 ,四川农业大学,畜禽遗传资源发掘与创新利用四川省重点实验室,成都 611130
,四川农业大学,畜禽遗传资源发掘与创新利用四川省重点实验室,成都 611130Regulatory mechanism for lncRNAs in skeletal muscle development and progress on its research in domestic animals
Rui Zhou, Yixin Wang, Keren Long, Anan Jiang, Long Jin,Farm Animal Genetic Resources Exploration and Innovation Key Laboratory of Sichuan Province, Sichuan Agricultural University, Chengdu 611130, China通讯作者:
编委: 任军
收稿日期:2017-12-18修回日期:2018-03-1网络出版日期:2018-04-20
| 基金资助: |
Editorial board:
Received:2017-12-18Revised:2018-03-1Online:2018-04-20
| Fund supported: |
作者简介 About authors
周瑞,硕士研究生,专业方向:动物遗传育种与繁殖E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (577KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
周瑞, 王以鑫, 龙科任, 蒋岸岸, 金龙. LncRNA调控骨骼肌发育的分子机制及其在家养动物中的研究进展. 遗传[J], 2018, 40(4): 292-304 doi:10.16288/j.yczz.17-358
Rui Zhou, Yixin Wang, Keren Long, Anan Jiang, Long Jin.
长链非编码RNA (long non-coding RNA, lncRNA)是一类长度大于200 nt的转录本,主要由RNA聚合酶II转录,具有5°端帽子结构和3°端polyA尾巴,能发生可变剪接。过去认为lncRNA因缺乏典型的开放阅读框(open reading frames, ORF)而无法翻译产生蛋白质[1],但是近年来的研究发现lncRNA具有一类能编码小肽的短开放阅读框(small open reading frames, sORF),并且sORF编码的小肽具有重要生物学功能[2,3,4]。这些研究拓展了人们对基因组遗传编码潜力的传统认知,并丰富了lncRNA的种类和功能的多样性。
lncRNA广泛存在于哺乳动物中,其表达具有时空和组织特异性,在表观调控、转录调控及转录后调控等多个层面发挥重要作用[5]。lncRNA在不同生物学过程中扮演重要角色,包括机体抗病免疫、胚胎发育、脂肪代谢以及肿瘤发生等生理和病理过程。同时也在骨骼肌增殖和分化的过程中扮演重要角色(图1)[6]。近年来,高通量测序技术以及新兴分子生物学技术的不断发展极大地促进了骨骼肌lncRNA的研究,尤其是在人(Homo sapiens)和小鼠(Mus musculus)等模式动物上。在农业动物研究领域,虽然已有相当数量的研究对不同家养动物骨骼肌lncRNA进行了鉴定,但目前已明确具有重要功能的lncRNA仍然相对较少。因此,加速家养动物重要经济性状相关主效lncRNA的鉴定及其分子调控机制的解析将为畜禽重要经济性状的遗传改良提供必要的理论基础。本文综述了lncRNA在人和小鼠骨骼肌发育中的作用机制,lncRNA的研究方法以及lncRNA在家养动物骨骼肌发育中的相关研究,并对未来研究方向进行了展望,以期为深入理解和挖掘lncRNA在家养动物骨骼肌发育中的作用提供参考。
图1
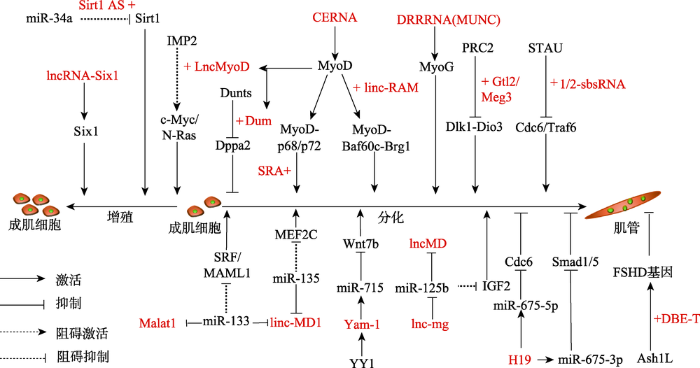 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1lncRNA在骨骼肌增殖和分化中的作用
lncRNAs、肌肉特异性转录因子、MyoD和MyoG组成的信号通路调控成肌细胞增殖以及成肌细胞分化为肌管,其中lncRNAs用红色字体表示。参考文献[6]修改绘制。
Fig. 1Role of lncRNAs in skeletal muscle proliferation and differentiation
1 lncRNA在骨骼肌发育中的分子调控机制
转录组测序分析(RNA-seq)已在多个物种中鉴定出大量与骨骼肌生成相关的lncRNAs。研究发现,这些lncRNAs可以通过影响表观遗传修饰,作为分子海绵以及编码功能性小肽等多种方式调控基因的表达(表1)。Table 1
表1
表1 骨骼肌发育调控相关主要lncRNAs
Table 1
| lncRNA | 肌肉分化调控 | 种类 | 主要定位 | 功能 | 效应分子 | 参考文献 |
|---|---|---|---|---|---|---|
| Malat1 | 上调 | 基因间 | 细胞核 | miR-133海绵,增加SRF水平,促进成肌细胞分化 | SRF;SR蛋白 | [9~11, 25] |
| linc-MD1 | 上调 | 基因间 | 细胞质 | miR-133、miRNA-135海绵;促进肌源性分化 | HuR;MEF2C; MAML1 | [12, 13] |
| H19 | 上调 | 基因间 | 细胞质和细胞核 | 表观遗传抑制;let-7 miRNA海绵;miRNA前体 | PCR2;let-7 | [15, 20] |
| lnc-mg | 上调 | - | - | miRNA-125b海绵,调控IGF2水平 | miR-125b | [16] |
| LncMyoD | 上调 | - | 细胞质 | 促进成肌细胞分化和早期肌肉再生;竞争性结合IMP2 | IMP2 | [8] |
| Dum | 上调 | 基因间 | 细胞核和细胞质 | 甲基化修饰;通过募集Dnmts沉默Dppa2基因 | Dnmts;Dppa2 | [17] |
| DBE-T | 上调 | - | - | 增加FSHD候补基因的表达 | - | [18] |
| Gtl2/Meg3 | 上调 | - | 细胞核 | 招募PRC2来抑制Dlk1-Dio3区域转录出的许多基因 | PCR2 | [21] |
| 1/2-sbsRNAs | 上调 | - | 细胞质 | 介导mRNA降解;抑制成肌细胞分化 | STUA1; STUA2 | [24] |
| Msx1 AS | - | 反义链 | - | 干扰pre-mRNA剪接 | Msx1 | [26, 27] |
| Sirt1 AS | 下降 | 反义链 | 细胞质 | 竞争性内源RNA (ceRNA) | miR-34a | [28, 29] |
| DRRRNA(MUNC) | 上调 | 基因间 | 细胞核 | 作为MyoG位点的增强子;促进肌源性分化 | MyoG | [30, 31] |
| CERNA | 上调 | 基因间 | 细胞核 | 作为 MyoD位点的增强子;促进肌源性分化 | MyoD | [31] |
| SRA | 上调 | - | 细胞核和细胞质 | 转录辅激活因子;骨架分子;促进肌源性分化 | MyoD | [32, 33] |
| Yam-1 | 上调/ 下调 | - | 细胞核和细胞质 | 抑制肌源性分化;激活转录miR-715靶向Wnt7b发挥作用 | miR-715 | [34] |
| linc-RAM | 上调 | 基因间 | - | linc-RAM增强MyoD表达;促进肌源性分化 | MyoD | [35] |
新窗口打开|下载CSV
1.1 lncRNA作为分子海绵调节骨骼肌分化
细胞质中的lncRNAs可以作为microRNA (miRNA)海绵(miRNA sponges)和蛋白海绵(protein sponges)[6]发挥功能。作为miRNA海绵的lncRNA通常被称为内源竞争性RNA (competing endogenous RNA, ceRNA),其通过竞争性结合miRNA,降低miRNA对目标mRNA的影响从而正向调控靶基因的表达[7];而lncRNA作为蛋白海绵是通过与靶基因的mRNA竞争性结合某一类蛋白,从而反向调节靶基因的表达[8]。lncRNA Malat1 (metastasis-associated lung adenocarcinoma transcript 1)在成肌细胞分化过程中上调表达,沉默Malat1会抑制成肌细胞增殖和分化[9]。众所周知,血清应答因子SRF (serum response factor)是肌肉增殖和分化的重要转录因子[10]。Han等[11]研究发现,Malat1和SRF具有同一miRNA靶位点,Malat1作为ceRNA竞争性结合miR-133,促进SRF的表达和肌细胞的分化。同样,linc-MD1 (long intergenic noncoding RNA muscle differentiation 1)也通过ceRNA机制调控骨骼肌分化。在小鼠和人的成肌细胞中,linc-MD1竞争性结合miR-133和miR-135,从而分别激活靶基因MAML1和MEF2C,加速肌肉过渡到分化后期[12](图2A)。同时,linc-MD1对miR-133的海绵作用也能上调RNA结合蛋白HuR的表达,HuR与linc-MD1结合可以阻碍Drosha酶对linc-MD1的分解。因此,HuR与linc-MD1可形成一个正反馈通路促进成肌细胞的早期分化。伴随着分化的进行,miR-133的合成增多并抑制HuR的表达,结束这一正反馈通路,成肌细胞进入下一步的分化[13]。H19是研究较早的lncRNA之一,在动物机体内发挥着广泛的作用,可以通过顺式和反式作用于靶基因参与哺乳动物胚胎各组织生长发育的调控[14]。lncRNA H19有多种作用机制,其中一种就是作为let-7 miRNA的分子海绵,阻碍let-7对靶基因Hmga2和 Dicer的抑制作用,进而阻止肌肉分化的过早发育[15](图2A)。暨南大学王晓刚课题组新发现了一个可以促进骨骼肌干细胞分化和肌肉再生的lncRNA并命名为lnc-mg (myogenesis-associated lncRNA),其作为ceRNA竞争性结合miR-125b,提高胰岛素样生长因子2 (insulin-like growth factor 2, IGF2)的蛋白水平,激活骨骼肌干细胞分化和再生的下游信号通路,从而促进肌肉生成。以上研究证明lncRNA作为内源竞争性RNA在肌肉发育中发挥重要作用[16]。
图2
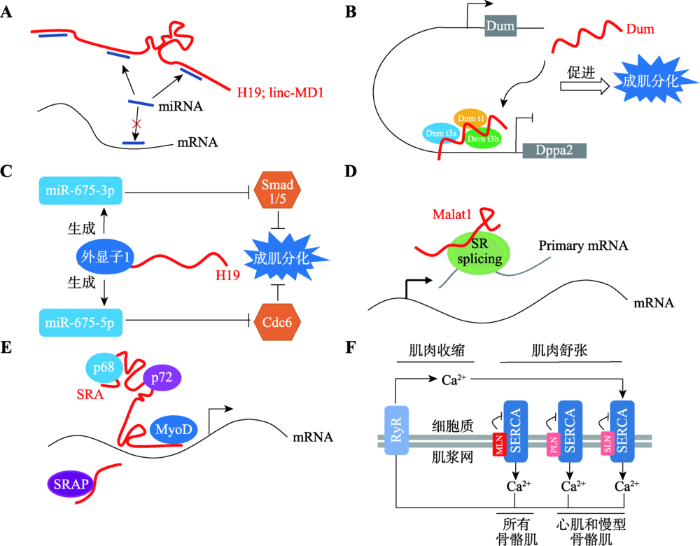 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2lncRNA调控基因表达的不同分子机制
