 ,中国科学院微生物研究所,微生物资源前期开发国家重点实验室,北京 100101
,中国科学院微生物研究所,微生物资源前期开发国家重点实验室,北京 100101Systematic discovery of novel prokaryotic defense systems: progress and prospects
Ming Li, Feiyue Cheng, Luyao Gong, Hua Xiang,State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China通讯作者:
编委: 韩玉波
收稿日期:2018-03-21修回日期:2018-04-7网络出版日期:2018-04-20
| 基金资助: |
Editorial board:
Received:2018-03-21Revised:2018-04-7Online:2018-04-20
| Fund supported: |
作者简介 About authors
李明,博士,副研究员,研究方向:CRISPR分子机制E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (304KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
李明, 程飞跃, 龚路遥, 向华. 微生物新型防御系统的系统性发现与展望. 遗传[J], 2018, 40(4): 259-265 doi:10.16288/j.yczz.18-075
Ming Li, Feiyue Cheng, Luyao Gong, Hua Xiang.
几乎所有的细胞生命都会时刻面临着病毒的威胁,它们在长期的选择压力下进化出了一系列复杂而精巧的病毒防御机制[1,2,3]。原核生物包括细菌和古菌,它们的病毒防御机制十分多样,按照其直接靶向的组分(病毒组分或宿主自身组分)大致可分为两大类(表1)。第一大类是主动型防御(active defense)机制,例如,人们所熟知的限制-修饰(restriction- modification, R-M)系统,它通过识别特定DNA序列(一般具有回文特征)的甲基化状态区分并特异地切割病毒DNA[4]。CRISPR-Cas是另一种靶向病毒DNA/RNA的核酸免疫系统,也是原核生物中已知的唯一一种适应性免疫机制,它通过获取病毒的DNA序列信息而获得记忆性免疫,利用转录加工产生的CRISPR RNA指导Cas核酸酶切割病毒基因 组[5,6]。第二大类是自杀型防御(suicidal defense)机制,包括流产感染(abortive infection, Abi)系统[7]和毒素-抗毒素(toxin-antitoxin, TA)系统[8]。流产感染实际上是一种表型定义,可以泛指各类诱发被感染细胞死亡或代谢停滞的防御机制,也特指各种休眠的毒性蛋白,它们在侵染发生时被病毒组分活化并阻断宿主细胞的关键生理过程,导致进入宿主细胞的病毒DNA无法产生(或产生极少量的)子代病毒颗粒,从而保护菌群中的其他个体免于病毒侵染,因此是一种群体水平的防御策略。而毒素-抗毒素系统编码一对稳定的毒素蛋白和不稳定的抗毒素蛋白或RNA,病毒侵染往往会导致后者(抗毒素)快速降解而释放前者(毒素),最终引发流产感染。
Table 1
表1
表1 原核生物中几种主要的防御机制
Table 1
| 类别 | 防御机制/系统 | 标志性或关键性组分 | 分子机理 | 参考文献 |
|---|---|---|---|---|
| 主动防御机制 | 限制-修饰系统(restriction-modification, R-M) | 限制性内切酶和DNA甲基化酶 | 切割病毒DNA中未甲基化的特定识别序列(通常有回文特征) | [4] |
| CRISPR-Cas系统 | CRISPR重复序列和Cas1等Cas蛋白 | RNA指导特异性切割病毒核酸 | [5,6] | |
| 原核Argonautes (pAgos) | PIWI结构域蛋白 | 可能由DNA或RNA指导特异性切割外源核酸 | [17,18] | |
| BREX (bacteriophage exclusion) | pglZ;pglX;brxA-B-C-L | 抑制病毒DNA复制,具体分子机制未知 | [19] | |
| DISARM (defense island system associated with restriction-modification) | DNA甲基化酶;解旋酶;DUF1998,PLD结构域 | 多基因的限制-修饰系统 | [12] | |
| Zorya系统 | zorA-B-C,核酸酶基因(zorD或zorE) | 可能以未知机制靶向病毒DNA,并引起条件性流产感染 | [13] | |
| Thoeris系统 | thsA(NAD+结合结构域) thsB(TIR结构域) | 机制未知,ThsB可能负责识别病毒组分 | [13] | |
| Druantia系统 | DUF1998结构域 | 机制未知 | [13] | |
| Wadjet系统 | jetA-B-C-D | 可能在细胞分裂过程中干扰质粒分配 | [13] | |
| 自杀防御机制 | 流产感染系统(abortive infection, Abi) | 组分多样,一般为休眠的 蛋白 | 休眠的蛋白被病毒组分活化,并阻断宿主细胞的关键生理过程 | [7] |
