全文HTML
--> --> -->
原子间的相互作用通常是很弱且短程的范德瓦耳斯作用, 即使Feshbach共振技术可以对相互作用强度进行任意调控[3], 也并不能改变这一特性. 原子间相互作用的这些特性带来了一些限制, 很多凝聚态体系中非常重要的问题, 目前在超冷原子体系中还很难实现. 比如量子磁性问题, 由于原子间相互作用强度太小, 为了在光晶格中实现和观测磁性相变, 需要远远低于现有超冷技术能达到的温度. 目前, 很多研究组还在持续探索在超冷原子中获得更低温度的方法. 另一方面, 如果可以找到一种更强的、长程的相互作用, 也可以帮助解决这一问题. 超冷极性分子间的电偶极相互作用就可以满足这样的要求[4-11](一些特殊的镧系和过渡金属元素, 包括铬[12]、铒[13]和镝[14], 有很大的磁偶极矩, 因而可以有磁偶极相互作用; 磁性原子也是超冷物理研究的热点之一, 但不在本文讨论范围之内).
本文讨论的极性分子是指最简单的由两种不同原子结合形成的异核中性双原子分子, 比如钠(23Na)原子和铷(87Rb)原子. 由于两种原子对电子的亲和力不同, 对整个分子而言, 电荷的分布是不对称的, 导致分子中的正负电荷中心不重合, 因而产生了一个沿着分子轴向的永久电偶极矩. 需要注意的是, 由于电偶极算符是奇宇称的, 在没有外加电场的情况下, 极性分子在实验室坐标系中的电偶极矩的期望值为零, 因而分子间电偶极相互作用平均值为零. 外加电场可以将分子内部不同宇称的态混合, 或者说将分子极化, 从而诱导出沿着电场方向的有效电偶极矩


 图 1 电场诱导23Na87Rb分子产生的有效电偶极矩
图 1 电场诱导23Na87Rb分子产生的有效电偶极矩Figure1. The induced effective dipole moments of the first two rotational states versus the electric field (23Na87Rb).
如图2所示, 两个在电场中被极化的极性分子间的相互作用可以表示为[8]
 图 2 极性分子在电场中可以产生电偶极相互作用
图 2 极性分子在电场中可以产生电偶极相互作用Figure2. Dipole-dipole interaction between polar molecules in an external electric field.







相比范德瓦耳斯作用, Udd的强度要大很多. 例如, 把

总之, 对超冷极性分子体系的理论研究目前已经进行得非常深入, 但实验研究却大大滞后. 造成这种状况的原因主要有两个: 1)基态超冷极性分子非常难制备; 2)基态分子间的碰撞也比预期的复杂很多. 下面几个部分, 我们将围绕这两个问题, 对过去10年间的一些进展进行总结.
需要指出的是, 目前实验上已经有很多不同制备基态冷分子的方法. 近年, 分子的直接冷却, 特别是对一些特殊的自由基分子的直接激光冷却, 取得了长足的进步[20-24], 但目前得到的分子样品温度还没有达到

2.1.研究现状
目前实验中用到的超冷原子, 都是首先通过激光冷却然后再进行蒸发冷却来获得的. 由于原子的能级结构简单, 比较容易找到接近闭合的两能级体系, 在它们之间进行连续的光子散射, 可以很快把原子从高温冷却到mK以下, 然后装载到磁阱或光阱中进行蒸发冷却. 由于磁阱和光阱的阱深都很浅, 必须经过激光冷却之后原子才能被囚禁住. 分子的情况就复杂得多, 由于分子有很多振动和转动能级, 一般来讲, 不存在闭合的两能级体系, 因此每一个基态分子吸收和散射几个光子之后, 就会转移到暗态, 导致其与冷却光的作用停止. 由于散射的光子数目太少, 不会使分子的温度显著降低, 以致磁阱和光阱无法囚禁.为了克服这一问题, 过去的20年间, 很多研究组已经进行了不同的尝试, 也发展了几种技术. 大体上讲, 可以将这些技术分为两大类: 1)对已经存在的基态分子直接用各种方法冷却; 2)用间接的方法, 从超冷原子开始, 利用光或者磁缔合技术合成超冷分子[27,28].
3
2.1.1.基态分子直接冷却
在自然状态下, 绝大多数分子都处于基态, 因此只要把它们的温度降低就可以获得冷分子. 在各种冷却技术中, 由Harvard大学Doyle研究组[29]发展的缓冲气体冷却(buffer gas cooling)是一种通用的技术, 可以对各种分子进行冷却, 但这种技术得到的温度由缓冲气体决定, 一般在4 K左右(液氦), 对这些分子直接进行保守势阱装载并进一步蒸发冷却仍很困难. 另一方面, 利用这种技术可以很快地冷却大量的处于最低振转能级的分子, 将它们引出缓冲气体冷却装置后, 可以获得亮度很高的分子束[30], 为冷分子光谱提供了非常好的条件. 其他实验如直接激光冷却, 通常也需要先以此对分子进行预冷却. 目前, 国内华东师范大学和浙江大学的两个研究组已经掌握了这一技术, 并成功制备了基态MgF[31]和BaF[32]冷分子.另一项研究得很广泛的技术是Stark(斯塔克)减速技术[33]. 这种技术利用与分子束运动同步的交变高电场产生随时间变化的Stark效应来降低分子的运动速率, 因而只对极性分子有效. 由于这一方法涉及对高电场的快速操控, 技术上很有挑战性. 利用这一技术, 目前分子的温度可以达到几十mK量级, 而样品的密度还较低[34]. 国内华东师范大学的研究组在这一方向做了多年的理论和实验工作, 取得了一系列成果[35,36]. 南通大学的研究组近期也开始了这方面的探索[37].
最近几年, 对一些特殊分子的直接激光冷却这一方向发展非常迅猛. 如SrF, YO, CaF等自由基分子, 可以找到与振动基态(v" = 0)闭合性很高的分子振动激发态(Franck-Condon因子在95%以上), 因而在光子散射后, 分子从激发态经自发辐射回到基态只占据少数几个振动态[38]; 另一方面, 对处于振动基态的第一激发转动态(J′′ = 1)的分子, 由于角动量的选择定则, 冷却跃迁 J′′ = 1 ? J = 0对转动能级是完全闭合的, 即基态 J′′ = 1的分子被光子激发到J = 0后, 自发辐射只能回到基态J′′ = 1[39]. 由于这两个原因, 对这些特殊分子, 只需利用少数几个回泵激光, 就可以获得准闭环系统, 使分子能够散射足够多的光子, 从而实现冷却.
分子的直接激光冷却最早是由Yale大学的DeMille研究组[20]实现的. 目前已经有很多研究组在这个方向开展工作, 包括美国的JILA[24], Harvard[23], 以及英国的Imperial College London[40]等的研究组最近都取得了很多突破. 例如在最新发表的文章中, Harvard大学的研究组已经可以将CaF分子的温度冷却到20