A:lncRNA作为分子海绵调控基因表达。以lncRNA H19和linc-MD1为例,其作为miRNA海绵,阻碍miRNA对mRNA表达的抑制作用;B:lncRNA通过影响DNA甲基化调控基因表达。以lncRNA Dum为例,其通过招募多种DNA甲基化转移酶到Dppa2基因启动子上导致Dppa2基因沉默;C:lncRNA作为印记基因调控基因表达。以lncRNA H19为例,H19基因第一外显子产生的miR-675-3p和miR-675-5p可分别抑制Smad和Cdc6基因的表达;D:lncRNA通过调节靶mRNA的剪切、稳定性和丰度调控基因表达,以lncRNA Malat1为例,其通过招募SR剪接因子影响可变剪接体的形成;E:lncRNA通过影响染色质修饰和转录因子活性调控基因表达。以lncRNA SRA为例,其可作为 p68/p72和MyoD的分子支架介导MyoD对靶基因的转录激活;F:lncRNA通过编码小肽调控基因表达。以小肽MLN为例,其通过直接作用于肌浆网上的SERCA,阻止Ca2+进入肌浆网。参考文献[3,17,37,38]绘制。
Fig. 2lncRNAs regulate gene expression through diverse modes of action
lncRNA以这种海绵吸附机制发挥调控作用还体现在lncRNA竞争性结合RNA结合蛋白上。例如,Gong等[8]研究发现LncMyoD是一条由MyoD临近基因编码的lncRNA,在成肌细胞分化过程中MyoD基因直接介导LncMyoD转录激活从而影响LncMyoD的表达水平。LncMyoD竞争性结合IGF2 mRNA结合蛋白2(IGF2-mRNA-binding protein 2, IMP2)使得IMP2介导的N-Ras和c-Myc两种增殖基因的翻译受阻。敲除LncMyoD将导致细胞周期循环无法结束,进而强有力地抑制成肌细胞终末分化的发生。
1.2 lncRNA通过影响DNA甲基化和组蛋白修饰调控肌肉发育
lncRNA可以通过介导DNA甲基化和组蛋白修饰进而调控染色质状态,实现对基因转录的调控,最终导致表型的变化。在小鼠成肌细胞中,Dppa2基因上游的转录产物lncRNA Dum (developmental pluripotency-associated 2 (Dppa2) upstream binding muscle lncRNA)通过招募多种DNA甲基化转移酶—Dnmts (Dnmt1、Dnmt3a和Dnmt3b)到Dppa2基因启动子区域,导致Dppa2基因沉默,进而促进成肌细胞的分化和肌肉再生(图2B)[17]。此外,DBE-T (DBE-transcript)也是一种在面肩肱肌营养不良症(facioscapulohumeral muscular dystrophy, FSHD)患者中异常表达的lncRNA。DBE-T通过招募Trithorax家族的Ash1L蛋白到第4号染色体第35区(4q35),使组蛋白H3K36甲基化及染色质重塑从而激活4q35区域基因的转录并诱发FSHD[18]。
1.3 lncRNA作为印记基因在骨骼肌生长发育中的作用
印记基因在机体生长和发育中起着关键的调节作用。大部分基因印记区域可编码至少一种lncRNAs,这些lncRNAs通过顺式或反式作用改变染色质状 态[19]。lncRNA H19 转录自H19/IGF2基因座母体等位基因,在发育的胚胎和成年机体的肌肉中高表达。H19通过结合PCR2 (polycomb repressive complex 2)复合物,抑制IGF2的转录。此外,H19基因第1外显子产生的miR-675-3p和miR-675-5p,可分别抑制Smad和Cdc6基因的表达,表明H19能通过自身编码的miRNAs介导的反式调节作用影响骨骼肌的分化和再生活动(图2C)[20]。Gtl2是Dlk1-Dio3印记区域内一个母源性表达的lncRNA,Meg3是Gtl2在人上的同源异型基因。Glt2/Meg3 lncRNA通过招募PRC2复合物到Dlk1-Dio3区域,直接沉默来自Dlk1-Dio3区域的许多基因,如Dlk1[21]。此外,敲除Gtl2/Meg3将导致小鼠围产期的死亡以及骨骼肌发育的缺陷[22]。在小鼠和绵羊(Ovis aries)中,Dlk1- Dio3印记簇的异常激活是双肌臀形成的原因[23]。1.4 lncRNA通过调节靶mRNA的剪切、稳定性和丰度影响骨骼肌的生长发育
RNA结合蛋白STAU1与mRNA的3°UTR中的Staufen1蛋白结合位点相互作用可以介导SMD (Staufen1(STAU1)-mediated mRNA decay)降解途径。含SINE (short interspersed element)序列的lncRNA通过与mRNA的3°UTR中的Alu元件结合,招募STAU1蛋白来启动SMD降解途径。例如,1/2-sbsRNA lncRNA通过结合Cdc6 mRNA和Traf6 mRNA 3'UTR区域,导致Cdc6 mRNA和Traf6 mRNA降解,从而降低C2C12细胞中Cdc6和Traf6蛋白水平[24]。在前体mRNA (pre-mRNA)的加工过程中,核内核斑(nuclear speckles)的SR (serine/arginine-rich)蛋白家族具有重要作用。Malat1通过与SR蛋白特异性结合,影响剪接因子的分布。敲除Malat1或者过表达SR蛋白都会改变pre-mRNA的可变剪接,并且发现Malat1还能调节SR蛋白的磷酸化水平,从而调控其活性(图2D)[25]。有研究报道,过表达Msx1会阻碍成肌细胞的分化甚至使肌管逆转成增殖的成肌细胞[26]。这说明Msx1基因在肌肉发育中发挥重要的调控作用。lncRNA—Msxl AS转录自Msx1基因的反义链。Wakkach等[27]研究发现Msx1 AS通过与Msx1基因转录的pre-mRNA配对结合,干扰pre-mRNA的剪接过程,阻碍Msx1蛋白的表达。已知Sirt1是一种依赖NAD的去乙酰酶,参与平衡成肌细胞的增殖和分化[28]。Wang等[29]研究发现,Srit1 AS通过与miR- 34a竞争性结合Sirt1 mRNA 3'UTR区域来促进Sirt1的表达并提高Sirt1 mRNA稳定性,进而促进C2C12细胞增殖、抑制其分化。1.5 lncRNA通过调控染色质修饰和转录因子活性调节肌肉发育
研究表明,部分核内lncRNAs具有类似增强子的作用,称为增强子相关RNA (enhancer-associated RNA, eRNA),它们可以激活邻近蛋白编码基因的表达。例如,MyoD基因的远端调控区域(distal regulatory regions, DRR)和核心增强子区域(core enhancer, CE)产生两种eRNA—CERNA和DRRRNA(也称为MUNC)[30],CERNA通过增加染色质的“粘性”使RNA聚合酶II (RNAP II)容易结合,以顺式作用方式促进MyoD的表达;而DRRRNA通过反式作用促进MyoG的表达和肌肉的分化[31]。此外,有些核内lncRNAs还可以通过影响序列特异性转录因子的活性控制基因表达,lncRNA SRA (steroid receptor RNA activator)就是其中一种。在肌肉分化时,SRA lncRNA作为p68/p72和MyoD的分子支架介导MyoD对靶基因的转录激活[32]。SRA转录本的可变剪接体还能编码一种SRAP蛋白,SRAP富集在成肌细胞中,会阻碍SRA介导的MyoD的转录活性和肌肉的分化(图2E)[33]。因此,适当的SRA lncRNA与SARP蛋白水平对于维持肌肉的正常分化是非常重要的。Lu等[34]发现一种受转录因子YY1正向调控的lncRNA— Yam-1,Yam-1通过激活miR-715、抑制Wnt7b的表达从而发挥肌细胞抑制因子的功能。Yu等[35]发现在小鼠肌细胞分化过程中,MyoD会激活linc-RAM (linc-RNA activator of myogenesis)的表达,随后linc-RAM与调控因子MyoD结合,促进表观遗传复合物MyoD-Baf60c-Brg1的装配,改变MyoD靶基因染色体的构象,进而影响下游肌源性分化基因的表达。并且linc-RAM的这种功能独立于linc-RAM编码的小肽—myoregulin(MLN)。1.6 lncRNA通过编码小肽调节肌肉性能
过去对lncRNA的定义是不编码蛋白的RNA,但随着研究的深入,研究人员发现部分lncRNAs不但能翻译小肽,还能利用其编码的小肽行使生物学功能。MLN是lncRNA linc-RAM (LINC00948 in humans and 2310015B20Rik in mice)编码的小肽[3, 35]。MLN的功能类似于受磷蛋白(PLN)、肌脂蛋白(SLN),通过直接作用于肌浆网上的肌浆网钙泵(SERCA)并阻止Ca2+进入肌浆网。MLN几乎在所有骨骼肌中表达,敲除MLN将增强Ca2+的传递效率并提升骨骼肌运动能力(图2F)[3]。最近,Matsumoto等[36]发现,lncRNA LINC00961可以翻译一个由90个氨基酸组成的小肽,并命名为SPAR。SPAR定位于晚期核内体和溶酶体中,负调控mTORC1的活性。在骨骼肌急性损伤后,LINC00961表达下调,激活mTORC1并促进肌肉再生。2 lncRNA功能研究方法
lncRNA通过多种作用方式调控基因表达,而lncRNA分子作用机制的揭示需要依赖于多种分子生物学研究方法。目前,探索lncRNA功能的方法主要包括:(1) lncRNA的亚细胞的定位;(2) lncRNA靶基因预测;(3) lncRNA与蛋白质的相互作用; (4) lncRNA基因敲除、RNAi(RNA interference)和基因过表达。2.1 亚细胞定位
亚细胞定位是指某一种表达产物在细胞内的具体存在位置,例如在核内、胞质内或者细胞膜上存在。lncRNA的功能很大程度上决定于其亚细胞定位的特异性。细胞质中lncRNAs的主要作用包括:作为ceRNA竞争性结合miRNA;影响mRNA的稳定性;部分lncRNAs通过编码小肽行使功能等。细胞核内lncRNAs的主要功能有:作为eRNA招募RNAPII;影响转录因子活性;影响RNA的可变剪接等[39]。RNA-FISH技术能对lncRNA进行检测和定位。Tripathi等[25]利用该技术确定了Malat1主要位于细胞分裂间期的核斑结构中。此外,RNA-FISH与DNA-FISH结合可以验证转录本与染色质序列的共定位[40]。FISSEQ (fluorescent in situ sequencing)是一种FISH和RNA-seq相结合的新测序方案,能在完整的细胞和组织中获得全基因组范围内的基因表达图谱,具有更高的分辨率,并且比RNA-FISH鉴定更多的靶标[40,41]。除此之外,利用RNA亚细胞定位检索工具RNALocate (http://www.rna-society.org/ rnalocate),有助于预测lncRNA在细胞中的位置,为进一步的研究提供重要参考[42]。2.2 lncRNA的靶标预测
通过整合lncRNA序列和功能的信息,国内外的研究机构建立了多个集查阅、共享、注释和分析等功能为一体的lncRNA数据库,为lncRNA的研究提供了有效工具。lncRNA数据库大致可分为:lncRNA原始资源整合数据库;lncRNA筛选及鉴定数据库;lncRNA功能分析数据库等几大类。在对lncRNA的靶标基因进行研究时,利用相关数据库预测lncRNA的靶标,将大大减少工作量和花费。常见的lncRNA靶标预测数据库详细信息见表2。Table 2