| 毒素-抗毒素系统(toxin-antitoxin, TA) | 一对毒性蛋白和抗毒性蛋白/RNA | 抗毒素快速降解,从而释放毒素的细胞毒性 | [8] |
新窗口打开|下载CSV
人们基于对上述防御机制,尤其是核酸免疫机制的研究,开发出了一系列重要的分子生物学工具,推动了生命科学的发展。例如,来源于细菌限制-修饰系统的限制性内切酶几乎是分子克隆的必备工具,而CRISPR-Cas9基因组编辑技术也是起源于人们对CRISPR-Cas适应性免疫过程的认识和改造[9,10,11]。因此,发掘和解析原核生物中新型病毒防御机制无论在理论上还是应用上都具有重要意义。近期,以色列的Rotem Sorek研究团队通过分析海量的微生物基因组信息,预测并鉴定了一系列新型的防御系统[12,13],拓展了人们对于微生物防御系统复杂性和多样性的认识,并有望为科学家们开发新型分子工具提供丰富的遗传元件。
1 微生物新型防御系统的系统性发掘
不同防御系统的编码基因往往在细菌或古菌基因组上聚簇存在并形成一个或多个“防御岛(defense island)”[14,15]。2009年,Makarova等[16]发现原核Argonaute(简称pAgos)的编码基因往往位于各种防御基因(如限制性内切酶、CRISPR-Cas系统)附近,推测可能是一种新型的防御系统。这一推测后来由van der Oost J团队在细菌嗜热栖热菌(Thermus thermophilus)[17]和古菌激烈火球菌(Pyrococcus furiosus)[18]的pAgo研究中得到证实。类似地,Sorek团队通过分析两种防御基因在微生物基因组中的分布,分别发现了BREX(bacteriophage exclusion)[19]和DISARM(defense island system associated with restriction-modification)[12]这两个新型防御系统。上述系统的发现说明通过分析与已知防御基因相关联的保守未知基因,能够有效预测新型的防御系统。近期,Sorek实验室进一步沿着这一思路,分析了大量已知防御基因在海量微生物基因组中的分布,并结合合成生物学手段系统性地鉴定了10种新型的防御系统,其中的9种是抗病毒的,另外1种是抵御质粒的[13]。首先,根据Makarova等[14]于2011年统计的参与抗噬菌体免疫防御的基因家族信息,以及BREX和DISARM的信息,获得已知参与抗噬菌体免疫的pfams(蛋白家族,即一组具有相同结构域的蛋白)和COGs(直系同源蛋白簇)数据集。然后,他们分析了14 083个pfam 的成员基因在45 000个细菌和古菌的基因组中的分布,并引入了两个统计学概念——防御相关性(defense score)和防御背景复杂度(defense context variability score)。一个pfam的防御相关性是指其成员基因“邻近”(比如距离10个基因以内)已知防御基因的比例,而防御背景复杂度反映了其成员基因在不同已知防御系统附近出现的情况。通过参考已知防御系统设定参数阈值,该团队获得了277个可能参与病毒防御的pfam,其中大部分从未报道与防御过程相关。加上2011年Makarova等[18]预测的35个非pfam(non-pfam)基因家族和23个未通过上述实验阈值的pfams,共得到335个候选的防御基因家族(图1)。
图1
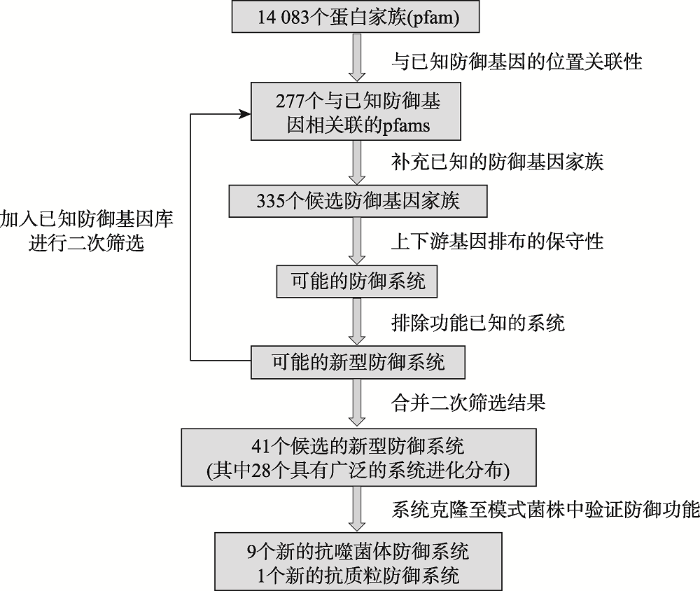 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1Sorek研究中新型原核防御系统的预测和鉴定流程
Fig. 1Schematic illustration of the pipeline to identify novel prokaryotic defense systems
如同CRISPR-Cas、BREX、DISARM等,防御系统通常并非是一个单一基因,而是由来自多个家族的若干基因构成,且这些基因通常形成操纵子,在不同物种之间具有比较保守的基因排布[20,21]。因此,Sorek团队将每个候选基因家族的所有成员及其上下游基因选出,通过序列同源性进行聚簇分析,进一步筛选出在不同基因组中具有保守基因排布的基因簇作为可能的多基因防御系统。通过分析注释信息,他们人工剔除了一些假阳性结果(可能参与其他生理过程的系统)和一些已知的病毒防御系统。另外,为了获得更多的结果,他们将这些新鉴定的候选防御系统添加到已知数据集中进行二次筛选(图1),最终获得了41个单基因或多基因的候选系统,其中28个具有广泛的系统进化分布,可能代表在原核生物中比较普遍存在的防御机制。
要将上述生物信息学预测的候选系统鉴定为新型的防御系统需要更直接的实验证据。Sorek团队借助合成生物学手段,直接合成它们的全基因序列(包括上下游基因间区以保持其启动子、终止子等调控序列的完整性),并在原有免疫系统失活的模式菌株大肠杆菌(Escherichia coli)MG1655和枯草芽孢杆菌(Bacillus subtilis)BEST7003中对其病毒防御能力进行验证。由于在选择每个候选防御系统的具体基因序列时尽量选择了与模式菌株亲缘关系较近的原生宿主菌,这一合成生物学的策略非常成功,在28个候选防御系统中,26个系统有至少一个候选序列在模式菌株中成功表达。接下来他们选择了多种不同类型的噬菌体做侵染实验,结果发现,成功表达的26种系统中有9种对至少一种噬菌体有防御作用,而1种在枯草芽孢杆菌中对外源质粒DNA有明显的抵御作用,他们将这10种防御系统分别以各种神话中保护神的名字命名。这些新发现的防御系统具有不同的进化分布、基因构成和病毒抗性特征,暗示了它们在不同的微生物类群中采取了非常多样化的分子防御机制。通过关键基因突变等分析,Sorek团队进一步探讨了其中4个新型系统(Zorya, Thoeris, Druantia和Wadjet)的功能机制(表1)。
