Min Chen1, Zheng Zhang2, Ziyuan Meng1, Xuejun Zhang,1 1. Department of Dermatology and Venerology, the First Affiliated Hospital of Anhui Medical University, Heifei 230022, China 2. Department of Dermatology and Venerology, Huashan Hospital Affiliated to Fudan University, Shanghai 201100, China
Supported by the International Regional Cooperation and Exchange Programs No.81320108016
作者简介 About authors 陈敏,硕士研究生,专业方向:皮肤遗传。E-mail:626074054@qq.com。
摘要 染色质转座酶可及性测序(assay for transposase-accessible chromatin with high-throughput sequencing,ATAC-seq)是利用Tn5转座酶研究染色质可及性的高通量测序技术。ATAC-seq可以在全基因组范围内绘制染色质可及性图谱,揭示转录因子结合位点以及核小体的位置。在医学领域,ATAC-seq技术是研究重大疾病发病机制、药物作用机制、新药研发和生物标志物功能等的新一代有力工具。本文对ATAC-seq技术的优势及其在复杂疾病研究中的应用和前景进行了综述,以期为人类复杂疾病基因表达调控机制等相关研究的开展提供借鉴与参考。 关键词:ATAC-seq;染色质可及性;Tn5转座酶;转录调控水平
Abstract ATAC-seq is a high-throughput technology that defines and quantifies chromatin accessibility by analyzing Tn5 transposase enzymes. ATAC-seq is used to map chromatin accessibility genome-wide and to identify regions of transcription-factor binding and nucleosome position. As such, ATAC-seq is a new generation tool used in biomedical research to measure and articulate the pathogenesis of major diseases, to demonstrate the pharmacology of current drugs, and to guide the development of new drugs and the function of biomarkers. In this review, we summarize the current applications and advantages of ATAC-seq, and define its prospective contributions related to the regulatory mechanism of gene expression to identify and manage complex disease while elucidating and guiding future research references and strategies. Keywords:ATAC-seq;chromatin accessibility;Tn5 transposase;transcriptional level
PDF (474KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文 本文引用格式 陈敏, 张峥, 孟紫媛, 张学军. ATAC-seq在复杂疾病研究中的应用进展. 遗传[J], 2020, 42(4): 347-353 doi:10.16288/j.yczz.19-282 Min Chen. ATAC-seq and its applications in complex disease. Hereditas(Beijing)[J], 2020, 42(4): 347-353 doi:10.16288/j.yczz.19-282
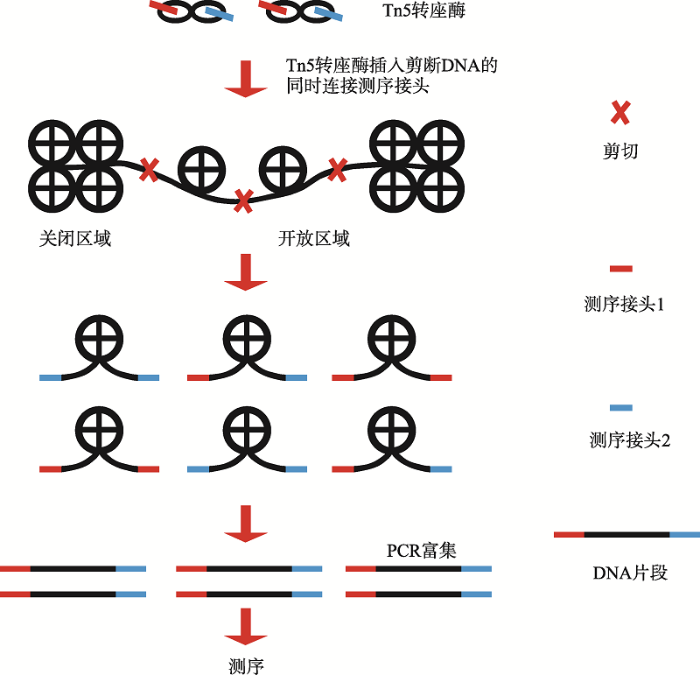
表观遗传学在许多重要的细胞调控过程中发挥关键的作用,表观遗传学水平的失调可能是导致严重疾病的根源。控制特定基因表达谱和表型的所有启动子和增强子的全基因组特性是理解生物体内调控过程的关键。传统的技术手段分析表观基因组需要大量的细胞,而在临床稀缺样本中很难满足这一要求。近年来,随着高通量测序法的快速发展,尤其是染色质转座酶可及性测序法(assay for transposase-accessible chromatin with high-throughput sequencing, ATAC-seq)仅利用少量细胞就可以识别基因组中所有调控序列,极大增加了人们对基因表达调控机制的理解。本文对ATAC-seq技术及其在复杂疾病中的应用进展进行了综述,为促进复杂疾病的基因表观遗传学发病机制、药物靶标搜寻、新药开发等领域的研究提供参考。
BuenrostroJD, GiresiPG, ZabaLC, ChangHY, GreenleafWJ . Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position Nat Methods, 2013,10(12):1213-1218. [本文引用: 5]
DoganliC, SandovalM, ThomasS, HartD . Assay for Transposase-accessible chromatin with high-throughput sequencing (ATAC-Seq) protocol for zebrafish embryos Methods Mol Biol, 2017,1507:59-66. [本文引用: 2]
SongLY, Crawford GE. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells Cold Spring Harb Protoc, 2010, 2010(2): pdb prot5384. [本文引用: 1]
HanJL, LiZJ, WangK . Progress on genome-wide identification and analysis of transcriptional regulatory elements based on open-chromatin signatures J Fujian Agric Fore Uni (Nat Sci Edi), 2017,46(1):1-8. [本文引用: 1]
WillemzeR, CerroniL, KempfW, BertiE, FacchettiF, SwerdlowSH, JaffeES . The 2018 update of the WHO- EORTC classification for primary cutaneous lymphomas Blood, 2019,133(16):1703-1714. [本文引用: 1]
NewM, OlzschaH, La ThangueNB . HDAC inhibitor- based therapies: Can we interpret the code? Mol Oncol, 2012,6(6):637-656. [本文引用: 1]
QuK, ZabaLC, SatpathyAT, GiresiPG, LiR, JinYH, ArmstrongR, JinC, SchmittN, RahbarZ, UenoH, GreenleafWJ, KimYH, Chang HY, . Chromatin accessibility landscape of cutaneous T cell lymphoma and dynamic response to HDAC inhibitors Cancer Cell, 2017, 32(1): 27-41. e4. [本文引用: 3]