表2
表2 常见预测lncRNA靶标的数据库信息
Table 2
| 数据库名称 | 网址 | 说明 | 参考文献 |
|---|---|---|---|
| ChIPBase | http://rna.sysu.edu.cn/chipbase/ | 利用ChIP-seq数据分析miRNA-lncRNA的转录调节关系。 | [43] |
| DIANA-LncBase | www.microrna.gr/LncBase | CLIP-seq实验验证的和计算机预测的miRNA- lncRNA相互作用信息。 | [44] |
| lncRNA2Target | http://mlg.hit.edu.cn/lncrna2target/ | lncRNA靶基因数据库,提供lncRNA敲除或过表达后引起表达差异的基因。 | [45] |
| lncRNASNP | http://bioinfo.life.hust.edu.cn/lncRNASNP/ | 预测lncRNA上的SNP位点,对miRNA-lncRNA互作结合位点处的SNP进行排序,预测miRNA- lncRNA相互作用。 | [46] |
| NPInter | http://www.bioinfo.org/NPInter/ | 预测ncRNA与其他生物分子(蛋白质、DNA和RNA)之间的相互作用。 | [47] |
| Predicted RNA-RNA interactions | http://rtools.cbrc.jp/cgi-bin/RNARNA/index.pl | 预测lncRNA和mRNA的相互作用。 | [48] |
| starBase v2.0 | http://starbase.sysu.edu.cn/ | 利用CLIP-seq数据中分析miRNA-ceRNA、miRNA- ncRNA和蛋白质-RNA相互作用网络。 | [49] |
新窗口打开|下载CSV
2.3 lncRNA与蛋白质的相互作用
lncRNA在基因表达调控中的作用已经毋庸置疑,其通过RNA-蛋白复合物的形式行使功能,如染色质修饰复合物,转录因子和RNP复合物。因此,研究人员开发了多种鉴定lncRNA-蛋白质相互作用的技术,用于揭示lncRNA在生物学过程中的调控机理。RNA-pulldown实验是在体外初步对RNA和蛋白质的相互作用进行鉴定的方法。通常用于发现与已知RNA结合的未知蛋白质分子[50]。RIP (RNA immunoprecipitation)技术的基本原理类似于ChIP技术,可用于检测单个的目的蛋白质与特定非编码RNA分子之间的相互作用。RIP下游结合微阵列和高通量测序技术被称为RIP-Chip[51]和RIP-seq[21]。RIP-Chip技术的缺点是识别特异性较差,分辨率较低,因此应用范围有限。ChIRP (chromatin isolation by RNA purification)和CHART (capture hybridization analysis of RNA targets)都是基于同样的理念[52]。但两者主要的不同在于lncRNA靶标探针的设计,ChIRP采用lncRNA序列全长覆盖探针设计,靶向所有潜在位点,探针的设计是简单的RNA序列,无需事先的RNA的结构或功能域的知识[53];而CHART通过RNase H试验寻找合适的靶标位点设计探针[54]。CLIP (UV-crosslinking and immunoprecipitation)技术能揭示RNA 结合蛋白质在体内的RNA结合位点。CLIP 技术的优势在于能捕获体内原位蛋白质和RNA的相互作用,紫外交联的复合物能通过严格的纯化,因此实验结果更为真实可靠。但CLIP实验因紫外交联的效率低(1%~5%)、易受非特异RNA污染、实验步骤复杂和RNA容易降解等因素的影响,使用受到限制。针对以上不足研究人员开发了提高紫外交联效率的PAR-CLIP (photoactivatable-ribonucleoside-enhanced CLIP)技术[55]。传统方法的新发展拓展了其应用范围,上述方法的联合应用更有利于揭示lncRNA的生物学功能(表3)。
Table 3
表3
表3 研究lncRNA与蛋白质相互作用的技术方法
Table 3
| 方法 | 研究内容 | 特点 | 应用 | 参考文献 |
|---|---|---|---|---|
| RNA pulldown | 研究与目的RNA互作的蛋白质 | (1)体外转录合成单链RNA探针 (2)标记的RNA捕获RBPs (3)富集内源、过表达和体外翻译的蛋白 | 在C2C12细胞中,利用体外生成的生物素标记的Dum转录本证实Dum与大量Dnmts蛋白结合[17] | [16] |
| RIP和RIP-Chip/seq | 研究与目的蛋白互作的RNA | (1)检测单个蛋白于特定RNA的相互作用 (2)利用微阵列或者高通量测序发现RNA的种类 (3)通过qRT-PCR检测RNA | 全基因组范围内筛选与polycomb相关的RNA,如Dlk1[21]。RIP实验证明LncMyoD直接与IMPs结合[8] | [8] |
| ChIRP和CHART | 研究与目的RNA互作的蛋白质和DNA | (1)短DNA寡核苷酸探针靶向RNA (2)高通量筛选RNA结合的蛋白和DNA | 运用改良的ChIRP技术(dChIRP),揭示了lncRNA Dum直接与Dpp2a基因的启动子结合[17] | [16] |
| CLIP | 研究与目的蛋白互作的RNA | (1)通过光反应或者化学反应交联RNA和蛋白质 (2)体内捕获RNA-蛋白质的相互作用 (3)高通量筛选与蛋白质结合的RNA | 利用改良的CLIP 技术(PAR-CLIP),筛选与AUF1蛋白互作的RNA,如lncRNA NEAT1[56] | [56] |
新窗口打开|下载CSV
2.4 基因敲除和过表达实验
在探究新鉴定的lncRNAs功能时,通过对候选lncRNAs进行过表达或敲除进而直接观察表型变化以推测其生物学功能是一种常用的手段。近年来,利用病毒(慢病毒、腺病毒等)为载体介导的外源性shRNA (short hairpin RNA)或者lncRNA在受体细胞中表达的方法因其转染效率高、目的基因可稳定表达等优势被广泛应用。值得注意的是,RNAi主要在细胞质中起作用,细胞核内lncRNAs具有高度二级结构而且常常与DNA、蛋白质相互结合形成复合体,影响RNAi与lncRNA的结合效率,因此采用反义寡核苷酸(antisense oligonucleotides, ASO)实现对细胞核内lncRNAs的敲除和干扰将更为有效。例如,Cui等[57]证明利用过表达质粒上调lncRNA HULC加速了HepG2和Huh7细胞中脂肪的生成,而HULC siRNA (small interfering RNA)降低了HepG2.2.15细胞中脂肪的生成。此外,基因编辑技术如CRISPR/ Cas9系统能以更精确、有效的手段对基因进行删除、插入,实现基因表达的上调、敲降。该技术的出现有望快速提高家养动物生产性能,例如,中国科学院动物研究所赵建国课题组利用CRISPR/Cas9技术构建了UCP1基因定点敲入的猪(Sus scrofa),实现了UCP1基因在猪白色脂肪组织的特异表达,敲入该基因的猪脂肪率和背膘厚显著降低,瘦肉率和抗寒能力显著增加[58]。基因敲除和过表达的技术多种多样,因此在实验中需根据实验目的、实验材料等内容选用合适的方法。
3 家养动物骨骼肌lncRNA研究进展
借助于高通量二代测序技术的迅猛发展,目前在家养动物的研究中已经鉴定出相当数量的lncRNAs。以下对家养动物骨骼肌生长相关的潜在调控lncRNAs以及家养动物骨骼肌中lncRNAs的筛选相关研究进行简要综述。3.1 家养动物中骨骼肌生长相关lncRNA的鉴定及功能
有研究报道[59],在猪胎儿滋养层中发现的lncRNA—TncRNA在猪胚胎骨骼肌中表达上调,在通城猪和长白猪妊娠第90天胎儿的骨骼肌中差异表达,说明其可能影响猪骨骼肌的胚胎发育。Sun等[60]揭示了牛(Bos taurus) lncRNA—lncMD的作用机制。lncMD作为miR-125b海绵,减少miR-125b对IGF2基因的抑制作用,进而促进骨骼肌分化。Cai等[61]在隐性白洛克鸡(Gallus gallus)和杏花鸡差异表达的lncRNAs中发现了lncRNA-Six1。该基因在鸡胸肉组织中高表达。同时,lncRNA-Six1可生成一个约7.26 kDa的小肽,该小肽在lncRNA-Six1顺式激活Six1基因的过程中起作用,但具体的机制尚不清晰。Yue等[62]对牛背最长肌、肩胛、肋间和臀肌四种肌肉组织进行转录组测序分析,发现lncYYW正向调节牛成肌细胞中GH1 (growth hormone 1)基因及其下游基因AKT1和PIK3CD的表达。lncYYW在成肌细胞分化过程中上调表达,过表达lncYYW能增加细胞周期中S期的细胞数目。3.2 家养动物中骨骼肌生长相关lncRNA的高通量筛选
Zhao等[63]对猪胎儿骨骼肌进行转录组测序分析,鉴定了570种包含多个外显子的基因间长链非编码RNA (long intergenic non-coding RNA, lincRNA)。Xing等[64]首次检测了阉割对猪肌肉中lncRNA的影响,构建了阉割和非阉割的淮南公猪背最长肌的转录组图谱,鉴定了8946种lncRNAs,其中有385个在非阉割组和去势组之间差异表达,这些差异表达的lncRNAs及其靶基因可能参与雌激素受体信号通路(ESR signaling)以及骨骼和肌肉发育过程。Li等[65]利用RNA-seq技术在鸡不同胚胎时期的骨骼肌中鉴定了281个新的lincRNAs。Ouyang等[66]通过iTRAQ技术获得了杏花鸡11日龄、16日龄和孵化后1日龄腿肌的蛋白质表达图谱。通过整合这三个阶段蛋白质组学数据和lncRNA数据[67],发现差异表达lncRNAs的一些靶基因(DMD、MYL3、TNNI2、TNNT3)与肌肉收缩功能相关,并推测lnc00068445,lnc00037615,lnc00037619的靶标分别是TNNI2、TNNT3,DMD以及MYL3。该研究为鸡胚胎期肌肉发育的分子机制研究提供了大量候选基因。Zhan等[68]对山羊(Capra hircus)45日龄、60日龄和105日龄胎儿和3日龄羔羊背最长肌组织进行转录组测序,鉴定出3981个在四个阶段都高度保守的lncRNAs,通过两两时间点比较发现了577个差异表达的lncRNAs,这些lncRNAs可能在山羊早期肌肉发育中具有特定的生物学作用。Ren等[69]利用strand-specific Ribo-Zero RNA技术对湖羊3个重要发育阶段(胎儿、羊羔、成年)的背最长肌进行测序,共获得6924个lncRNAs。其中差异表达的lncRNAs可能与胚胎期器官形态发生、骨骼系统发育和肌肉发生等生物学过程相关。该研究首次对湖羊肌肉中的lncRNAs进行系统分析并为绵羊的肌肉发育研究提供宝贵资源。Billerey等[70]使用paired-end RNA-seq对9头利木赞牛犊的胸最长肌组织进行转录组测序,发现418个高置信度的lincRNAs,其中一些lincRNAs 位于肉质量性状相关的QTL位点。上述研究为探究lncRNA在家养动物骨骼肌中发挥的功能提供了重要参考。4 结语与展望