2 微生物新型防御系统的功能机制多样性
微生物防御系统的功能解析是其实现技术创新转化的关键,如CRISPR从1987年首次发现到2007揭示其功能花了20年时间,而从机制解析到发展成为基因组编辑颠覆性技术只用了短短几年时间[22]。进入21世纪以来,CRISPR的研究热潮也带来了对微生物新型防御系统的高度关注,但总体而言人们对这些新系统的功能机制还知之甚少,因此未来新系统的功能解析需进一步加强。部分新型防御系统简介如下:2.1 pAgos系统
Argonaute蛋白在原核生物和真核生物中广泛分布[23],在真核生物中作为RNA干扰通路的关键组分参与基因的转录后调控等过程,而在原核生物中的生理功能直到近几年才逐渐被揭示。2014年,Swarts等[17]发现来源于嗜热栖热菌(T. thermophilus)的细菌Ago在体外实验中具有DNA指导的DNA干扰活性;2015年,该团队又进一步发现来源于激烈火球菌(P. furiosus)的古菌Ago同样可以在DNA指导下切割同源DNA[18]。重要的是,两者在宿主细胞中的表达都能在一定程度上降低外源质粒的转化效率,说明它们至少能够抵御质粒类型的外源DNA。由于大部分病毒防御系统(如限制-修饰系统、CRISPR- Cas)同样可作用于外源质粒,因此pAgos可能也具有病毒防御功能。2.2 BREX系统
天蓝色链霉菌中存在一种称为Pgl (phage growth limitation)的特殊噬菌体防御现象——噬菌体侵染携带Pgl系统的菌株后产生的子代无法再次侵染该类型菌株[24],而2014年Sorek团队通过分析该系统的pglZ基因(编码一个可能的碱性磷酸酶)在1 500个微生物基因组中的分布,发现该基因和pglX(编码一个DNA甲基化酶)往往出现在一个由6个基因构成的保守基因簇中,其他4个基因分别命名为brxA、-B、-C和-L[19]。有意思的是,这一基因簇在枯草芽孢杆菌中异源表达时并未导致Pgl现象,但确实可以使宿主菌产生对多种噬菌体的抗性,说明采取了一种新的防御机制,因此,他们将这一系统命名为BREX (bacteriophage exclusion)。Southern实验进一步表明BREX的防御功能可能通过某种未知机制抑制病毒DNA复制而实现。2.3 DISARM系统
含有DUF1998(pfam0939)结构域的蛋白具有未知功能,但它们的编码基因往往分布于“防御岛”内部[14],因此可能参与防御过程。Sorek团队分析了该基因在35 893个微生物基因组中的分布,发现其中1 273个基因组携带该基因(命名为drmB),并往往位于一个解旋酶基因(drmA)和一个PLD(磷脂酶D)结构域编码基因(drmC)之间[12]。这3个核心基因有时与腺嘌呤甲基化酶基因(drmMI)和SNF2家族解旋酶基因(drmD)组成一个基因簇,有时与胞嘧啶甲基化酶基因(drmMII)和一个未知基因(drmE)组成基因簇,因此,该系统被命名为DISARM (defense island system associated with restriction-modification),而上述两种形式的基因簇分别定义为Ⅰ型和Ⅱ型。作者将1株土壤来源芽孢杆菌(Bacillus paralicheniformis ATCC 9945a)的Ⅱ型DISARM系统克隆至模式菌株枯草芽孢杆菌BEST7003中,发现赋予了该宿主对多种双链DNA噬菌体的抗性。研究进一步发现,该系统貌似一种多基因的限制-修饰系统,可以对病毒DNA进行限制性切割,并通过甲基化修饰宿主DNA的CCWGG(W=A/T)序列避免自我免疫。2.4 Zorya系统
Zorya防御系统可能兼具主动防御和自杀防御两种分子机制。该系统只分布于细菌基因组中,有两种类型,Ⅰ型由4个基因(zorA、-B、-C和-D)组成,Ⅱ型由3个基因(zorA、-B和-E)组成。Ⅰ型的zorD基因编码产物C端有时具有Mrr家族限制性内切酶特征,而Ⅱ型系统的zorE基因编码一个HNH核酸内切酶结构域,暗示该系统可能靶向病毒DNA。有意思的是,两类系统的共有基因zorA和zorB的蛋白产物与鞭毛马达蛋白MotA和MotB(质子通道组分,可利用质子迁移为鞭毛旋转提供能量[25,26])分别具有较高同源性,而且当ZorA和ZorB中可能参与质子迁移的关键残基被突变时,该防御系统失活。因此,ZorD和ZorE等可能参与主动防御并靶向病毒DNA,而ZorA和ZorB可能在主动防御失败时打开质子通道,使细胞膜电势去极性化,并最终导致细胞死亡。2.5 Thoeris系统
分布于细菌和古菌中的Thoeris防御系统可能采取与真核生物天然免疫系统相类似的外源分子识别方式。该系统由必需基因thsA(编码一个NAD+结合结构域)和thsB构成,其中thsB经常以多拷贝的形式存在,并编码一个常常与pAgos基因关联存在[16]的TIR (toll-interleukin receptor)结构域。事实上,该结构域在哺乳动物、植物和无脊椎动物的天然免疫系统中作为重要组分(如哺乳动物Toll样受体的胞内部分)参与病原分子模式识别[27],因此,该结构域可能负责识别噬菌体的不同组分,决定了Thoeris系统的免疫特异性。这一发现也说明TIR结构域在真核生物和原核生物的免疫系统中非常保守地参与了病原分子的特异性识别。2.6 Druantia系统