3
2.1.2.利用超冷原子缔合获得超冷分子
由于超冷原子的制备技术非常成熟, 结合光或者磁缔合技术, 就可以合成超冷分子[27]. 其中光缔合技术非常适合用于进行超精细分子光谱的研究[42], 国内山西大学的研究组多年来在此方向做了很多的工作[43,44]. 光缔合之后的自发辐射可以形成电子基态的分子, 但这些分子一般分布在很多个振动态, 难以进行分子态的纯化. 而利用磁缔合技术形成的分子都处于同一能级, 因而更加适合进行后续的研究. 利用磁缔合技术获得的分子温度等于超冷原子的温度, 而且分子密度也较高, 但分子处于弱束缚态, 需要用受激拉曼过程才能转移到基态. 目前, 从相空间密度的角度(即最高密度和最低温度), 超冷原子磁缔合结合受激拉曼转移是最成功的制备基态超冷极性分子的方法[45]. 利用这一方法, 最近JILA的研究组已经成功实现了钾铷(40K87Rb)分子的费米简并[26,45], 而奥地利的Innsbruck (87Rb133Cs)[46], 英国的Durham (87Rb133Cs)[47], 美国麻省理工学院(MIT)(23Na40K)[48], 德国马普研究所(MPQ)(23Na40K)[49], 以及中国的香港中文大学(23Na87Rb)[50]和中国科学技术大学(23Na40K)[51]都已经成功地实现了基态超冷极性分子的制备. 下面我们将以23Na87Rb分子为例对这一方法进行更详细的讨论.2
2.2.Feshbach共振和磁缔合
两个碰撞原子间的Feshbach共振通常可按图3(a)所示的两通道模型来理解. 对碱金属, 这里的通道对应于两种原子的超精细能级的不同组合. 如果在不同通道中的两个原子以及它们形成的分子对磁场的响应不同, 在一定的磁场下, 在通道1中的两个原子的散射阈值就有可能和通道2的束缚态能级交叉, 从而引起共振散射, 即Feshbach共振. 在共振磁场B0附近, 散射长度a随磁场B的变化为[3] 图 3 (a)Feshbach共振的两通道模型; (b)利用Feshbach共振进行磁缔合产生弱束缚分子
图 3 (a)Feshbach共振的两通道模型; (b)利用Feshbach共振进行磁缔合产生弱束缚分子Figure3. (a) Two-channel model for a Feshbach resonance; (b) formation of a weakly bound Feshbach molecule by magnetoassociation.

Feshbach共振的另一个重要应用是磁缔合. 如图3(b)所示, 磁缔合可以用Landau-Zener过程来理解. 在共振附近, 由于两个通道之间的耦合, 在能量交叉处产生了一个劈裂, 劈裂的大小由耦合强度决定, 一般和Feshbach共振的性质有关. 如果初始时磁场处于共振之上, 当把磁场绝热地扫描经过共振后, 两个原子就会缔合为一个Feshbach分子. 注意在共振的两端, 散射长度的符号相反, 分子处于散射长度为正的一端. 磁缔合是一种非常强大的技术, 它可以将各种相同或不同的玻色子、费米子及其混合物缔合为分子, 特别是在两分量的费米子中, a > 0和a < 0两端分别对应BEC(两个费米子合成的分子为玻色子)和BCS(Bardeen-Cooper-Schrieffer)区, 可以用来研究BEC和BCS crossover(跨接效应)的相关物理.
就超冷分子的研究而言, 磁缔合需要在两种原子的混合气体中进行, 这是一个很复杂的过程, 目前还很难达到很高的转换效率. 比如在23Na87Rb分子的实验中, 初始时23Na和87Rb原子的数目都约为2 × 105, 而经过磁缔合后获得的Feshbach分子只有104, 对应的转换效率只有5%. 其他类似实验上得到的转换效率也都在5%到10%[45-49]. 如何提高转换效率, 从而得到更多的分子是一个有待解决的问题. 另外, 剩余的原子可以和分子发生碰撞, 从而引起分子的损耗, 因而必须尽快把它们从光阱中移除.
23Na和87Rb之间的Feshbach共振位于347.75 G附近[52,53], 磁场从高到低扫描经过共振后, 一部分原子被缔合为分子, 如图4(a)所示. 利用分子和原子对磁场的相互作用不同, 可以加一个磁场梯度将剩余的原子从阱中移除, 而分子则不受影响. 探测是在磁场关断后, 对原子进行吸收成像. 在实验中, 为了确认磁场扫描共振后形成了Feshbach分子, 可以用如图4(a)所示的时序, 在把剩余的原子移除后直接关掉磁场, 利用吸收成像探测看不到信号, 这是由于探测光是与原子跃迁共振的, Feshbach分子对它的散射截面很小. 只有当再做一个反向的磁场扫描, 把不能直接探测到的分子离解成原子后, 才能重新看到信号, 如图4(b)所示, 这样就证明磁缔合形成了Feshbach分子.
 图 4 利用磁缔合制备23Na87Rb Feshbach分子 (a)磁场改变的时序; (b)分别在离解和不离解的情况下探测钠和铷原子[52]
图 4 利用磁缔合制备23Na87Rb Feshbach分子 (a)磁场改变的时序; (b)分别在离解和不离解的情况下探测钠和铷原子[52]Figure4. Creation of 23Na87Rb Feshbach molecule via magnetoassociation: (a) The magnetic field sequence; (b) absorption images of 23Na and 87Rb with and without dissociation procedure[52].
在共振附近, Feshbach分子的结合能随磁场失谐的增大而增加. 一般而言, 对于两通道耦合较强的共振, 亦即所谓的宽共振, Feshbach分子的结合能Eb随散射长度的变化可以近似地表示为
 图 5 23Na87Rb Feshbach分子的结合能随磁场的变化, 插图表示离共振越远, 分子的closed channel成分越多[52]
图 5 23Na87Rb Feshbach分子的结合能随磁场的变化, 插图表示离共振越远, 分子的closed channel成分越多[52]Figure5. Binding energy of 23Na87Rb Feshbach molecules versus magnetic field near 347.7 Gauss. The inset shows the closed-channel fraction of the Feshbach molecule versus magnetic field[52].
通过磁缔合形成的Feshbach分子处于分子的最低电子能态, 但处于振转能级的高激发态. 比如23Na87Rb分子的电子基态,