对模式动物骨骼肌lncRNA进行的研究使人们认识到lncRNA在调控骨骼肌生长发育过程中调控作用的重要性以及调控方式的复杂性。在此基础上,近年来畜禽研究者在家养动物上开展了大量lncRNA的研究工作,尤其是对重要经济性状—骨骼肌生长的研究最为广泛。通过鉴定骨骼肌相关主效lncRNA,揭示其分子作用机理,有助于农业动物产肉性状的改良。但目前研究工作还停留在lncRNA鉴定上,对作用机制缺乏进一步的研究,其主要原因有: (1) lncRNA在物种间的序列保守性不高,导致物种之间lncRNA作用机制的相互借鉴的意义不大; (2)农业动物少有稳定的细胞系,因此很难对鉴定出的lncRNA进行深入的功能研究;(3) lncRNA的表达具有极强的时空特异性,其表达动态变化过程不易捕获,对相关研究造成困难;(4) lncRNA高级结构(二级结构、三级结构和四级结构)影响着lncRNA的作用方式,但目前鲜有研究报道lncRNA的高级结构,这也加大了lncRNA功能研究的难度;(5)农业动物缺乏完整准确的lncRNA数据库。因此,整合不同家养动物lncRNA信息的可靠集合,构建数据库迫在眉睫,同时将不断更新的相关生物学研究技术应用到家养动物lncRNA的挖掘上有助于促进家养动物重要经济性状相关lncRNA功能的研究。特别是基因组编辑技术的出现给农业动物的遗传改良带来了全新的契机,将可能大大缩短农业动物改良的周期,为农业动物的分子育种提供了崭新的思路。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:16141072 [本文引用: 1]

This study describes comprehensive polling of transcription start and termination sites and analysis of previously unidentified full-length complementary DNAs derived from the mouse genome. We identify the 5' and 3' boundaries of 181,047 transcripts with extensive variation in transcripts arising from alternative promoter usage, splicing, and polyadenylation. There are 16,247 new mouse protein-coding transcripts, including 5154 encoding previously unidentified proteins. Genomic mapping of the transcriptome reveals transcriptional forests, with overlapping transcription on both strands, separated by deserts in which few transcripts are observed. The data provide a comprehensive platform for the comparative analysis of mammalian transcriptional regulation in differentiation and development.
URLPMID:4892890 [本文引用: 1]
Abstract Muscle contraction depends on release of Ca(2+) from the sarcoplasmic reticulum (SR) and reuptake by the Ca(2+)adenosine triphosphatase SERCA. We discovered a putative muscle-specific long noncoding RNA that encodes a peptide of 34 amino acids and that we named dwarf open reading frame (DWORF). DWORF localizes to the SR membrane, where it enhances SERCA activity by displacing the SERCA inhibitors, phospholamban, sarcolipin, and myoregulin. In mice, overexpression of DWORF in cardiomyocytes increases peak Ca(2+) transient amplitude and SR Ca(2+) load while reducing the time constant of cytosolic Ca(2+) decay during each cycle of contraction-relaxation. Conversely, slow skeletal muscle lacking DWORF exhibits delayed Ca(2+) clearance and relaxation and reduced SERCA activity. DWORF is the only endogenous peptide known to activate the SERCA pump by physical interaction and provides a means for enhancing muscle contractility. Copyright 脗漏 2016, American Association for the Advancement of Science.
URLPMID:25640239 [本文引用: 3]
Abstract Functional micropeptides can be concealed within RNAs that appear to be noncoding. We discovered a conserved micropeptide, which we named myoregulin (MLN), encoded by a skeletal muscle-specific RNA annotated as a putative long noncoding RNA. MLN shares structural and functional similarity with phospholamban (PLN) and sarcolipin (SLN), which inhibit SERCA, the membrane pump that controls muscle relaxation by regulating Ca(2+) uptake into the sarcoplasmic reticulum (SR). MLN interacts directly with SERCA and impedes Ca(2+) uptake into the SR. In contrast to PLN and SLN, which are expressed in cardiac and slow skeletal muscle in mice, MLN is robustly expressed in all skeletal muscle. Genetic deletion of MLN in mice enhances Ca(2+) handling in skeletal muscle and improves exercise performance. These findings identify MLN as an important regulator of skeletal muscle physiology and highlight the possibility that additional micropeptides are encoded in the many RNAs currently annotated as noncoding. Copyright 脗漏 2015 Elsevier Inc. All rights reserved.
URLPMID:27923914 [本文引用: 1]
Abstract Micropeptides function as master regulators of calcium-dependent signaling in muscle. Sarco/endoplasmic reticulum Ca0562 ATPase (SERCA), the membrane pump that promotes muscle relaxation by taking up Ca0562 into the sarcoplasmic reticulum, is directly inhibited by three muscle-specific micropeptides: myoregulin (MLN), phospholamban (PLN), and sarcolipin (SLN). The widespread and essential function of SERCA across diverse cell types has raised questions as to how SERCA is regulated in cells that lack MLN, PLN, and SLN. We identified two transmembrane micropeptides, endoregulin (ELN) and another-regulin (ALN), that share key amino acids with their muscle-specific counterparts and function as direct inhibitors of SERCA pump activity. The distribution of transcripts encoding ELN and ALN mirrored that of SERCA isoform-encoding transcripts in nonmuscle cell types. Our findings identify additional members of the SERCA-inhibitory micropeptide family, revealing a conserved mechanism for the control of intracellular Ca0562 dynamics in both muscle and nonmuscle cell types.