WuJ, WoodGS . Reduction of Fas/CD95 promoter methylation, upregulation of Fas protein, and enhancement of sensitivity to apoptosis in cutaneous T-cell lymphoma Arch. Dermatol, 2011,147(4):443-449. [本文引用: 1]
DechassaML, Tryndyak V, deConti A, XiaoW, BelandFA, PogribnyIP . Identification of chromatin-accessible domains in non-alcoholic steatohepatitis-derived heaptocellular carcinoma Mol Carcinog, 2018,57(8):978-987. [本文引用: 1]
BrittonE, RogersonC, MehtaS, LiY, LiX, FitzgeraldRC, AngYS, SharrocksAD . Open chromatin profiling identifies AP1 as a transcriptional regulator in oesophageal adenocarcinoma PLoS Genetics, 2017,13(8):e1006879. [本文引用: 1]
WangZF, TuKL, XiaL, LuoK, LuoWX, TangJ, LuKY, HuXL, HeYJ, QiaoWL, ZhouYZ, ZhangJ, CaoF, DaiSP, TianPW, WangY, LiuLX, CheGW, ZhouQH, XieD, LiWM . The open chromatin landscape of non-small cell lung carcinoma Cancer Res, 2019,79(19):4840-4854. [本文引用: 1]
ZhuL, YinZJ, JuBM, ZhangJ, WangYH, LvXH, HaoZM, HeL . Altered frequencies of memory B cells in new-onset systemic lupus erythematosus patients Clin Rheumatol, 2018,37(1):205-212. [本文引用: 1]
VaughnSE, KottyanLC, MunroeME, HarleyJB . Genetic susceptibility to lupus: the biological basis of genetic risk found in B cell signaling pathways J Leukoc Biol, 2012,92(3):577-591. [本文引用: 1]
FarhKK, MarsonA, ZhuJ, KleinewietfeldM, HousleyWJ, BeikS, ShoreshN, WhittonH, RyanRJ, ShishkinAA, HatanM, Carrasco-AlfonsoMJ, MayerD, LuckeyCJ, PatsopoulosNA, De JagerPL, KuchrooVK, EpsteinCB, DalyMJ, HaflerDA, BernsteinBE . Genetic and epigenetic fine mapping of causal autoimmune disease variants Nature, 2015,518(7539):337-343. [本文引用: 1]
ScharerCD, BlalockEL, BarwickBG, HainesRR, WeiC, SanzI, BossJM . ATAC-seq on biobanked specimens defines a unique chromatin accessibility structure in na?ve SLE B cells Sci Rep, 2016,6(1):27030. [本文引用: 1]
KariukiSN, KirouKA, MacDermott EJ, Barillas-AriasL, CrowMK, NiewoldTB . Cutting edge: autoimmune disease risk variant of STAT4 confers increased sensitivity to IFN-α in lupus patients in vivo J Immunol, 2009,182(1):34-38. [本文引用: 1]
GensterblumE, RenauerP, CoitP, StricklandFM, KilianNC, MillerS, OgnenovskiM, WrenJD, TsouPS, LewisEE, Maksimowicz-McKinnon K, McCuneWJ, RichardsonBC, SawalhaAH, . CD4+CD28+KIR+CD11a hi T cells correlate with disease activity and are characterized by a pro-inflammatory epigenetic and transcriptional profile in lupus patients J Autoimmun, 2018,86:19-28. [本文引用: 1]
BacosK, GillbergL, VolkovP, OlssonAH, HansenT, PedersenO, GjesingAP, EibergH, TuomiT, AlmgrenP, GroopL, EliassonL, VaagA, DayehT, LingC . Blood-based biomarkers of age-associated epigenetic changes in human islets associate with insulin secretion and diabetes Nat Commun, 2016,7:11089.
BysaniM, AgrenR, Daveg?rdhC, VolkovP, R?nnT, UnnebergP, BacosK, LingC . ATAC-seq reveals alterations in open chromatin in pancreatic islets from subjects with type 2 diabetes Sci Rep, 2019,9(1):7785.
AckermannAM, WangZ, SchugJ, NajiA, KaestnerKH . Integration of ATAC-seq and RNA-seq identifies human alpha cell and beta cell signature genes Mol Metab, 2016,5(3):233-244.
TakYG, FarnhamPJ . Making sense of GWAS: using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome Epigenetics Chromatin, 2015,8(1):57. [本文引用: 1]
HendricksonDG, SoiferI, WranikBJ, BotsteinD, Scott McIsaac R,. Simultaneous profiling of DNA accessibility and gene expression dynamics with ATAC-Seq and RNA-Seq Methods Mol Biol, 2018,1819:317-333. [本文引用: 1]
 ,1
,1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT
{kind=link}
{kind=link}