 图 6 异核碱金属双原子分子的电偶极矩和核间距(a0为玻尔半径)的关系, 图中红色空心三角为23Na87Rb分子
图 6 异核碱金属双原子分子的电偶极矩和核间距(a0为玻尔半径)的关系, 图中红色空心三角为23Na87Rb分子Figure6. Electric-dipole moment of heteronuclear molecules as a function of internuclear distance (a0 is the Bohr radius). The red up-pointing triangle is 23Na87Rb molecule.
2
2.3.受激拉曼转移的方案选择及分子光谱研究
以23Na87Rb分子为例, 图7(a)为其相关能态的势能曲线, 其中Feshbach分子和振转基态分子的结合能差约为5000 cm–1, 为了在完成转态的同时把这部分能量带走, 通用的方法是利用一个以Feshbach分子和基态分子为初态


 图 7 (a) 23Na87Rb分子的相关势能曲线; (b)分子中间激发态的超高分辨谱; (c)基态转动能级; 其中
图 7 (a) 23Na87Rb分子的相关势能曲线; (b)分子中间激发态的超高分辨谱; (c)基态转动能级; 其中




Figure7. (a) 23Na87Rb molecule potential energy curves and the two-photonRaman process forpopulation transfer,




考虑到分子初末态的一些性质, 中间态能级的选取需要满足以下条件: 1)基于Franck-Condon原理, 分子能级跃迁强度正比于上下能级振动波函数重叠的平方







方案确定之后就需要进行分子光谱的研究以及找到具体的能级位置. 用超冷分子做光谱实验可以达到极高的分辨率, 包括所有跃迁定则允许的超精细结构(图7(b)), 这是之前的传统分子光谱学无法达到的. 但如果已经有一些其他方法获得的分子光谱数据和理论计算, 对加快这一十分耗时的步骤有很大帮助. 另外, 原则上可以从自由原子开始, 用光缔合技术研究激发态的光谱, 但Feshbach分子的束缚-束缚跃迁更强, 找到能级信号更快.
找到可能的中间态之后, 就可以利用Autler-Townes效应来确定振转基态的能级位置. 首先将耦合Feshbach分子和中间态的pump光(抽运光)保持共振, 这时应该看到Feshbach分子的损耗, 然后同时打开链接中间态和基态的dump光(倾泻光), 并扫描其频率. 当dump光与基态振动能级共振时, 如果其光强较大, 由于AC-Stark效应, 会导致激发态能级产生劈裂, 从而使pump光失谐. 在这种情况下, 基态能级的信号表现为Feshbach分子损耗的减少(图7(c)) .
2
2.4.窄线宽激光器和受激拉曼转移
在超精细结构完全可分辨的情况下, 图7(a)所示的受激拉曼转移过程是一个标准的由









在超冷分子中实现态转移, 目前通用的技术为受激拉曼绝热通道(stimulated Raman adiabatic passage, STIRAP), 这种受激拉曼技术需要对


 图 8 利用STIRAP制备23Na87Rb基态分子 (a) STIRAP过程中Feshbach分子数目随时间的变化; (b)同一实验中pump和dump激光器的拉比频率随时间的变化; 为了探测基态分子, 需要一个逆过程将分子从基态转移回Feshbach 能级[50]
图 8 利用STIRAP制备23Na87Rb基态分子 (a) STIRAP过程中Feshbach分子数目随时间的变化; (b)同一实验中pump和dump激光器的拉比频率随时间的变化; 为了探测基态分子, 需要一个逆过程将分子从基态转移回Feshbach 能级[50]Figure8. Creation of 23Na87Rb molecules in the rovibrational ground state via STIRAP: (a) Time evolution of the 23Na87Rb Feshbach molecule number during a round-trip STIRAP, the reversed STIRAP is necessary for detection; (b) the pump and dump beam Rabi frequency during the STIRAP pulse sequence[50].


为了实现高效率(接近100%)的分子转移, STIRAP过程必须满足绝热条件. 这需要双光子的有效拉比频率







由于


图8为利用STIRAP技术从Feshbach分子开始制备23Na87Rb基态分子的过程. 这里的实验探测手段为标准的吸收成像技术, 由于散射截面过小, 这一技术不能直接探测基态分子, 因此所有的信号都是通过探测Feshbach分子获得的. 如图8(a)所示, 初始时全部分子都处于Feshbach能级, 这时打开dump光对分子数没有影响, 但在dump光关断的同时打开pump光时, Feshbach分子数随之减少, 直到为零. 这是由于分子被转移到了基态, 不能被吸收成像探测到. 而之后再进行一个逆过程, 将分子从基态转移回Feshbach能级, 就可以看到分子数大部分恢复了. 一般可以假设两个过程的转换效率一样, 因此可以从初始的分子数N0和恢复的分子数N估计出单次的转移效率和基态分子数分别为


