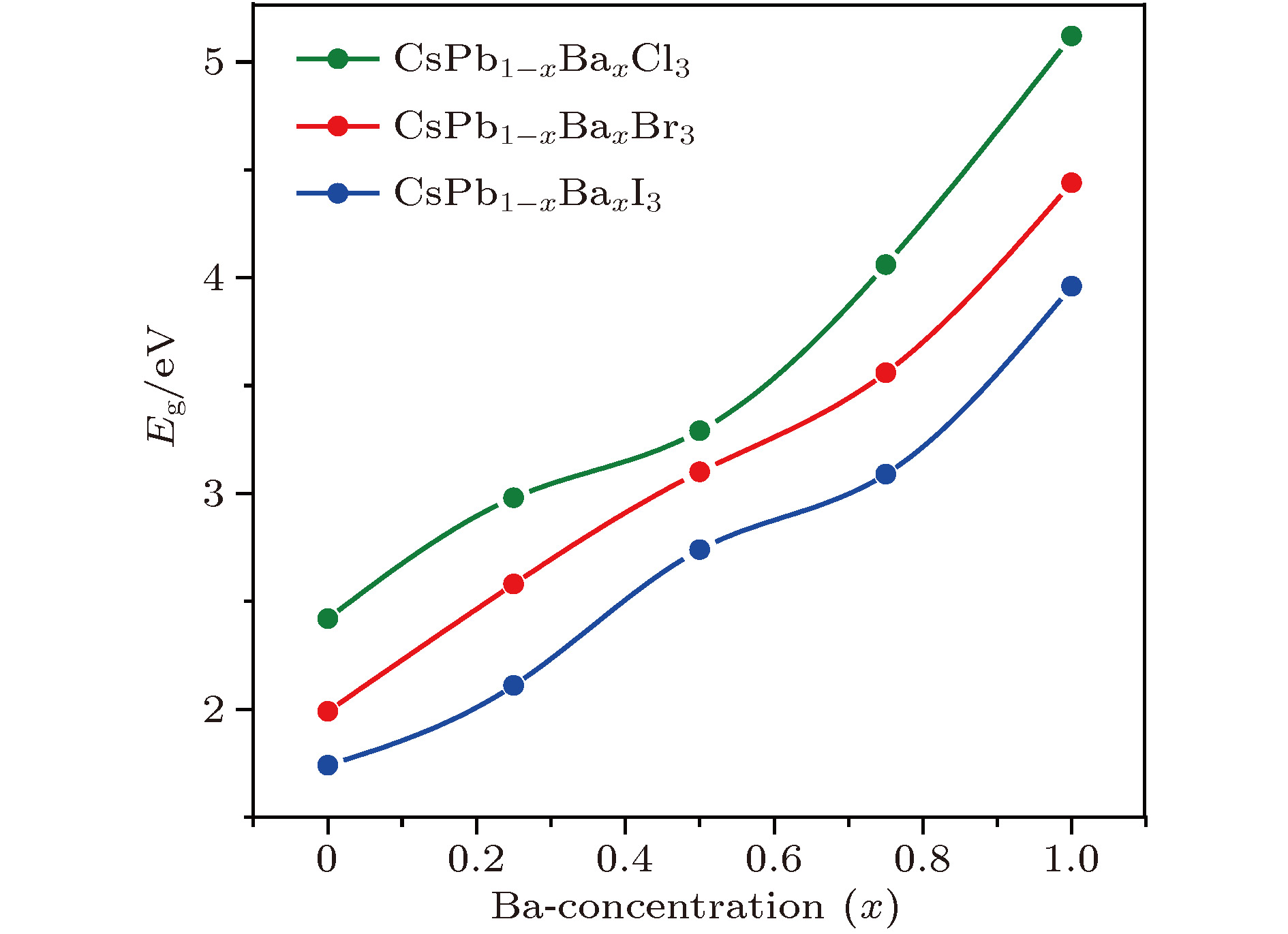 图 3 计算得到的CsPb1–xBaxX3 (X = Cl, Br, I; x = 0, 0.25, 0.5, 0.75, 1)合金钙钛矿体系的带隙变化规律
图 3 计算得到的CsPb1–xBaxX3 (X = Cl, Br, I; x = 0, 0.25, 0.5, 0.75, 1)合金钙钛矿体系的带隙变化规律Figure3. Calculated band gaps of the alloyed perovskite CsPb1–xBaxX3 (X = Cl, Br, I; x = 0, 0.25, 0.5, 0.75, 1) varied with Ba concentration.