URLPMID:4394487 [本文引用: 1]
Abstract High-throughput sequencing has revealed that the majority of RNAs have no capacity to encode protein. Among these non-coding transcripts, recent work has focused on the roles of long noncoding RNAs (lncRNAs) of >200 nucleotides. Although many of their attributes, such as patterns of expression, remain largely unknown, lncRNAs have key functions in transcriptional, post-transcriptional, and epigenetic gene regulation; Also, new work indicates their functions in scaffolding ribonuclear protein complexes. In plants, genome-wide identification of lncRNAs has been conducted in several species, including Zea mays, and recent research showed that lncRNAs regulate flowering time in the photoperiod pathway, and function in nodulation. In this review, we discuss the basic mechanisms by which lncRNAs regulate key cellular processes, using the large body of knowledge on animal and yeast lncRNAs to illustrate the significance of emerging work on lncRNAs in plants.
URLPMID:29253523 [本文引用: 3]
Abstract Skeletal muscle myogenesis during development and the injury induced regeneration contribute to the formation and maintenance of muscle tissue. Emerging studies have demonstrated that long non-coding RNAs (lncRNAs) participate in the regulation of gene expression during skeletal myogenesis and their aberrant expression is associated with several muscular diseases. In this review, we summarize recent studies of lncRNAs in the regulation of myogenesis and muscle diseases with mechanistic characterization. These findings have greatly enhanced our understanding of gene regulatory mechanisms governing muscle formation and regeneration, which will eventually lead to novel therapeutics against various muscle diseases.
URLPMID:3235919 [本文引用: 1]
Here, we present a unifying hypothesis about how messenger RNAs, transcribed pseudogenes, and long noncoding RNAs "talk" to each other using microRNA response elements (MREs) as letters of a new language. We propose that this "competing endogenous RNA" (ceRNA) activity forms a large-scale regulatory network across the transcriptome, greatly expanding the functional genetic information in the human genome and playing important roles in pathological conditions, such as cancer.
URLPMID:26143994 [本文引用: 3]
Long non-coding RNAs are regulators of various biological functions. Gong and Li et聽al. show that LncMyoD is a LncRNA target of MyoD during myogenesis and is required for myoblast differentiation by affecting IMP2-mediated mRNA translation. LncMyoD is functionally conserved between mouse and human, despite limited sequence homology.
URLPMID:23485710 [本文引用: 1]
Abstract Myostatin, a member of the transforming growth factor-0205 (TGF-0205) superfamily of secreted proteins, is a potent negative regulator of myogenesis. Free myostatin induces the phosphorylation of the Smad family of transcription factors, which, in turn, regulates gene expression, via the canonical TGF-0205 signaling pathway. There is, however, emerging evidence that myostatin can regulate gene expression independent of Smad signaling. As such, we acquired global gene expression data from the gastrocnemius muscle of C57BL/6 mice following a 6-day treatment with recombinant myostatin compared with vehicle-treated animals. Of the many differentially expressed genes, the myostatin-associated decrease (-11.20-fold; P < 0.05) in the noncoding metastasis-associated lung adenocarcinoma transcript 1 (Malat1) was the most significant and the most intriguing because of numerous reports describing its novel role in regulating cell growth. We therefore sought to further characterize the role of Malat1 expression in skeletal muscle myogenesis. RT-PCR-based quantification of C2C12 and primary human skeletal muscle cells revealed a significant and persistent upregulation (4- to 7-fold; P < 0.05) of Malat1 mRNA during the differentiation of myoblasts into myotubes. Conversely, targeted knockdown of Malat1 using siRNA suppressed myoblast proliferation by arresting cell growth in the G(0)/G(1) phase. These results reveal Malat1 as novel downstream target of myostatin with a considerable ability to regulate myogenesis. The identification of new targets of myostatin will have important repercussions for regenerative biology through inhibition and/or reversal of muscle atrophy and wasting diseases.
URLPMID:15647354 [本文引用: 1]
Abstract Serum response factor (SRF) controls the transcription of muscle genes by recruiting a variety of partner proteins, including members of the myocardin family of transcriptional coactivators. Mice lacking SRF fail to form mesoderm and die before gastrulation, precluding an analysis of the roles of SRF in muscle tissues. To investigate the functions of SRF in skeletal muscle development, we conditionally deleted the Srf gene in mice by skeletal muscle-specific expression of Cre recombinase. In mice lacking skeletal muscle SRF expression, muscle fibers formed, but failed to undergo hypertrophic growth after birth. Consequently, mutant mice died during the perinatal period from severe skeletal muscle hypoplasia. The myopathic phenotype of these mutant mice resembled that of mice expressing a dominant negative mutant of a myocardin family member in skeletal muscle. These findings reveal an essential role for the partnership of SRF and myocardin-related transcription factors in the control of skeletal muscle growth and maturation in vivo.
URLPMID:25868726 [本文引用: 1]
Metastasis-associated lung adenocarcinoma transcript 1 (Malat1) is an example of a functional long noncoding RNA involved in many biologic processes. However, the mechanisms for Malat1 in myogenesis are unclear. Serum response factor (SRF) is a pivotal transcription factor for muscle proliferation and differentiation and is reported to be a target gene for muscle-specific microRNA-133 (miR-133). In this study, we initially found that silencing Malat1 in the mouse myoblast C2C12 cell line inhibited myocyte differentiation and decreased Srf at both the RNA and protein levels. Srf silencing decreased Malat1 expression as well. Further study revealed that Malat1 contained an miR-133 functional target site, and the interplay between Malat1 and Srf was miR-133 dependent. We demonstrated that Malat1 modulates Srf through miR-133 as a competing endogenous RNA and established a novel connection among Malat1, miR-133, and Srf in myoblast differentiation.
URLPMID:22000014 [本文引用: 1]
Abstract Recently, a new regulatory circuitry has been identified in which RNAs can crosstalk with each other by competing for shared microRNAs. Such competing endogenous RNAs (ceRNAs) regulate the distribution of miRNA molecules on their targets and thereby impose an additional level of post-transcriptional regulation. Here we identify a muscle-specific long noncoding RNA, linc-MD1, which governs the time of muscle differentiation by acting as a ceRNA in mouse and human myoblasts. Downregulation or overexpression of linc-MD1 correlate with retardation or anticipation of the muscle differentiation program, respectively. We show that linc-MD1 "sponges" miR-133 and miR-133 [corrected] to regulate the expression of MAML1 and MEF2C, transcription factors that activate muscle-specific gene expression. Finally, we demonstrate that linc-MD1 exerts the same control over differentiation timing in human myoblasts, and that its levels are strongly reduced in Duchenne muscle cells. We conclude that the ceRNA network plays an important role in muscle differentiation. Copyright 漏 2011 Elsevier Inc. All rights reserved.
URLPMID:3919156 [本文引用: 1]
The muscle-specific long noncoding RNA linc-MD1 was shown to be expressed during early phases of muscle differentiation and to trigger the switch to later stages by acting as a sponge for miR-133 and miR-135. Notably, linc-MD1 is also the host transcript of miR-133b, and their biogenesis is mutually exclusive. Here, we describe that this alternative synthesis is controlled by the HuR protein, which favors linc-MD1 accumulation through its ability to bind linc-MD1 and repress Drosha cleavage. We show that HuR is under the repressive control of miR-133 and that the sponging activity of linc-MD1 consolidates HuR expression in a feedforward positive loop. Finally, we show that HuR also acts in the cytoplasm, reinforcing linc-MD1 sponge activity by cooperating for miRNA recruitment. An increase in miR-133 synthesis, mainly from the two unrelated miR-133a coding genomic loci, is likely to trigger the exit from this circuitry and progression to later differentiation stages.
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:3843377 [本文引用: 1]
Abstract Abundantly expressed in fetal tissues and adult muscle, the developmentally regulated H19 long noncoding RNA (lncRNA) has been implicated in human genetic disorders and cancer. However, how H19 acts to regulate gene function has remained enigmatic, despite the recent implication of its encoded miR-675 in limiting placental growth. We noted that vertebrate H19 harbors both canonical and noncanonical binding sites for the let-7 family of microRNAs, which plays important roles in development, cancer, and metabolism. Using H19 knockdown and overexpression, combined with in vivo crosslinking and genome-wide transcriptome analysis, we demonstrate that H19 modulates let-7 availability by acting as a molecular sponge. The physiological significance of this interaction is highlighted in cultures in which H19 depletion causes precocious muscle differentiation, a phenotype recapitulated by let-7 overexpression. Our results reveal an unexpected mode of action of H19 and identify this lncRNA as an important regulator of the major let-7 family of microRNAs. Copyright 脗漏 2013 Elsevier Inc. All rights reserved.
URLPMID:28281528 [本文引用: 1]
Recent studies indicate important roles for long noncoding RNAs (lncRNAs) as essential regulators of myogenesis and adult skeletal muscle regeneration. However, the specific roles of lncRNAs in myogenic differentiation of adult skeletal muscle stem cells and myogenesis are still largely unknown. Here we identify a lncRNA that is specifically enriched in skeletal muscle (myogenesis-associated lncRNA, in short, lnc-mg). In mice, conditional knockout of lnc-mg in skeletal muscle results in muscle atrophy and the loss of muscular endurance during exercise. Alternatively, skeletal muscle-specific overexpression of lnc-mg promotes muscle hypertrophy. In vitro analysis of primary skeletal muscle cells shows that lnc-mg increases gradually during myogenic differentiation and its overexpression improves cell differentiation. Mechanistically, lnc-mg promotes myogenesis, by functioning as a competing endogenous RNA (ceRNA) for microRNA-125b to control protein abundance of insulin-like growth factor 2. These findings identify lnc-mg as a novel noncoding regulator for muscle cell differentiation and skeletal muscle development.