目前已知的几个实验中, STIRAP的单次转移效率都在90%以上[46-48, 50]. 转换效率没有达到100%的原因可能是由于激光器的一些没有被压制的噪声, 例如在普遍采用的半导体激光器中, 增益介质的背景频谱很宽, 不能被反馈完全压制. 没有被转移到基态的10%左右的分子, 有可能是在转移过程中被激发到了中间态, 通过自发辐射, 这部分分子会分布到电子基态的很多振转能级. 实验上已经证实, 这一小部分分子对基态分子的动力学并不产生可观测的影响, 因此90%以上的转换效率, 已经完全可以接受.
2
3.1.极化率
改变制备和探测这两个STIRAP过程中间的时间, 记录分子在光阱中的质心位置变化, 就可以测量基态分子在光阱中的振荡频率, 如图9所示. 再结合光阱的光强, 就可以计算出在光阱波长下基态分子的极化率[58], 并与ab initio计算结果比较. 图 9 通过质心振荡运动测量基态23Na87Rb分子的囚禁频率
图 9 通过质心振荡运动测量基态23Na87Rb分子的囚禁频率Figure9. Center-of-mass motion of the absolute ground-state molecules along the horizontal direction (Xc) and the vertical direction (Yc) in the optical dipole trap.
2
3.2.永久电偶极矩
另外一个非常重要的参数是基态分子的永久电偶极矩, 它可以通过测量基态分子在电场中的Stark效应来获得. 在23Na87Rb实验中, 电场是由一对加在真空玻璃腔外有ITO镀膜的导电玻璃平板电极提供的. 由于Feshbach分子的电偶极矩为零, 其能级不随电场变化, 基态分子的Stark频移就表现为pump和dump光频率差的变化. 图10所示为NaRb分子在电场中从0到1.5 kV/cm的Stark频移, 由拟合得到的永久电偶极矩为3.2(1) Debye. 另外也可以看到, 在1.5 kV/cm时, 有效电偶极矩已经达到了1.06 Debye .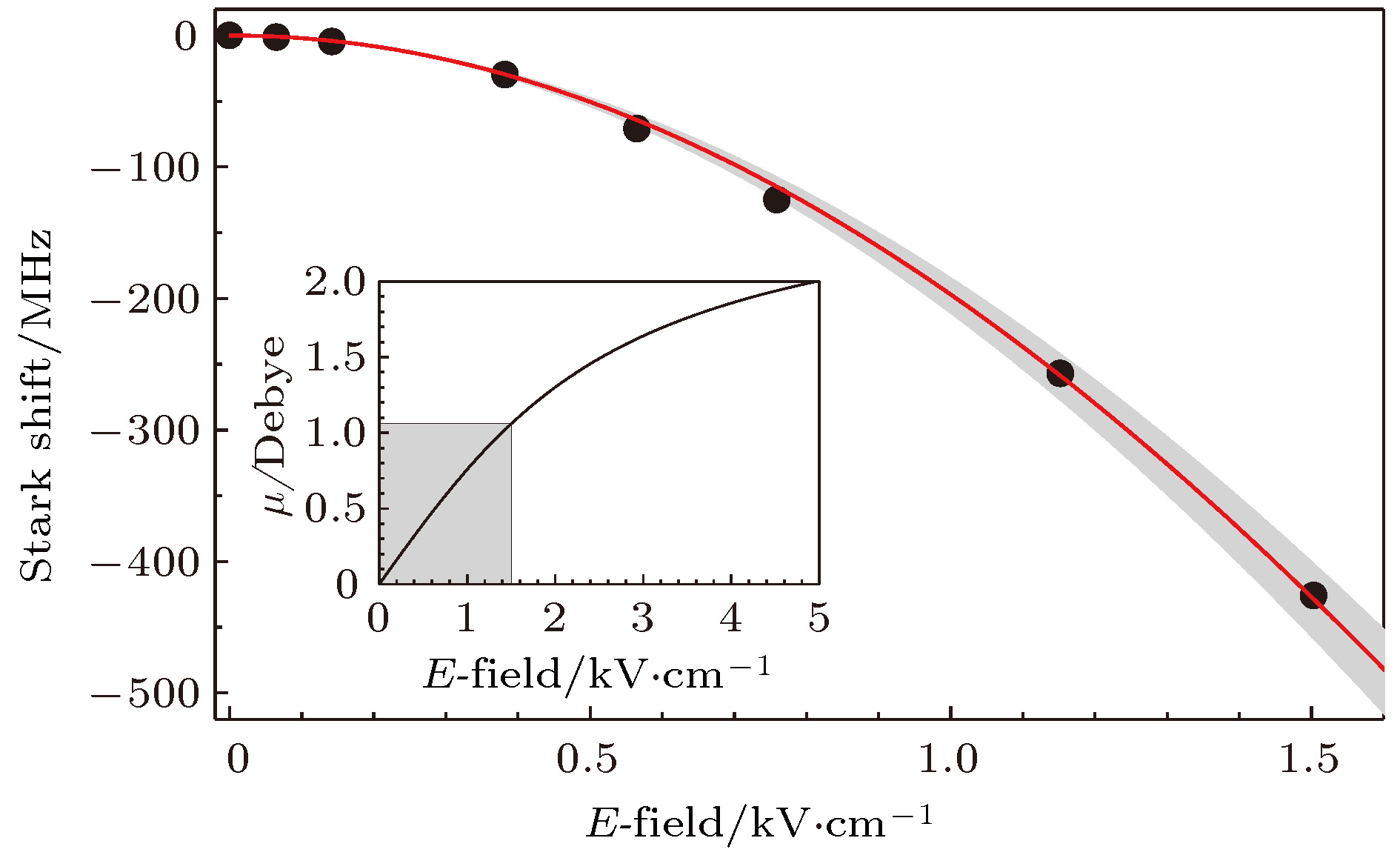 图 10 基态23Na87Rb分子在直流电场中的Stark频移. 红色曲线为对数据点的拟合. 插图为有效电偶极矩和电场的关系[50]
图 10 基态23Na87Rb分子在直流电场中的Stark频移. 红色曲线为对数据点的拟合. 插图为有效电偶极矩和电场的关系[50]Figure10. Stark shift of the rovibrational ground state 23Na87Rb molecule in electric field. The red curve is the fit to a model including contributions from several higher rotational levels. The inset shows the induced dipole moment vs the electric field with the currently accessible region marked by the shading area[50].
2
3.3.振动和超精细能级操控
由于
 图 11 (a) 基态23Na87Rb分子在340 Gauss磁场中的超精细结构, 其中
图 11 (a) 基态23Na87Rb分子在340 Gauss磁场中的超精细结构, 其中