病毒防御系统Druantia非常有意思,该系统的编码序列约12 kb,除了其特征基因编码一个已知的防御相关结构域DUF1998(该结构域也是DISARM系统的核心组分[12])以外,其他基因几乎不含有任何已知的功能结构域。该系统可分为3种类型,它们除了编码特征基因以外,还分别编码3个350~600 氨基酸(amino acid, AA)的(Ⅰ型),2个700~900 AA的(Ⅱ型),或者1个1000~1200 AA的(Ⅲ型)未知基因。可见,Druantia系统采取一种复杂而独特的未知机制。2.7 Wadjet系统
不同于上述几种新型防御系统,Wadjet系统虽然不能防御病毒侵染,但能够抵御质粒类型的外源DNA。该系统由jetA、-B、-C和-D四个必需基因组成,广泛分布于细菌和古菌中。虽然该系统的作用机制尚不清楚,但JetA、JetB和JetC与凝缩蛋白MukF、MukE和MukB[28,29]分别具有显著的结构相似性,因此可能通过干扰质粒在子代细胞中的正常分离实现其防御功能。如前所述,由于大部分病毒防御系统一般可同样作用于外源质粒,Wadjet或许能够防御特定的病毒类群,比如某类单链DNA病毒(质粒往往以单链DNA形式进入宿主细胞)。3 微生物新型防御系统的研究和应用展望
原核生物防御系统的功能实现依赖于其对外来因子的特异性识别,如限制-修饰系统通过识别特定序列(一般具有回文特征)的甲基化状态区分外源DNA,而CRISPR-Cas系统凭借CRISPR RNA序列以及PAM(protospacer adjacent motif)识别[6,30,31]等分子机制特异性靶向病毒DNA。正是基于对这些特异性分子识别机制的认识,科学家们开发出了限制性内切酶和CRISPR-Cas9等重要的分子生物学工具。Sorek团队借助生物信息学和海量的微生物基因组数据,预测了大量新型防御系统,并利用合成生物学手段验证了BREX、DISARM和其他10种系统的病毒(或质粒)防御能力,这远远超出了已知防御系统的数量。虽然针对其中大部分新型系统,Sorek团队尚未深入解析它们具体如何发挥其免疫功能,但可以肯定的是,这些复杂多样的防御系统蕴藏着丰富的特异性分子靶向机制。尤其是其中可能靶向DNA或RNA的新型核酸免疫系统,或将为科学家们开发新型的基因组编辑等工具提供重要元件。值得注意的是,除了作者已验证防御功能的10余种新型系统(包括BREX和DISARM)以外,他们预测的其他候选系统也可能本身具有防御功能,只不过在选择的两种模式菌株中表达时可能由于缺乏某种辅助因子或调控元件等未知原因,其防御功能没有得以异源重建,抑或是其防御能力特异性地靶向实验设定以外的某种(类)特定病毒,因此,未来在其他模式菌株中开展更多的病毒侵染实验或将有望进一步鉴定其中更多的候选系统。另外,在生物信息学预测过程中作者设定的一些标准可能会导致部分真实防御系统的遗漏,例如,作者剔除了一些分布于单一进化枝(如古菌、蓝细菌等)的候选系统,但这些系统可能实际上具有防御功能,有待于进一步验证。
总之,Sorek团队报道的这一系列新型防御系统暗示了更多原核生物新型防御系统的存在,这项工作使得人们对于原核生物防御系统和免疫机制的复杂性和多样性有了更深刻的认识。但由于目前预测方法还存在一定局限性,检测系统相对较少,可以相信微生物中蕴藏着的丰富抗病毒机制还远未得到充分挖掘。因此未来如何继续创新该类核酸靶向系统的高通量发现,建立高效的功能评价体系,并对具有重要应用前景的新型系统开展分子机制研究,将是开发新一代颠覆性分子工具(包括新型基因组编辑系统)面临的重要课题。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URL [本文引用: 1]
URLPMID:20979102 [本文引用: 1]

Bacteria, the most abundant organisms on the planet, are outnumbered by a factor of 10 to 1 by phages that infect them. Faced with the rapid evolution and turnover of phage particles, bacteria have evolved various mechanisms to evade phage infection and killing, leading to an evolutionary arms race. The extensive co-evolution of both phage and host has resulted in considerable diversity on the part of both bacterial and phage defensive and offensive strategies. Here, we discuss the unique and common features of phage resistance mechanisms and their role in global biodiversity. The commonalities between defense mechanisms suggest avenues for the discovery of novel forms of these mechanisms based on their evolutionary traits.