URLPMID:256866992 [本文引用: 3]
Abstract Emerging studies document the roles of long non-coding RNAs (LncRNAs) in regulating gene expression at chromatin level but relatively less is known how they regulate DNA methylation. Here we identify an lncRNA, Dum (developmental pluripotency-associated 2 (Dppa2) Upstream binding Muscle lncRNA) in skeletal myoblast cells. The expression of Dum is dynamically regulated during myogenesis in vitro and in vivo. It is also transcriptionally induced by MyoD binding upon myoblast differentiation. Functional analyses show that it promotes myoblast differentiation and damage-induced muscle regeneration. Mechanistically, Dum was found to silence its neighboring gene, Dppa2, in cis through recruiting Dnmt1, Dnmt3a and Dnmt3b. Furthermore, intrachromosomal looping between Dum locus and Dppa2 promoter is necessary for Dum/Dppa2 interaction. Collectively, we have identified a novel lncRNA that interacts with Dnmts to regulate myogenesis.Cell Research advance online publication 17 February 2015; doi:10.1038/cr.2015.21.
URLPMID:12020 [本文引用: 1]
Repetitive sequences account for more than 50% of the human genome. Facioscapulohumeral muscular dystrophy (FSHD) is an autosomal-dominant disease associated with reduction in the copy number of the D4Z4 repeat mapping to 4q35. By an unknown mechanism, D4Z4 deletion causes an epigenetic switch leading to de-repression of 4q35 genes. Here we show that the Polycomb group of epigenetic repressors targets D4Z4 in healthy subjects and that D4Z4 deletion is associated with reduced Polycomb silencing in FSHD patients. We identify DBE-T, a chromatin-associated noncoding RNA produced selectively in FSHD patients that coordinates de-repression of 4q35 genes. DBE-T recruits the Trithorax group protein Ash1L to the FSHD locus, driving histone H3 lysine 36 dimethylation, chromatin remodeling, and 4q35 gene transcription. This study provides insights into the biological function of repetitive sequences in regulating gene expression and shows how mutations of such elements can influence the progression of a human genetic disease.
URLPMID:23498939 [本文引用: 1]
Abstract X chromosome inactivation and genomic imprinting are classic epigenetic processes that cause disease when not appropriately regulated in mammals. Whereas X chromosome inactivation evolved to solve the problem of gene dosage, the purpose of genomic imprinting remains controversial. Nevertheless, the two phenomena are united by allelic control of large gene clusters, such that only one copy of a gene is expressed in every cell. Allelic regulation poses significant challenges because it requires coordinated long-range control in cis and stable propagation over time. Long noncoding RNAs have emerged as a common theme, and their contributions to diseases of imprinting and the X chromosome have become apparent. Here, we review recent advances in basic biology, the connections to disease, and preview potential therapeutic strategies for future development. Copyright 脗漏 2013 Elsevier Inc. All rights reserved.
URL [本文引用: 1]
URLPMID:21172659 [本文引用: 3]
Polycomb proteins play essential roles in stem cell renewal and human disease. Recent studies of HOX genes and X inactivation have provided evidence for RNA cofactors in Polycomb repressive complex 2 (PRC2). Here we develop a RIP-seq method to capture the PRC2 transcriptome and identify聽a genome-wide pool of >9000 PRC2-interacting RNAs in embryonic stem cells. The transcriptome includes antisense, intergenic, and promoter-associated transcripts, as well as many unannotated RNAs. A large number of transcripts occur within imprinted regions, oncogene and tumor suppressor loci, and stem cell-related bivalent domains. We provide evidence for direct RNA-protein interactions, most likely via the Ezh2 subunit. We also identify Gtl2 RNA as a PRC2 cofactor that directs PRC2 to the reciprocally imprinted Dlk1 coding gene. Thus, Polycomb proteins interact with a genome-wide family of RNAs, some of which may be used as biomarkers and therapeutic targets for human disease.
URLPMID:20610486 [本文引用: 1]
The Dlk1-Gtl2 imprinting locus is located on mouse distal chromosome 12 and consists of multiple maternally expressed non-coding RNAs and several paternally expressed protein-coding genes. The imprinting of this locus plays a crucial role in embryonic development and postnatal growth. At least one cis-element, the intergenic differentially methylated region (IG-DMR) is required for expression of maternally expressed genes and repression of silenced paternally expressed genes. The mechanism by which the IG-DMR functions is largely unknown. However, it has been suggested that the unmethylated IG-DMR acts as a positive regulator activating expression of non-coding RNAs. Gtl2 is the first non-coding RNA gene downstream of the IG-DMR. Although its in vivo function in the mouse is largely unknown, its human ortholog MEG3 has been linked to tumor suppression in human tumor-derived cell lines. We generated a knockout mouse model, in which the first five exons and adjacent promoter region of the Gtl2 gene were deleted. Maternal deletion of Gtl2 resulted in perinatal death and skeletal muscle defects, indicating that Gtl2 plays an important role in embryonic development. The maternal deletion also completely abolished expression of downstream maternally expressed genes, activated expression of silenced paternally expressed genes and resulted in methylation of the IG-DMR. By contrast, the paternal inherited deletion did not have this effect. These data strongly indicate that activation of Gtl2 and its downstream maternal genes play an essential role in regulating Dlk1-Gtl2 imprinting, possibly by maintaining active status of the IG-DMR.
URLPMID:15498495 [本文引用: 1]
The callipyge ( CLPG ) phenotype is an inherited skeletal muscle hypertrophy described in sheep. It is characterized by an unusual mode of inheritance (鈥減olar overdominance鈥) in which only heterozygous individuals having received the CLPG mutation from their father ( + MAT /CLPG PAT ) express the phenotype [1] . + MAT /CLPG PAT individuals are born normal and develop the muscular hypertrophy at approximately 1 month of age. The CLPG mutation was identified as an A to G transition in a highly conserved dodecamer motif located between the imprinted DLK1 and GTL2 genes 2 and 3 . This motif is thought to be part of a long-range control element (LRCE) because the CLPG mutation was shown, in postnatal skeletal muscle, to enhance the transcript levels of the DLK1, PEG11, GTL2 , and MEG8 genes in cis without altering their imprinting status [4] . As a result, the + MAT /CLPG PAT individuals have a unique expression profile thought to underlie the callipyge phenotype: an overexpression of the paternally expressed protein encoding DLK1 (Figure 1A) and PEG11 transcripts in the absence of an overexpression of the maternally expressed noncoding GTL2 and MEG8 transcripts [4] . However, the way in which this distinct expression profile causes the callipyge muscular hypertrophy has remained unclear. Herein, we demonstrate that the callipyge phenotype is perfectly correlated with ectopic expression of DLK1 protein in hypertrophied muscle of + MAT /CLPG PAT sheep. We demonstrate the causality of this association by inducing a generalized muscular hypertrophy in transgenic mice that express DLK1 in skeletal muscle. The absence of DLK1 protein in skeletal muscle of CLPG/CLPG animals, despite the presence of DLK1 mRNA, supports a trans inhibition mediated by noncoding RNAs expressed from the maternal allele.
URLPMID:23558772 [本文引用: 1]
Staufen1-mediated mRNA decay (SMD) degrades mRNAs that harbor a Staufen1-binding site (SBS) in their 3' untranslated regions (UTRs). Human SBSs can form by intermolecular base-pairing between a 3' UTR Alu element and an Alu element within a long noncoding RNA (lncRNA) called a 1/2-sbsRNA. Since Alu elements are confined to primates, it was unclear how SMD occurs in rodents. Here we identify mouse mRNA 39 UTRs and lncRNAs that contain a B1, B2, B4, or identifier (ID) element. We show that SMD occurs in mouse cells via mRNA-lncRNA base-pairing of partially complementary elements and that mouse 1/2-sbsRNA (m 1/2-sbsRNA)triggered SMD regulates C2C12 cell myogenesis. Our findings define new roles for lncRNAs as well as B and ID short interspersed elements (SINEs) in mice that undoubtedly influence many developmental and homeostatic pathways.
URLPMID:4158944 [本文引用: 2]
Alternative splicing (AS) of pre-mRNA is utilized by higher eukaryotes to achieve increased transcriptome and proteomic complexity. The serine/arginine (SR) splicing factors regulate tissue- or cell-type-specific AS in a concentration- and phosphorylation-dependent manner. However, the mechanisms that modulate the cellular levels of active SR proteins remain to be elucidated. In the present study, we provide evidence for a role for the long nuclear-retained regulatory RNA (nrRNA), MALAT1 in AS regulation. MALAT1 interacts with SR proteins and influences the distribution of these and other splicing factors in nuclear speckle domains. Depletion of MALAT1 or overexpression of an SR protein changes the AS of a similar set of endogenous pre-mRNAs. Furthermore, MALAT1 regulates cellular levels of phosphorylated forms of SR proteins. Taken together, our results suggest that MALAT1 regulates AS by modulating the levels of active SR proteins. Our results further highlight the role for an nrRNA in the regulation of gene expression.Highlights? Nuclear speckle-localized MALAT1 interacts with SR splicing factors ? MALAT1 influences the nuclear speckle distribution of splicing factors ? MALAT1 depletion changes alternative splicing of endogenous pre-mRNAs ? MALAT1 regulates the cellular levels of phosphorylated SR proteins
URLPMID:11163185 [本文引用: 1]
Abstract The process of cellular differentiation culminating in terminally differentiated mammalian cells is thought to be irreversible. Here, we present evidence that terminally differentiated murine myotubes can be induced to dedifferentiate. Ectopic expression of msx1 in C2C12 myotubes reduced the nuclear muscle proteins MyoD, myogenin, MRF4, and p21 to undetectable levels in 20%-50% of the myotubes. Approximately 9% of the myotubes cleave to produce either smaller multinucleated myotubes or proliferating, mononucleated cells. Finally, clonal populations of the myotube-derived mononucleated cells can be induced to redifferentiate into cells expressing chondrogenic, adipogenic, myogenic, and osteogenic markers. These results suggest that terminally differentiated mammalian myotubes can dedifferentiate when stimulated with the appropriate signals and that msx1 can contribute to the dedifferentiation process.