Figure11. (a) The calculated hyperfine Zeeman structures of the lowest rovibrational level of NaRb molecule at 340 Gauss,


在23Na87Rb分子实验中, 由于使用的磁场较高, 再加上光偏振和跃迁选择定则的限制, 能量最低的超精细结构和相邻能级间的劈裂有几百kHz, 如图11(b)所示. 在两束拉曼激光的拉比频率都为1 MHz左右时, 实验中获得的双光子跃迁的半高宽为200 kHz, 足以分辨这些能级从而制备100%纯度的绝对基态分子(即处于最低电子、振动、转动、和超精细能级的分子).
在后续实验中, 经常需要制备处于转动激发态J = 1的分子或者J = 1和J = 0的叠加态. 在目前的几个实验中, 由于选择定则的限制, STIRAP过程不能直接制备J = 1的分子, 这一问题可以通过微波操控来解决. 对极性分子而言, 从J = 0到J = 1能级间的跃迁是由微波的电场和分子的永久电偶极矩直接作用来驱动的, 具有较高的跃迁强度, 因而用标准的微波器件就可以获得较大的拉比频率. 在23Na87Rb分子中, J = 0到J = 1的能量差为4.179 GHz, 对应的微波信号可以方便地用一个信号发生器和标准的微波天线获得.
微波的另外一个重要应用是操控分子的超精细结构. 一般而言, 由于核自旋与微波磁场的作用很微弱, 微波操控核自旋需要很大的微波功率. 但如图12所示, 由于电子四极矩作用以及核自旋和转动角动量的耦合, 使得不同的



 图 12 (a) 23Na87Rb分子的J = 0和J = 1转动态具有不同核自旋态的成分; (b)利用单光子微波(上), 和双光子微波(中和下)操控, 可以实现对转动能级和核自旋的操控[59]
图 12 (a) 23Na87Rb分子的J = 0和J = 1转动态具有不同核自旋态的成分; (b)利用单光子微波(上), 和双光子微波(中和下)操控, 可以实现对转动能级和核自旋的操控[59]Figure12. (a) 23Na87Rb molecule rotational states with J = 0, 1 consist of different nuclear spin components; (b) coherent manipulation with microwave pulses shows the observed Rabi oscillations for the three microwave transitions in (a)[59].
4.1.超冷基态分子的稳定性
如前所述, 超冷原子到Feshbach分子磁缔合的效率不高, 这就导致虽然初始原子混合物的相空间密度很高, 甚至是两团气体都为量子简并, 由此产生的Feshbach分子和基态分子的相空间密度还是远低于量子简并. 因此, 为了获得量子简并的超冷极性分子, 需要进一步降低温度和增加密度, 这些要求只能通过分子的蒸发冷却来实现.蒸发冷却需要通过分子间的两体碰撞来进行. 与原子的情况类似, 分子的碰撞也可分为好的弹性碰撞和坏的非弹性碰撞, 只有弹性碰撞才可能在损耗粒子数目的同时增加相空间密度, 而非弹性碰撞在损耗粒子数目的同时有加热效应 (anti-evaporation effect), 因此并不能引起相空间密度的增加. 一般而言, 在超冷气体中, 弹性和非弹性碰撞总是同时存在, 但只要弹性碰撞对非弹性碰撞的比值足够大, 高效率的蒸发冷却就可以进行.
基于这样的认识, 在超冷分子实验的早期, 研究者们就已经认识到分子必须被制备到绝对基态才可能避免由于激发态弛豫引起的非弹性损耗, 可能的过程包括激发态振动、转动能级的弛豫以及超精细能级弛豫. 绝对基态的分子没有这些可能的损耗通道, 但2008年超冷基态极性分子第一次在JILA的40K87Rb实验中被成功制备后, 还是发现有很大的损耗. 随后的研究表明, 虽然对每一个分子来说, 绝对基态是其能量的最低点, 两个分子却可能发生化学反应.
两个相同的分子AB碰撞后, 从组合来说, 可能发生的过程有以下几种(图13):
 图 13 (a) 40K87Rb分子和(b) 23Na87Rb分子体系两体反应的相关能级示意图, 两个基态40K87Rb分子间可以发生(6c)式中的化学反应, 两个基态23Na87Rb分子是化学稳定的, 将23Na87Rb分子制备到振动激发态, 可以允许化学反应发生
图 13 (a) 40K87Rb分子和(b) 23Na87Rb分子体系两体反应的相关能级示意图, 两个基态40K87Rb分子间可以发生(6c)式中的化学反应, 两个基态23Na87Rb分子是化学稳定的, 将23Na87Rb分子制备到振动激发态, 可以允许化学反应发生Figure13. Schematic energy-level diagram for chemical reactivity of (a) 40K87Rb molecules and (b) 23Na87Rb molecules. The schematic reaction coordinates for the 40K87Rb + 40K87Rb → 40K2 + 87Rb2 process is exothermic and thus allowed. But the same process is endothermic for 23Na87Rb and thus forbidden. For 23Na87Rb in the first excited rovibrational level (v = 1, J = 0), the same reaction is already exothermic and thus allowed.
超冷实验常用的5种碱金属原子6Li, 23Na, 40K, 87Rb, 133Cs, 可以两两组合出10种异核双原子体系. 经过比较它们和5种同核分子的基态结合能, 可以发现化学反应(6c)式可以在40K87Rb分子间发生[60-62], 而不能在v = 0态的23Na87Rb分子间发生.
2
4.2.费米40K87Rb分子间的化学反应
如图13(a)所示, 40K87Rb + 40K87Rb的结合能比40K2 + 87Rb2要小约17 cm–1, 即两个基态40K87Rb分子碰撞的阈值比基态40K2 + 87Rb2的能量高约17 cm–1, 40K87Rb (v = 0) + 40K87Rb (v = 0) → 40K2 (v = 0) + 87Rb2(v = 0)为放热反应, 可以发生.在40K87Rb分子的碰撞实验中, 如图14(a)所示, 观察到基态分子在光阱中的密度n随囚禁时间损耗很快, 而且损耗曲线与两体碰撞速率方程
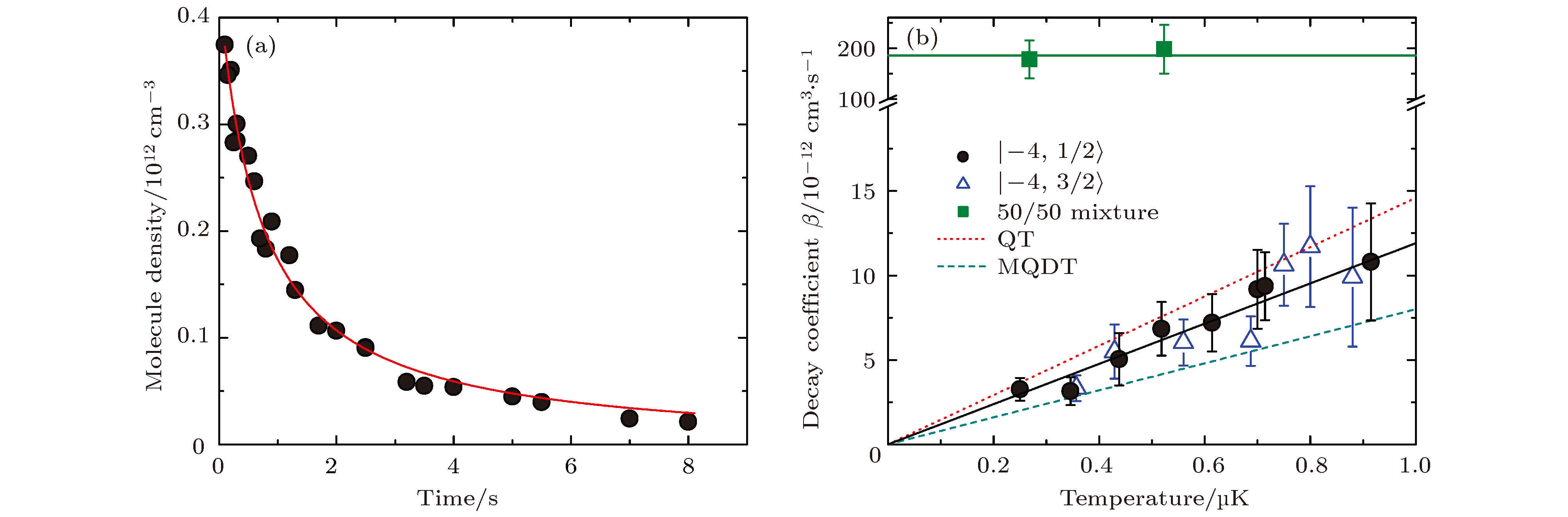 图 14 基态费米40K87Rb分子的碰撞研究 (a) 分子密度随时间的变化, 红色曲线为用(7)式做的两体损耗拟合, 从中可以提取出损耗速率常数
图 14 基态费米40K87Rb分子的碰撞研究 (a) 分子密度随时间的变化, 红色曲线为用(7)式做的两体损耗拟合, 从中可以提取出损耗速率常数