URLPMID:26958724 [本文引用: 1]
Abstract Bacteriophages (phages) specifically infect bacteria and are the most abundant biological entities on Earth. The constant exposure to phage infection imposes a strong selective pressure on bacteria to develop viral resistance strategies that promote prokaryotic survival. Thus, this parasite-host relationship results in an evolutionary arms race of adaptation and counteradaptation between the interacting partners. The evolutionary outcome is a spectrum of remarkable strategies used by the bacteria and phages as they attempt to coexist. These approaches include adsorption inhibition, injection blocking, abortive infection, toxin-antitoxin, and CRISPR-Cas systems. In this review, we highlight the diverse and complementary antiphage systems in bacteria, as well as the evasion mechanisms used by phages to escape these resistance strategies.
URLPMID:15979932 [本文引用: 1]
The phenomena of prokaryotic restriction and modification, as well as anti-restriction, were first discovered five decades ago but have yielded only gradually to rigorous analysis. Work presented at the 5 th New England Biolabs Meeting on Restriction-Modification (available on REBASE, http://www.rebase.com ) and several recently published genetic, biochemical and biophysical analyses indicate that these fields continue to contribute significantly to basic science. Recently, there have been several studies that have shed light on the still developing field of restriction-modification and on the newly re-emerging field of anti-restriction.
URLPMID:17379808 [本文引用: 1]
Clustered regularly interspaced short palindromic repeats (CRISPR) are a distinctive feature of the genomes of most Bacteria and Archaea and are thought to be involved in resistance to bacteriophages. We found that, after viral challenge, bacteria integrated new spacers derived from phage genomic sequences. Removal or addition of particular spacers modified the phage-resistance phenotype of the cell. Thus, CRISPR, together with associated cas genes, provided resistance against phages, and resistance specificity is determined by spacer-phage sequence similarity.
URLPMID:18703739 [本文引用: 2]
Abstract Prokaryotes acquire virus resistance by integrating short fragments of viral nucleic acid into clusters of regularly interspaced short palindromic repeats (CRISPRs). Here we show how virus-derived sequences contained in CRISPRs are used by CRISPR-associated (Cas) proteins from the host to mediate an antiviral response that counteracts infection. After transcription of the CRISPR, a complex of Cas proteins termed Cascade cleaves a CRISPR RNA precursor in each repeat and retains the cleavage products containing the virus-derived sequence. Assisted by the helicase Cas3, these mature CRISPR RNAs then serve as small guide RNAs that enable Cascade to interfere with virus proliferation. Our results demonstrate that the formation of mature guide RNAs by the CRISPR RNA endonuclease subunit of Cascade is a mechanistic requirement for antiviral defense.
URLPMID:7601848 [本文引用: 1]
Abstract Lactococcal phage abortive infection (AbiD1) determined by plasmid pIL105 is active on both prolate- and small-isometric-head phages of the C6A and 936 phage groups, respectively, which are considered two different species. The Abi phenotype was found to be encoded by a single gene, designated abiD1. The abiD1-encoded protein (351 amino acids) does not show homology with any known protein and has a deduced isoelectric point of 10. It also possesses two helix-turn-helix structures and an unusually high content of asparagine, isoleucine, and lysine. A consensual promoter with a TGy extension to the -10 box was mapped 76 bp upstream of the start codon. Transcription initiated at this strong promoter stops at a terminator located 48 bp downstream from the promoter. The termination process is very efficient, and transcripts corresponding to the abiD1 gene were not visible in our experimental conditions with or without phage infection. Expression of abiD1 under the control of a T7 promoter induced a lag phase in Lactococcus lactis cell growth, suggesting that overproduction of AbiD1 could be toxic for the cells. AbiD1 protein was visualized in Escherichia coli by using a tightly controlled expression system.