URLPMID:11390985 [本文引用: 1]
Msx1 is a key factor for the development of tooth and craniofacial skeleton and has been proposed to play a pivotal role in terminal cell differentiation. In this paper, we demonstrated the presence of an endogenous Msx1 antisense RNA (Msx1-AS RNA) in mice, rats, and humans. In situ analysis revealed that this RNA is expressed only in differentiated dental and bone cells with an inverse correlation with Msx1 protein. These in vivo data and overexpression of Msx1 sense and AS RNA in an odontoblastic cell line (MO6-G3) showed that the balance between the levels of the two Msx1 RNAs is related to the expression of Msx1 protein. To analyze the impact of this balance in the Msx-Dlx homeoprotein pathway, we analyzed the effect of Msx1, Msx2, and Dlx5 overexpression on proteins involved in skeletal differentiation. We showed that the Msx1-AS RNA is involved in crosstalk between the Msx-Dlx pathways because its expression was abolished by Dlx5. Msx1 was shown to down-regulate a master gene of skeletal cells differentiation, Cbfa1. All these data strongly suggest that the ratio between Msx1 sense and antisense RNAs is a very important factor in the control of skeletal terminal differentiation. Finally, the initiation site for Msx1-AS RNA transcription was located by primer extension in both mouse and human in an identical region, including a consensus TATA box, suggesting an evolutionary conservation of the AS RNA-mediated regulation of Msx1 gene expression.
URLPMID:3117458 [本文引用: 1]
Skeletal muscle aging is associated with increased inflammation and oxidative stress, a decrease in the ability to rebuild muscle after injury and in response to exercise. In this perspective, we discuss the mechanisms regulating Sirt1 activity and expression in skeletal muscles, emphasizing their implications in muscle physiology and the impairment of muscle function with age.
URLPMID:26902620 [本文引用: 1]
Recent studies demonstrate the functions of long non-coding RNAs (lncRNAs) in mediating gene expression at the transcriptional or translational level. Our previous study identified a Sirt1 antisense (AS) lncRNA transcribed from the Sirt1 AS strand. However, its role and regulatory mechanism is still unknown in myogenesis. Here, functional analyses showed that Sirt1 AS lncRNA overexpression promoted myoblast proliferation, but inhibited differentiation. Mechanistically, Sirt1 AS lncRNA was found to activate its sense gene, Sirt1. The luciferase assay provided evidences that Sirt1 AS lncRNA interacted with Sirt1 3鈥 UTR and rescued Sirt1 transcriptional suppression by competing with miR-34a. In addition, RNA stability assay showed that Sirt1 AS lncRNA prolonged Sirt1 mRNA half-life from 2 to 10鈥塰. Ribonuclease protection assay further indicated that it fully bound to Sirt1 mRNA in the myoblast cytoplasm. Moreover, Sirt1 AS overexpression led to less mouse weight than the control because of less lean mass and greater levels of Sirt1, whereas the fat mass and levels of miR-34a were not altered. Based on the findings, a novel regulatory mechanism was found that Sirt1 AS lncRNA preferably interacted with Sirt1 mRNA forming RNA duplex to promote Sirt1 translation by competing with miR-34a, inhibiting muscle formation.
URLPMID:4285423 [本文引用: 1]
Abstract An in silico screen for myogenic long noncoding RNAs (lncRNAs) revealed nine lncRNAs that are upregulated more than 10-fold in myotubes versus levels in myoblasts. One of these lncRNAs, MyoD upstream noncoding (MUNC, also known as DRR(eRNA)), is encoded 5 kb upstream of the transcription start site of MyoD, a myogenic transcription factor gene. MUNC is specifically expressed in skeletal muscle and exists as in unspliced and spliced isoforms, and its 5' end overlaps with the cis-acting distal regulatory region (DRR) of MyoD. Small interfering RNA (siRNA) of MUNC reduced myoblast differentiation and specifically reduced the association of MyoD to the DRR enhancer and myogenin promoter but not to another MyoD-dependent enhancer. Stable overexpression of MUNC from a heterologous promoter increased endogenous MyoD, Myogenin, and Myh3 (myosin heavy chain, [MHC] gene) mRNAs but not the cognate proteins, suggesting that MUNC can act in trans to promote gene expression but that this activity does not require an induction of MyoD protein. MUNC also stimulates the transcription of other genes that are not recognized as MyoD-inducible genes. Knockdown of MUNC in vivo impaired murine muscle regeneration, implicating MUNC in primary satellite cell differentiation in the animal. We also discovered a human MUNC that is induced during differentiation of myoblasts and whose knockdown decreases differentiation, suggesting an evolutionarily conserved role of MUNC lncRNA in myogenesis. Although MUNC overlaps with the DRR enhancer, our results suggest that MUNC is not a classic cis-acting enhancer RNA (e-RNA) acting exclusively by stimulating the neighboring MyoD gene but more like a promyogenic lncRNA that acts directly or indirectly on multiple promoters to increase myogenic gene expression. Copyright 漏 2015, American Society for Microbiology. All Rights Reserved.
URLPMID:3786356 [本文引用: 1]
Transcription factors and DNA regulatory binding motifs are fundamental components of the gene regulatory network. Here, by using genome-wide binding profiling, we show extensive occupancy of transcription factors of myogenesis (MyoD and Myogenin) at extragenic enhancer regions coinciding with RNA synthesis (i.e., eRNA). In particular, multiple regions were transcribed to eRNA within the regulatory region of MYOD1, including previously characterized distal regulatory regions (DRR) and core enhancer (CE). While (CE)RNA enhanced RNA polymerase II (Pol II) occupancy and transcription at MYOD1, DRR RNA acted to activate the downstream myogenic genes. The deployment of transcriptional machinery to appropriate loci is contingent on chromatin accessibility, a rate-limiting step preceding Pol II assembly. By nuclease sensitivity assay, we found that eRNAs regulate genomic access of the transcriptional complex to defined regulatory regions. In conclusion, our data suggest that eRNAs contribute to establishing a cell-type-specific transcriptional circuitry by directing chromatin-remodeling events.
URL [本文引用: 1]
URLPMID:3025577 [本文引用: 1]
Abstract The steroid receptor RNA activator (SRA) has the unusual property to function as both a non-coding RNA (ncRNA) and a protein SRAP. SRA ncRNA is known to increase the activity of a range of nuclear receptors as well as the master regulator of muscle differentiation MyoD. The contribution of SRA to either a ncRNA or a protein is influenced by alternative splicing of the first intron, the retention of which disrupts the SRAP open reading frame. We reported here that the ratio between non-coding and coding SRA isoforms increased during myogenic differentiation of human satellite cells but not myotonic dystrophy patient satellite cells, in which differentiation capacity is affected. Using constructs that exclusively produce SRA ncRNA or SRAP, we demonstrated that whereas SRA ncRNA was indeed an enhancer of myogenic differentiation and myogenic conversion of non-muscle cells through the co-activation of MyoD activity, SRAP prevented this SRA RNA-dependant co-activation. Interestingly, the SRAP inhibitory effect is mediated through the interaction of SRAP with its RNA counterpart via its RRM-like domain interacting with the functional sub-structure of SRA RNA, STR7. This study thus provides a new model for SRA-mediated regulation of MyoD transcriptional activity in the promotion of normal muscle differentiation, which takes into account the nature of SRA molecules present.
URLPMID:23942234 [本文引用: 1]
Skeletal muscle differentiation is orchestrated by a network of transcription factors, epigenetic regulators, and non-coding RNAs. The transcription factor Yin Yang 1 (YY1) silences multiple target genes in myoblasts (MBs) by recruiting Ezh2 (Enhancer of Zeste Homologue2). To elucidate genome-wide YY1 binding in MBs, we performed chromatin immunoprecipitation (ChIP)-seq and found 1820 specific binding sites in MBs with a large portion residing in intergenic regions. Detailed analysis demonstrated that YY1 acts as an activator for many loci in addition to its known repressor function. No significant co-occupancy was found between YY1 and Ezh2, suggesting an additional Ezh2-independent function for YY1 in MBs. Further analysis of intergenic binding sites showed that YY1 potentially regulates dozens of large intergenic non-coding RNAs (lincRNAs), whose function in myogenesis is underexplored. We characterized a novel muscle-associated lincRNA (Yam-1) that is positively regulated by YY1. Yam-1 is downregulated upon differentiation and acts as an inhibitor of myogenesis. We demonstrated that Yam-1 functions through in cis regulation of miR-715, which in turn targets Wnt7b. Our findings not only provide the first genome-wide picture of YY1 association in muscle cells, but also uncover the functional role of lincRNA Yam-1.
URLPMID:28091529 [本文引用: 2]
Abstract Long non-coding RNAs (lncRNAs) are important regulators of diverse biological processes. Here we report on functional identification and characterization of a novel long intergenic non-coding RNA with MyoD-regulated and skeletal muscle-restricted expression that promotes the activation of the myogenic program, and is therefore termed Linc-RAM (Linc-RNA Activator of Myogenesis). Linc-RAM is transcribed from an intergenic region of myogenic cells and its expression is upregulated during myogenesis. Notably, in vivo functional studies show that Linc-RAM knockout mice display impaired muscle regeneration due to the differentiation defect of satellite cells. Mechanistically, Linc-RAM regulates expression of myogenic genes by directly binding MyoD, which in turn promotes the assembly of the MyoD-Baf60c-Brg1 complex on the regulatory elements of target genes. Collectively, our findings reveal the functional role and molecular mechanism of a lineage-specific Linc-RAM as a regulatory lncRNA required for tissues-specific chromatin remodelling and gene expression.