Figure14. Inelastic collisions of fermionic 40K87Rb molecules in the rovibronic ground state: (a) Sample data shows the time dependence of the molecule number density, the solid line is the fit based on a two-body decay model; (b) loss rate coefficient versus temperature[63].







这些现象可以用超冷化学反应的普适模型(universal model)来解释. 由于化学反应涉及反应物分子键的断裂和生成物新分子键的形成, 只能在分子间的距离很短时才能发生, 而分子的碰撞是从长程开始的. 因此, 如图15所示, 一般可将分子的相互作用分为长程和短程区, 分界为两个分子的长程相互作用决定的特征长度, 即范德瓦耳斯长度. 在普适模型中, 当能量允许时, 分子在短程区发生化学反应的概率接近100%, 因此化学反应的速率是由分子从长程到达短程的速率决定的.
 图 15 超冷化学反应的普适模型 (a) 全同费米分子通过p-波散射, 在长程有一个角动量引起的势垒; (b)非全同分子或全同玻色分子可以通过s-波散射, 没有长程势垒
图 15 超冷化学反应的普适模型 (a) 全同费米分子通过p-波散射, 在长程有一个角动量引起的势垒; (b)非全同分子或全同玻色分子可以通过s-波散射, 没有长程势垒Figure15. Universal model of the ultracold molecule reactivity: (a) Identical fermionic molecules react via p-wave scattering and the rate of chemical reactions is determined by the p-wave angular momentum barrier; (b) non-identical fermionic molecules and identical bosonic molecules react via s-wave scattering.
在没有外加电场的情况下, 分子还是通过范德瓦耳斯力作用, 因此在长程区, 两个距离为r的分子间的相互作用可以表示为
由于JILA的研究组实验中所用的40K87Rb分子为费米子, 当分子全部在同一量子态时, 它们之间的超冷碰撞只能通过l = 1的p-分波进行, 因此如图15(a)所示, 在长程会产生一个势垒. 这种情况下, 反应速率由分子对从长程隧穿到短程的速率决定[64]. 当分子不在同一量子态时, 分子也可以通过s-分波进行碰撞, 这种情况下, 没有长程的势垒, 这时反应速率由分子到达范德瓦耳斯长度的速率决定.
从图15(a)可以看到, 当分子为全同粒子时, p-分波势垒可以一定程度上压制化学反应的速率. 在非全同粒子中, 由于可以通过s-分波散射, 没有长程势垒, 因此化学反应要快得多. 其中