为了深入探究带隙增大的成因, 计算了CsPb1–xBaxX3 (X = Cl, Br, I; x = 0, 0.25, 0.5, 0.75, 1)合金钙钛矿体系的投影态密度(图4, 以及补充材料中的图S3和图S4 (online)). 以CsPb1–xBaxI3为例, 价带顶轨道电子贡献主要来源于I-5p和少量Pb-6s杂化(图4(a)—(e)). 在导带的低能区域, 随着Ba掺杂浓度增加, Pb-6p轨道的贡献逐渐减小, 使得s-p轨道耦合作用减小, 把导带底推向更高的能级, 最终导致带隙增大. 由图4(f)可以发现, 随着Ba掺杂浓度增加, 价带的态密度曲线变得越来越陡峭, 且导带边逐渐向高能区移动. 需要注意的是, 这里都以价带顶作为参考点, 能级设置为0. 实际的价带边的位置是各不相同的, 带隙增大的真实情况可能是价带下移和导带上移共同作用的结果. 同时发现, Ba元素对体系态密度的贡献很小. 但是, 由于Ba—X离子键的化学相互作用强于Pb—X共价键, 可以有效减少钙钛矿体系中普遍存在的卤素空位缺陷.
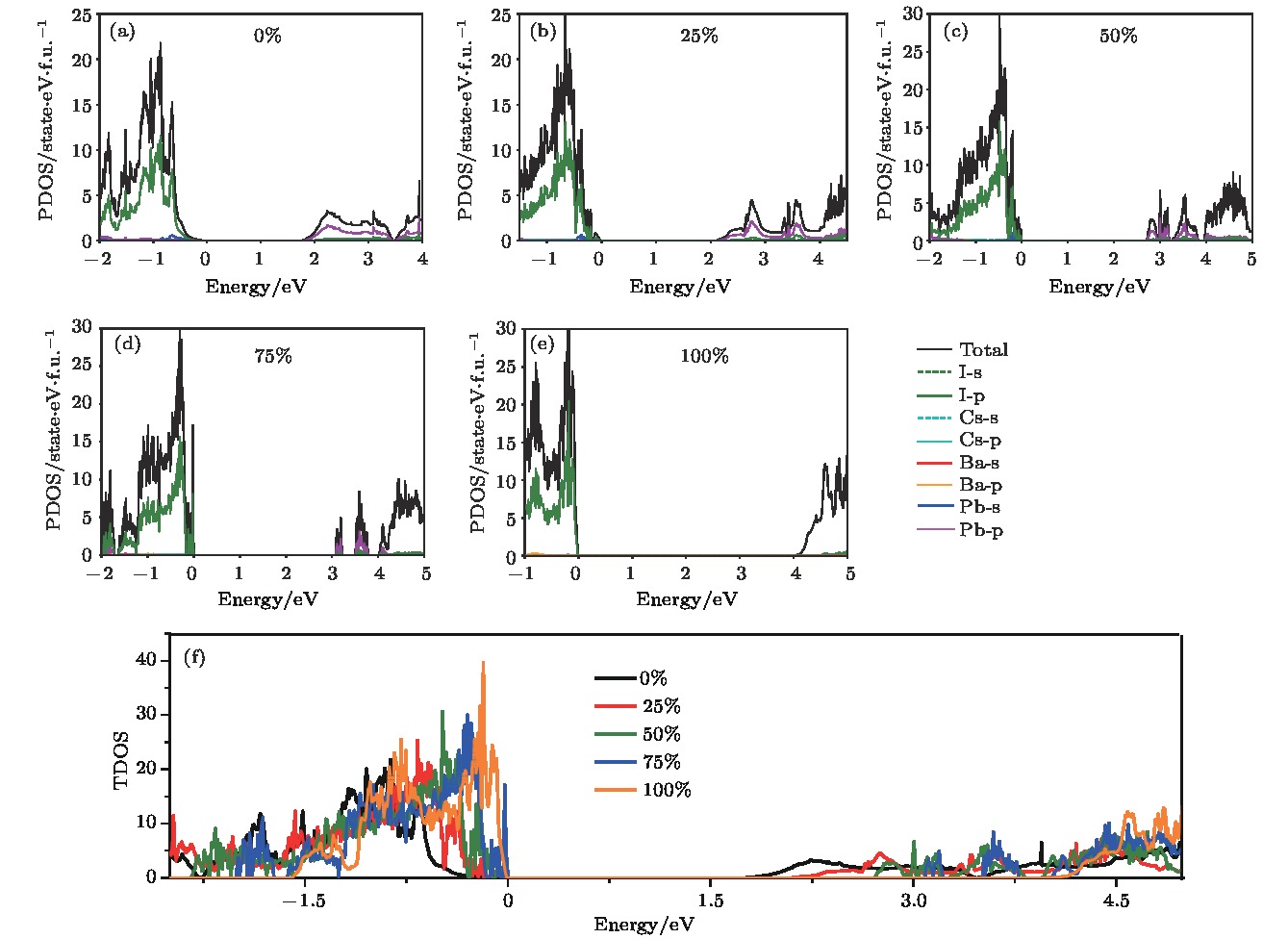 图 4 计算得到的CsPb1–xBaxI3 (x = 0, 0.25, 0.5, 0.75, 1)合金钙钛矿体系的(a)?(e)投影态密度, (f)总态密度
图 4 计算得到的CsPb1–xBaxI3 (x = 0, 0.25, 0.5, 0.75, 1)合金钙钛矿体系的(a)?(e)投影态密度, (f)总态密度Figure4. Calculated (a)?(e) Atomic-orbital-projected density of states (PDOS), (f) total density of states (TDOS) of the alloyed perovskite CsPb1–xBaxI3 (x = 0, 0.25, 0.5, 0.75, 1).