URLPMID:19124776 [本文引用: 1]
Various mechanisms exist that enable bacteria to resist bacteriophage infection. Resistance strategies include the abortive infection (Abi) systems, which promote cell death and limit phage replication within a bacterial population. A highly effective 2-gene Abi system from the phytopathogen Erwinia carotovora subspecies atroseptica, designated ToxIN, is described. The ToxIN Abi system also functions as a toxin-antitoxin (TA) pair, with ToxN inhibiting bacterial growth and the tandemly repeated ToxI RNA antitoxin counteracting the toxicity. TA modules are currently divided into 2 classes, protein and RNA antisense. We provide evidence that ToxIN defines an entirely new TA class that functions via a novel protein-RNA mechanism, with analogous systems present in diverse bacteria. Despite the debated role of TA systems, we demonstrate that ToxIN provides viral resistance in a range of bacterial genera against multiple phages. This is the first demonstration of a novel mechanistic class of TA systems and of an Abi system functioning in different bacterial genera, both with implications for the dynamics of phage-bacterial interactions.
URLPMID:23386978 [本文引用: 1]
Abstract Type II CRISPR immune systems in bacteria use a dual RNA-guided DNA endonuclease, Cas9, to cleave foreign DNA at specific sites. We show here that Cas9 assembles with hybrid guide RNAs in human cells and can induce the formation of double-strand DNA breaks (DSBs) at a site complementary to the guide RNA sequence in genomic DNA. This cleavage activity requires both Cas9 and the complementary binding of the guide RNA. Experiments using extracts from transfected cells show that RNA expression and/or assembly into Cas9 is the limiting factor for Cas9-mediated DNA cleavage. In addition, we find that extension of the RNA sequence at the 3' end enhances DNA targeting activity in vivo. These results show that RNA-programmed genome editing is a facile strategy for introducing site-specific genetic changes in human cells.DOI:http://dx.doi.org/10.7554/eLife.00471.001.
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:29085076 [本文引用: 4]
react-text: 399 Bacterial defense systems have made major contributions to biotechnology, in the past, at present, and in the future. 08 2015 The Author. Microbial Biotechnology published by John Wiley & Sons Ltd and Society for Applied Microbiology. /react-text react-text: 400 /react-text
URLPMID:29371424 [本文引用: 2]
The arms race between bacteria and phages led to the development of sophisticated antiphage defense systems, including CRISPR-Cas and restriction-modification systems. Evidence suggests that unknown defense systems are located in “defense islands” in microbial genomes. We comprehensively characterized the bacterial defensive arsenal by examining gene families that are clustered next to known defense genes in prokaryotic genomes. Candidate defense systems were systematically engineered and validated in model bacteria for their antiphage activities. We report nine previously unknown antiphage systems and one antiplasmid system that are widespread in microbes and strongly protect against foreign invaders. These include systems that adopted components of the bacterial flagella and condensin complexes. Our data also suggest a common, ancient ancestry of innate immunity components shared between animals, plants, and bacteria.
URLPMID:3194920 [本文引用: 3]
Abstract The arms race between cellular life forms and viruses is a major driving force of evolution. A substantial fraction of bacterial and archaeal genomes is dedicated to antivirus defense. We analyzed the distribution of defense genes and typical mobilome components (such as viral and transposon genes) in bacterial and archaeal genomes and demonstrated statistically significant clustering of antivirus defense systems and mobile genes and elements in genomic islands. The defense islands are enriched in putative operons and contain numerous overrepresented gene families. A detailed sequence analysis of the proteins encoded by genes in these families shows that many of them are diverged variants of known defense system components, whereas others show features, such as characteristic operonic organization, that are suggestive of novel defense systems. Thus, genomic islands provide abundant material for the experimental study of bacterial and archaeal antivirus defense. Except for the CRISPR-Cas systems, different classes of defense systems, in particular toxin-antitoxin and restriction-modification systems, show nonrandom clustering in defense islands. It remains unclear to what extent these associations reflect functional cooperation between different defense systems and to what extent the islands are genomic "sinks" that accumulate diverse nonessential genes, particularly those acquired via horizontal gene transfer. The characteristics of defense islands resemble those of mobilome islands. Defense and mobilome genes are nonrandomly associated in islands, suggesting nonadaptive evolution of the islands via a preferential attachment-like mechanism underpinned by the addictive properties of defense systems such as toxins-antitoxins and an important role of horizontal mobility in the evolution of these islands.