2
4.3.玻色23Na87Rb分子的碰撞
如图13(b)所示, 处于v = 0态的23Na87Rb +23Na87Rb的结合能比23Na2+87Rb2要大47 cm–1, 23Na87Rb (v = 0) + 23Na87Rb (v = 0) → 23Na2 (v = 0) + 87Rb2(v = 0)为吸热反应, 在超冷分子间由于碰撞能量太低而不可能发生[67]. 但如图16(a)所示, 即便在这种情况下, 23Na87Rb分子在光阱中的损耗仍很快, 如果用两体碰撞来进行拟合, 得到的损耗速率常数
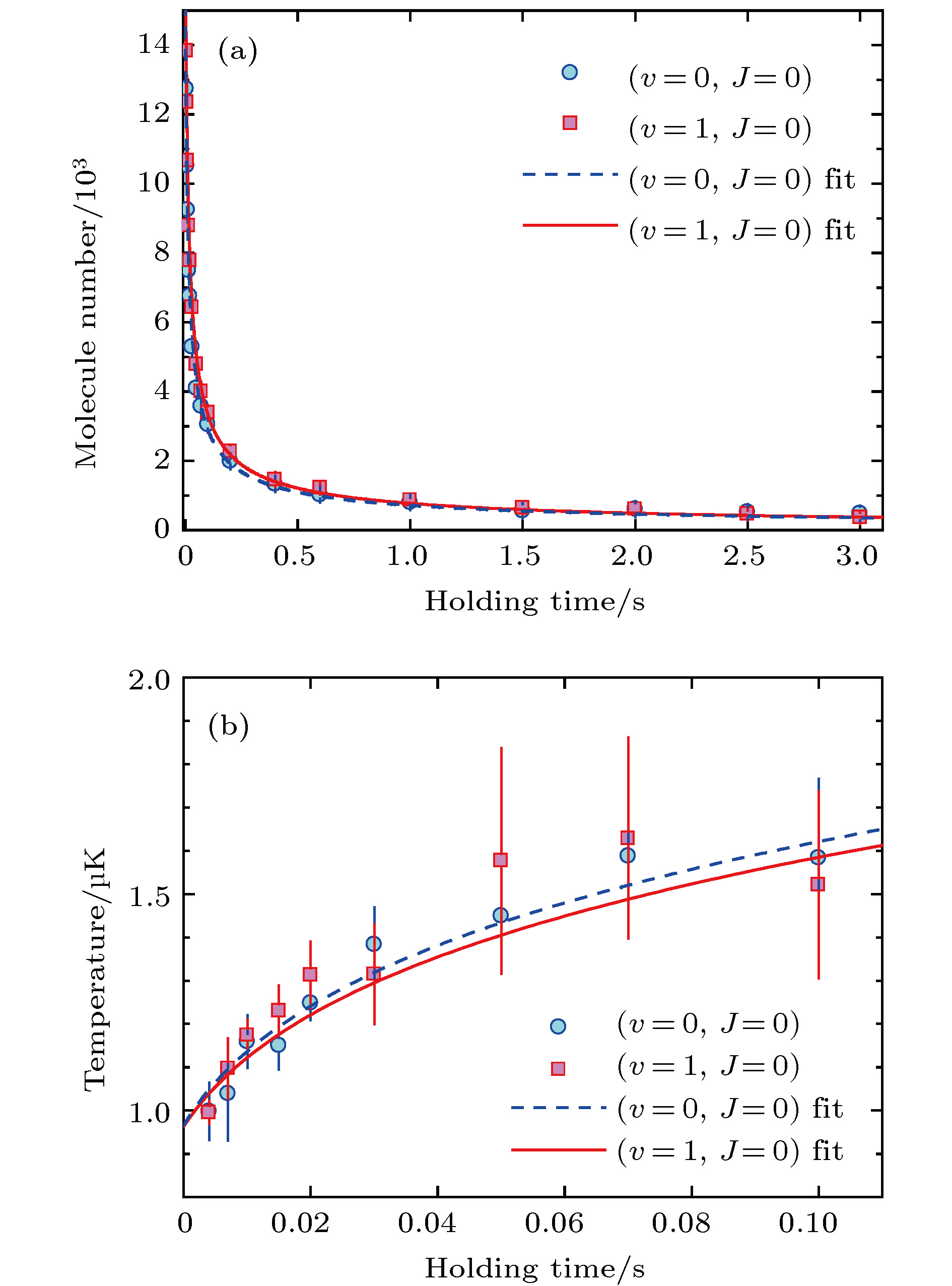 图 16 化学稳定(v = 0, J = 0)和有化学反应(v = 1, J = 0)的23Na87Rb分子在光阱中的损耗(a)和加热(b), 图中的曲线由通过对分子数目和温度同时拟合获得[68]
图 16 化学稳定(v = 0, J = 0)和有化学反应(v = 1, J = 0)的23Na87Rb分子在光阱中的损耗(a)和加热(b), 图中的曲线由通过对分子数目和温度同时拟合获得[68]Figure16. Inelastic collisions with different chemical reactivities of 23Na87Rb molecules. Time evolutions of (a) molecule numbers and (b) temperatures for both nonreactive (v = 0, J = 0)(filled circles) and reactive (v = 1, J = 0) (filled squares) samples in optical dipole trap. The blue dashed and redsolid curves are fitting resultsofmolecule number and temperature using Eq. (9)[68].
需要注意的是, 在分子损耗的测量中, 可以观察到比较明显的加热效应. 这是由于分子样品大致呈高斯分布, 中心的密度较高, 而碰撞损耗的速率和密度成正比, 因此损耗易于在分子样品的中心发生. 在样品中, 中心部分的分子的能量也是最低的, 当这一部分分子被从光阱中移除后, 剩余分子的平均能量就会上升, 导致加热现象. 这一过程和蒸发冷却正好相反, 因此也被称作反蒸发效应, 这种效应在所有的非弹性散射过程中都是存在的.
由于温度的增加会导致样品体积增大, 密度减小, 因此反蒸发效应对碰撞速率测量的影响不能忽略. 经过简单的推导, 可以得到以下两个耦合的速率方程来描述分子数目N和温度T随时间的演化[69-71]:


为了验证有无化学反应情况下23Na87Rb分子的损耗是否有区别, 利用23Na87Rb分子的化学反应活性可以操控的特性, 来打开化学反应通道. 例如, 如图13(b)所示, 当23Na87Rb分子被制备到v = 1时, 23Na87Rb + 23Na87Rb的结合能比23Na2 + 87Rb2要小108 cm–1, 因此23Na87Rb (v = 1) + 23Na87Rb (v = 1) → 23Na2 (v = 0) + 87Rb2(v = 0)为放热反应, 可以发生[72-74]. 在实验中, 制备v = 1态的23Na87Rb分子可以比较容易地通过调节dump光的频率来实现.
图16中比较了两种化学反应活性不同的23Na87Rb分子样品的数目和温度随时间的演化. 让人惊讶的是, 这样两种非常不同的情况下, 分子的损耗和加热几乎看不到区别. 图17是由损耗测量得到的


 图 17 化学稳定(v = 0)和有化学反应(v = 1)的23Na87Rb分子在光阱中的损耗速率常数
图 17 化学稳定(v = 0)和有化学反应(v = 1)的23Na87Rb分子在光阱中的损耗速率常数
Figure17. Temperature dependence of

在超冷温度下, 一般认为粒子的散射应该在Wigner区, 即








图17的结果还表明, 没有化学反应的分子间的碰撞损耗与温度的关系和有化学反应的情况确实很接近. 在相关的另外一些实验中, 如奥地利Innsbruck和英国Durham研究组的87Rb133Cs实验中, 以及MIT的23Na40K实验中, 都观察到没有化学反应的超冷极性分子中有类似的损耗. 这就引出了一个问题, 即在没有化学反应的情况下, 处于绝对基态的分子的损耗通道是什么?目前, 由于对四原子分子势能曲面的精确计算还很困难, 因此对于这一问题还没有确定的答案.
对于这一问题, 现阶段最流行的解释是损耗是由形成四体复合物(four-body complex)引起的[69,75]. 双原子分子的碰撞在短程涉及到四个原子, 由于系统的自由度较多, 描述它们之间的相互作用需要用到多维的势能曲面, 而且由于体系的质量较大, 势能曲面中的能态数目很大, 一般不能像双原子分子那样精确计算, 只能用态密度(density of states)描述. 在双原子分子的碰撞中, 两个分子由长程逐渐接近时, 可能会与很多个这些短程四体态共振, 在这种情况下, 两个分子会有很大的概率停留在一起, 形成一个不稳定的四体结构, 即四体复合物. 由于超冷分子实验的探测对四体复合物不敏感, 它们的形成就表现为分子的损耗. 图17中v = 0分子损耗的理论曲线, 就是基于这个模型, 可以看出, 理论上这个模型确实和化学反应是一样的.
目前的实验数据和这一模型是符合的, 但实验中并没有直接探测到四体复合物的任何信号, 这主要是由于一般超冷实验中探测手段的限制. 为了更好地回答化学稳定的超冷分子的损耗问题, 需要探测分子碰撞后的直接产物[76-78], 目前还没有任何实验装置具备这种能力. 最近, Harvard大学Ni研究组[79,80]利用光镊技术成功地形成了单个分子, 如果这一实验可以扩展到两个分子, 并能在单分子水平测量分子间的碰撞, 也可能为解决这一问题提供很好的答案.

本文对超冷分子的讨论主要着眼于研究超冷分子在长程作用下多体物理体系中的应用, 因而需要分子样品达到量子简并. 虽然JILA的研究组已经获得了KRb分子的简并样品[26], 但分子间的化学反应依然存在, 类似的方法是否可以应用到其他分子体系还需要进一步的研究. 目前看来, 分子间相互作用的复杂程度远远超出了最初的想象, 特别是化学稳定分子样品的损耗问题, 使得进一步的分子的蒸发冷却变得很困难, 因此下一步的研究重点应该是揭示损耗的微观机理[83]. 特别需要指出的是, 这些损耗机理的研究和化学反应的机理是有深刻联系的. 在超冷温度下, 由于分子的碰撞只能通过一个或少数几个分波进行, 化学反应的量子特性可以得到充分的体现, 因而这种极端条件下分子损耗机理的研究自身也是非常有价值的. 按照复合物形成模型, 超冷分子的损耗问题有可能是不可避免的, 为了获得长寿命的样品, 可以利用一些方法对损耗进行压制. 其中最直接的方法是把分子装载到三维光晶格中, 利用晶格势将分子隔绝开, 使得短程的碰撞不能发生. 这种方法已经在40K87Rb分子上得到了验证[84-86].
不同温度的极性分子在其他方面的应用也同等重要, 一个最重要的例子就是利用极性冷分子进行电子电偶极矩(eEDM)的测量. 非零的eEDM必然违反时间反演对称性(T-violation), 从而导致违反电荷共轭和宇称对称性(CP-violation), 也就预示着标准模型之外还有新的物理内容. 由于eEDM非常小, 要利用光谱技术测量它就需要很大的电场以增强灵敏度, 而极化的极性分子的内部电场可以达到几十个GV/cm, 因而非常适合用来寻找eEDM. 在Harvard大学和Yale大学合作的ACME项目中, 利用缓冲气体冷却技术得到的ThO分子束, 已经可以界定eEDM小于1.1 × 10–29 e·cm[87]. 目前, 已经有理论工作指出[88], 利用温度更低的分子, 例如激光冷却的分子, 可以进一步界定eEDM的值, 从而验证是否有超越标准模型的新物理内容.
总之, 超冷分子还是一个较新的研究方向, 随着分子冷却技术的成熟, 可用的、适合研究不同物理内容的超冷分子样品会越来越多. 虽然目前已经利用极性分子做了一些非常重要的工作, 但也只是超冷分子潜力的很小一部分, 因此这一方向未来还是大有可为的.