CsPb1–xBaxX3合金钙钛矿是离子键和共价键的混合体系. CsPb1–xBaxI3的价带顶和导带底处电荷密度分布如图5所示. 价带顶部(VBM), 电子主要集中在I和Pb原子周围; 导带底部(CBM), 电子主要聚集在Pb原子周围. Ba原子引入后, 由于Ba最外层电子的转移, I周围的电子浓度升高, 使得Pb—I键的库仑相互作用增强. 同时, 电子和空穴都倾向于局域在Pb原子周围, 可以形成类似于量子阱结构的局部势阱, 并进一步引起杂质周围的晶格变形以适应自陷激子. 这同样归因于Ba与Pb相比具有较大的原子半径和较小的电负性. 由于自陷激子的热猝灭激活能显著高于自由激子, 致使量子产率显著提高[47]. 这种结构与传统的二维钙钛矿量子阱结构相似, 具有较大的激子结合能和较高的量子产率[48-52]. 但是有所区别的是, 它不像二维钙钛矿量子阱结构有特定的排列规律和维度. 此外, 在CsPb1–xBaxX3合金体系中, 可以通过调整Ba掺杂浓度来对发光特性进行人为调控.
 图 5 计算得到的CsPb1-xBaxI3合金钙钛矿体系的部分电荷密度分布图样 (a), (f) 0%; (b), (g) 25%; (c), (h) 50%; (d), (i) 75%; (e), (j) 100%
图 5 计算得到的CsPb1-xBaxI3合金钙钛矿体系的部分电荷密度分布图样 (a), (f) 0%; (b), (g) 25%; (c), (h) 50%; (d), (i) 75%; (e), (j) 100%Figure5. Calculated partial charge distribution patterns of the alloyed perovskite CsPb1-xBaxI3: (a), (f) 0%; (b), (g) 25%; (c), (h) 50%; (d), (i) 75%; (e), (j) 100%.
图6显示了CsPbX3和CsPb0.25Ba0.75X3 (X = Cl, Br, I)合金钙钛矿的光吸收谱. 在吸收带边处, 光吸收系数均可达到105 cm–1, 当距离吸收阈值约0.5 eV处, 光吸收系数高达106 cm–1. 目前, CsPbBr3已经应用于太阳能电池器件中[10,53], 而CsPbI3-Pnma拥有比CsPbBr3更合适的带隙和更强的可见光吸收. 因此, 通过B位合金化形成的Pnma相的CsPb1–xBaxI3固溶体来提高能量转换效率和保持材料的长期稳定性是一种非常可行的方法.
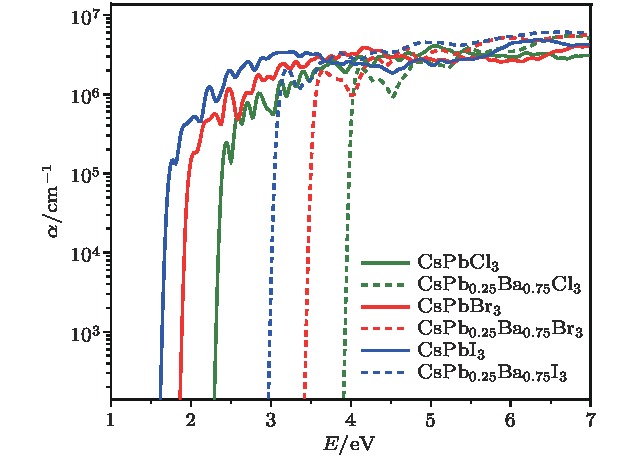 图 6 计算得到的CsPbX3和CsPb0.25Ba0.75X3 (X = Cl, Br, I)合金钙钛矿的光吸收谱
图 6 计算得到的CsPbX3和CsPb0.25Ba0.75X3 (X = Cl, Br, I)合金钙钛矿的光吸收谱Figure6. Calculated photo absorption spectra of the perovskite CsPbX3 and CsPb0.25Ba0.75X3 (X = Cl, Br, I).