URLPMID:3632139 [本文引用: 1]
Abstract Our knowledge of prokaryotic defense systems has vastly expanded as the result of comparative genomic analysis, followed by experimental validation. This expansion is both quantitative, including the discovery of diverse new examples of known types of defense systems, such as restriction-modification or toxin-antitoxin systems, and qualitative, including the discovery of fundamentally new defense mechanisms, such as the CRISPR-Cas immunity system. Large-scale statistical analysis reveals that the distribution of different defense systems in bacterial and archaeal taxa is non-uniform, with four groups of organisms distinguishable with respect to the overall abundance and the balance between specific types of defense systems. The genes encoding defense system components in bacterial and archaea typically cluster in defense islands. In addition to genes encoding known defense systems, these islands contain numerous uncharacterized genes, which are candidates for new types of defense systems. The tight association of the genes encoding immunity systems and dormancy- or cell death-inducing defense systems in prokaryotic genomes suggests that these two major types of defense are functionally coupled, providing for effective protection at the population level.
URL [本文引用: 2]
URLPMID:24531762 [本文引用: 2]
RNA interference is widely distributed in eukaryotes and has a variety of functions, including antiviral defence and gene regulation. All RNA interference pathways use small single-stranded RNA (ssRNA) molecules that guide proteins of the Argonaute (Ago) family to complementary ssRNA targets: RNA-guided RNA interference. The role of prokaryotic Ago variants has remained elusive, although bioinformatics analysis has suggested their involvement in host defence. Here we demonstrate that Ago of the bacterium Thermus thermophilus (TtAgo) acts as a barrier for the uptake and propagation of foreign DNA. In vivo, TtAgo is loaded with 5'-phosphorylated DNA guides, 13-25 nucleotides in length, that are mostly plasmid derived and have a strong bias for a 5'-end deoxycytidine. These small interfering DNAs guide TtAgo to cleave complementary DNA strands. Hence, despite structural homology to its eukaryotic counterparts, TtAgo functions in host defence by DNA-guided DNA interference.
[本文引用: 3]
URLPMID:25452498 [本文引用: 2]
Abstract The perpetual arms race between bacteria and phage has resulted in the evolution of efficient resistance systems that protect bacteria from phage infection. Such systems, which include the CRISPR-Cas and restriction-modification systems, have proven to be invaluable in the biotechnology and dairy industries. Here, we report on a six-gene cassette in Bacillus cereus which, when integrated into the Bacillus subtilis genome, confers resistance to a broad range of phages, including both virulent and temperate ones. This cassette includes a putative Lon-like protease, an alkaline phosphatase domain protein, a putative RNA-binding protein, a DNA methylase, an ATPase-domain protein, and a protein of unknown function. We denote this novel defense system BREX (Bacteriophage Exclusion) and show that it allows phage adsorption but blocks phage DNA replication. Furthermore, our results suggest that methylation on non-palindromic TAGGAG motifs in the bacterial genome guides self/non-self discrimination and is essential for the defensive function of the BREX system. However, unlike restriction-modification systems, phage DNA does not appear to be cleaved or degraded by BREX, suggesting a novel mechanism of defense. Pan genomic analysis revealed that BREX and BREX-like systems, including the distantly related Pgl system described in Streptomyces coelicolor, are widely distributed in ~10% of all sequenced microbial genomes and can be divided into six coherent subtypes in which the gene composition and order is conserved. Finally, we detected a phage family that evades the BREX defense, implying that anti-BREX mechanisms may have evolved in some phages as part of their arms race with bacteria. 漏 2014 The Authors.
URLPMID:28657885 [本文引用: 1]
Abstract Evolution of bacteria and archaea involves an incessant arms race against an enormous diversity of genetic parasites. Accordingly, a substantial fraction of the genes in most bacteria and archaea are dedicated to antiparasite defense. The functions of these defense systems follow several distinct strategies, including innate immunity; adaptive immunity; and dormancy induction, or programmed cell death. Recent comparative genomic studies taking advantage of the expanding database of microbial genomes and metagenomes, combined with direct experiments, resulted in the discovery of several previously unknown defense systems, including innate immunity centered on Argonaute proteins, bacteriophage exclusion, and new types of CRISPR-Cas systems of adaptive immunity. Some general principles of function and evolution of defense systems are starting to crystallize, in particular, extensive gain and loss of defense genes during the evolution of prokaryotes; formation of genomic defense islands; evolutionary connections between mobile genetic elements and defense, whereby genes of mobile elements are repeatedly recruited for defense functions; the partially selfish and addictive behavior of the defense systems; and coupling between immunity and dormancy induction/programmed cell death.
URLPMID:5426118 [本文引用: 1]
The evolution of CRISPR-cas loci, which encode adaptive immune systems in archaea and bacteria, involves rapid changes, in particular numerous rearrangements of the locus architecture and horizontal transfer of complete loci or individual modules. These dynamics complicate straightforward phylogenetic classification, but here we present an approach combining the analysis of signature protein families and features of the architecture of cas loci that unambiguously partitions most CRISPR-cas loci into distinct classes, types and subtypes. The new classification retains the overall structure of the previous version but is expanded to now encompass two classes, five types and 16 subtypes. The relative stability of the classification suggests that the most prevalent variants of CRISPR-Cas systems are already known. However, the existence of rare, currently unclassifiable variants implies that additional types and subtypes remain to be characterized.
URLPMID:27401123 [本文引用: 1]
Abstract Clustered regularly-interspaced short palindromic repeat (CRISPR) sequences cooperate with CRISPR-associated (Cas) proteins to form the basis of CRISPR-Cas adaptive immune systems in prokaryotes. For more than 20 years, these systems were of interest only to specialists, mainly molecular microbiologists, who tried to understand the properties of this unique defense mechanism. In 2012, the potential of CRISPR-Cas systems was uncovered and these were presented as genome-editing tools with an outstanding capacity to trigger targeted genetic modifications that can be applied to virtually any organism. Shortly thereafter, in early 2013, these tools were shown to efficiently drive specific modification of mammalian genomes. This review attempts to summarize, in a comprehensive manner, the key events and milestones that brought CRISPR-Cas technology from prokaryotes to mammals. Copyright 脗漏 2016 Elsevier Ltd. All rights reserved.
URLPMID:4691850 [本文引用: 1]
Argonaute proteins are conserved throughout all domains of life. Recently characterized prokaryotic Argonaute proteins (pAgos) participate in host defense by DNA interference, whereas eukaryotic Argonaute proteins (eAgos) control a wide range of processes by RNA interference. Here we review molecular mechanisms of guide and target binding by Argonaute proteins, and describe how the conformational changes induced by target binding lead to target cleavage. On the basis of structural comparisons and phylogenetic analyses of pAgos and eAgos, we reconstruct the evolutionary journey of the Argonaute proteins through the three domains of life and discuss how different structural features of pAgos and eAgos relate to their distinct physiological roles.
[本文引用: 1]
URLPMID:2154333 [本文引用: 1]
A number of mutants of motA , a gene necessary for flagellar rotation in E. coli, were isolated and characterized. Many mutations were dominant, owing to competition between functional and nonfunctional MotA for a limited number of sites on the flagellar motor. A new class of mutant was discovered in which flagellar torque is normal at low speeds but reduced at high speeds. Hydrogen isotope effects on these mutants indicate that MotA catalyzes proton transfer. We confirmed an earlier observation that overproduction of MotA leads to accumulation of the protein in the cytoplasmic membrane and to significant decreases in growth rate. When nonfunctional mutant variants of MotA were overproduced instead, they accumulated in the cytoplasmic membrane, but growth was not impaired. These results also suggest that MotA conducts protons. This was confirmed by measuring the proton permeabilities of vesicles containing wild-type or mutant MotA proteins.
[本文引用: 1]
URLPMID:28092811 [本文引用: 1]
Abstract TIR (Toll/interleukin-1 receptor/resistance protein) domains feature in animal, plant and bacterial proteins involved in innate immunity pathways and associated processes. They function through protein:protein interactions, in particular self-association and homotypic association with other TIR domains. Structures of TIR domains from all phyla have been determined, but common association modes have only emerged for plant and bacterial TIR domains, and not for mammalian TIR domains. Numerous attempts involving hybrid approaches, which have combined structural, computational, mutagenesis and biophysical data, have failed to converge onto common models of how these domains associate and function. We propose that the available data can be reconciled in the context of higher-order assembly formation, and that TIR domains function through signaling by cooperative assembly formation (SCAF). Copyright 漏 2016 Elsevier Ltd. All rights reserved.
URLPMID:15897176 [本文引用: 1]
Chromosome cohesion and condensation are essential prerequisites of proper segregation of genomes during mitosis and meiosis, and are supported by two structurally related protein complexes, cohesin and condensin, respectively. At the core of the two complexes lie members of the structural maintenance of chromosomes (SMC) family of ATPases. SMC proteins are also found in most bacterial and archaeal species, implicating the existence of an evolutionarily conserved theme of higher-order chromosome organization and dynamics. SMC dimers adopt a two-armed structure with an ATP-binding cassette (ABC)-like domain at the distal end of each arm. This article reviews recent work on the bacterial and eukaryotic SMC protein complexes, and discusses current understanding of how these uniquely designed protein machines may work at a mechanistic level. It seems most likely that the action of SMC proteins is highly dynamic and plastic, possibly involving a diverse array of intramolecular and intermolecular protein-protein interactions.
URLPMID:2020 [本文引用: 1]
The circular Escherichia coli chromosome is organized by bidirectional replication into two equal left and right arms (replichores). Each arm occupies a separate cell half, with the origin of replication (oriC) at mid-cell. E. coli MukBEF belongs to the ubiquitous family of SMC protein complexes that play key roles in chromosome organization and processing. In mukBEF mutants, viability is restricted to low temperature with production of anucleate cells, reflecting chromosome segregation defects. We show that in mukB mutant cells, the two chromosome arms do not separate into distinct cell halves, but extend from pole to pole with the oriC region located at the old pole. Mutations in topA, encoding topoisomerase I, do not suppress the aberrant positioning of chromosomal loci in mukB cells, despite suppressing the temperature-sensitivity and production of anucleate cells. Furthermore, we show that MukB and the oriC region generally colocalize throughout the cell cycle, even when oriC localization is aberrant. We propose that MukBEF initiates the normal bidirectional organization of the chromosome from the oriC region.
[本文引用: 1]
URL [本文引用: 1]

{kind=link}
{kind=link}