URLPMID:28024296 [本文引用: 1]
Abstract Although long non-coding RNAs (lncRNAs) are non-protein-coding transcripts by definition, recent studies have shown that a fraction of putative small open reading frames within lncRNAs are translated. However, the biological significance of these hidden polypeptides is still unclear. Here we identify and functionally characterize a novel polypeptide encoded by the lncRNA LINC00961. This polypeptide is conserved between human and mouse, is localized to the late endosome/lysosome and interacts with the lysosomal v-ATPase to negatively regulate mTORC1 activation. This regulation of mTORC1 is specific to activation of mTORC1 by amino acid stimulation, rather than by growth factors. Hence, we termed this polypeptide 'small regulatory polypeptide of amino acid response' (SPAR). We show that the SPAR-encoding lncRNA is highly expressed in a subset of tissues and use CRISPR/Cas9 engineering to develop a SPAR-polypeptide-specific knockout mouse while maintaining expression of the host lncRNA. We find that the SPAR-encoding lncRNA is downregulated in skeletal muscle upon acute injury, and using this in vivo model we establish that SPAR downregulation enables efficient activation of mTORC1 and promotes muscle regeneration. Our data provide a mechanism by which mTORC1 activation may be finely regulated in a tissue-specific manner in response to injury, and a paradigm by which lncRNAs encoding small polypeptides can modulate general biological pathways and processes to facilitate tissue-specific requirements, consistent with their restricted and highly regulated expression profile.
URLPMID:3973619
The vast majority of the mammalian genome is transcribed giving rise to many different types of noncoding RNAs. Among them, long noncoding RNAs are the most numerous and functionally versatile class. Indeed, the lncRNA repertoire might be as rich as the proteome. LncRNAs have emerged as key regulators of gene expression at multiple levels. They play important roles in the regulation of development, differentiation and maintenance of cell identity and they also contribute to disease. In this review, we present recent advances in the biology of lncRNAs in muscle development and differentiation. We will also discuss the contribution of lncRNAs to muscle disease with a particular focus on Duchenne and facioscapulohumeral muscular dystrophies.
URLPMID:4581346
A significant portion of the mammalian genome encodes numerous transcripts that are not translated into proteins, termed long non-coding RNAs. Initial studies identifying long non-coding RNAs inferred these RNA sequences were a consequence of transcriptional noise or promiscuous RNA polymerase II activity. However, the last decade has seen a revolution in the understanding of regulation and function of long non-coding RNAs. Now it has become apparent that long non-coding RNAs play critical roles in a wide variety of biological processes. In this review, we describe the current understanding of long non-coding RNA-mediated regulation of cellular processes: differentiation, development, and disease.
URLPMID:26996975 [本文引用: 1]
Transcriptome analysis of mammalian genomes has revealed widespread transcription, much of which does not encode protein. Long non-coding RNAs (lncRNAs) are a subset of the non-coding transcriptome that are emerging as critical regulators of various cellular processes. Differentiation of stem and progenitor cells requires a careful execution of specific genetic programs, and recent studies have revealed that lncRNA expression contributes to specification of cell identity. LncRNAs participate in regulating differentiation at multiple levels of gene expression through various mechanisms of action. In this review, functional roles of lncRNAs in regulating cellular differentiation of blood, muscle, skin, cardiomyocytes, adipocytes, and neurons are discussed.
URLPMID:26477492 [本文引用: 2]
It is known that more than 70% of mammalian genomes are transcribed, yet the vast majority of transcripts do not code for proteins. Are these noncoding transcripts merely transcriptional noise, or do they serve a biological purpose? Recent developments in genomic analysis technologies, especially sequencing methods, have allowed researchers to create a large atlas of transcriptomes, study subcellular localization, and investigate potential interactions with proteins for a growing number of transcripts. Here, we review the current methodologies available for discovering and investigating functions of long noncoding RNAs (lncRNAs), which require a wide variety of applications to study their potential biological roles. This article is part of a Special Issue entitled: Clues to long noncoding RNA taxonomy1, edited by Dr. Tetsuro Hirose and Dr. Shinichi Nakagawa.
URLPMID:25675209 [本文引用: 1]
RNA-sequencing (RNA-seq) measures the quantitative change in gene expression over the whole transcriptome, but it lacks spatial context. In contrast, in situ hybridization provides the location of gene expression, but only for a small number of genes. Here we detail a protocol for genome-wide profiling of gene expression in situ in fixed cells and tissues, in which RNA is converted into cross-linked cDNA amplicons and sequenced manually on a confocal microscope. Unlike traditional RNA-seq, our method enriches for context-specific transcripts over housekeeping and/or structural RNA, and it preserves the tissue architecture for RNA localization studies. Our protocol is written for researchers experienced in cell microscopy with minimal computing skills. Library construction and sequencing can be completed within 14 d, with image analysis requiring an additional 2 d.
URLPMID:28637286 [本文引用: 1]
Abstract RNA serves many functions in biology such as splicing, temperature sensing, and innate immunity. These functions are often determined by the structure of RNA. There is thus a pressing need to understand RNA structure and how it changes during diverse biological processes both in vivo and genome-wide. Here, we present Structure-seq2, which provides nucleotide-resolution RNA structural information in vivo and genome-wide. This optimized version of our original Structure-seq method increases sensitivity by at least 4-fold and improves data quality by minimizing formation of a deleterious by-product, reducing ligation bias, and improving read coverage. We also present a variation of Structure-seq2 in which a biotinylated nucleotide is incorporated during reverse transcription, which greatly facilitates the protocol by eliminating two PAGE purification steps. We benchmark Structure-seq2 on both mRNA and rRNA structure in rice (Oryza sativa). We demonstrate that Structure-seq2 can lead to new biological insights. Our Structure-seq2 datasets uncover hidden breaks in chloroplast rRNA and identify a previously unreported N1-methyladenosine (m1A) in a nuclear-encoded Oryza sativa rRNA. Overall, Structure-seq2 is a rapid, sensitive, and unbiased method to probe RNA in vivo and genome-wide that facilitates new insights into RNA biology. 漏 The Author(s) 2017. Published by Oxford University Press on behalf of Nucleic Acids Research.
URLPMID:3531181
Long non-coding RNAs (lncRNAs) and microRNAs (miRNAs) represent two classes of important non-coding RNAs in eukaryotes. Although these non-coding RNAs have been implicated in organismal development and in various human diseases, surprisingly little is known about their transcriptional regulation. Recent advances in chromatin immunoprecipitation with next-generation DNA sequencing (ChIP-Seq) have provided methods of detecting transcription factor binding sites (TFBSs) with unprecedented sensitivity. In this study, we describe ChIPBase (http://deepbase.sysu.edu.cn/chipbase/), a novel database that we have developed to facilitate the comprehensive annotation and discovery of transcription factor binding maps and transcriptional regulatory relationships of lncRNAs and miRNAs from ChIP-Seq data. The current release of ChIPBase includes high-throughput sequencing data that were generated by 543 ChIP-Seq experiments in diverse tissues and cell lines from six organisms. By analysing millions of TFBSs, we identified tens of thousands of TF-lncRNA and TF-miRNA regulatory relationships. Furthermore, two web-based servers were developed to annotate and discover transcriptional regulatory relationships of lncRNAs and miRNAs from ChIP-Seq data. In addition, we developed two genome browsers, deepView and genomeView, to provide integrated views of multidimensional data. Moreover, our web implementation supports diverse query types and the exploration of TFs, lncRNAs, miRNAs, gene ontologies and pathways.
URLPMID:23193281
Abstract Recently, the attention of the research community has been focused on long non-coding RNAs (lncRNAs) and their physiological/pathological implications. As the number of experiments increase in a rapid rate and transcriptional units are better annotated, databases indexing lncRNA properties and function gradually become essential tools to this process. Aim of DIANA-LncBase (www.microrna.gr/LncBase) is to reinforce researchers' attempts and unravel microRNA (miRNA)-lncRNA putative functional interactions. This study provides, for the first time, a comprehensive annotation of miRNA targets on lncRNAs. DIANA-LncBase hosts transcriptome-wide experimentally verified and computationally predicted miRNA recognition elements (MREs) on human and mouse lncRNAs. The analysis performed includes an integration of most of the available lncRNA resources, relevant high-throughput HITS-CLIP and PAR-CLIP experimental data as well as state-of-the-art in silico target predictions. The experimentally supported entries available in DIANA-LncBase correspond to >5000 interactions, while the computationally predicted interactions exceed 10 million. DIANA-LncBase hosts detailed information for each miRNA-lncRNA pair, such as external links, graphic plots of transcripts' genomic location, representation of the binding sites, lncRNA tissue expression as well as MREs conservation and prediction scores.
URLPMID:4331805
Abstract BACKGROUND: The GENCODE project has collected over 10,000 human long non-coding RNA (lncRNA) genes. However, the vast majority of them remain to be functionally characterized. Computational investigation of potential functions of human lncRNA genes is helpful to guide further experimental studies on lncRNAs. RESULTS: In this study, based on expression correlation between lncRNAs and protein-coding genes across 19 human normal tissues, we used the hypergeometric test to functionally annotate a single lncRNA or a set of lncRNAs with significantly enriched functional terms among the protein-coding genes that are significantly co-expressed with the lncRNA(s). The functional terms include all nodes in the Gene Ontology (GO) and 4,380 human biological pathways collected from 12 pathway databases. We successfully mapped 9,625 human lncRNA genes to GO terms and biological pathways, and then developed the first ontology-driven user-friendly web interface named lncRNA2Function, which enables researchers to browse the lncRNAs associated with a specific functional term, the functional terms associated with a specific lncRNA, or to assign functional terms to a set of human lncRNA genes, such as a cluster of co-expressed lncRNAs. The lncRNA2Function is freely available at http://mlg.hit.edu.cn/lncrna2function. CONCLUSIONS: The LncRNA2Function is an important resource for further investigating the functions of a single human lncRNA, or functionally annotating a set of human lncRNAs of interest.
URLPMID:4383871
Long non-coding RNAs (lncRNAs) play key roles in various cellular contexts and diseases by diverse mechanisms. With the rapid growth of identified lncRNAs and disease-associated single nucleotide polymorphisms (SNPs), there is a great demand to study SNPs in lncRNAs. Aiming to provide a useful resource about lncRNA SNPs, we systematically identified SNPs in lncRNAs and analyzed their potential impacts on lncRNA structure and function. In total, we identified 495,729 and 777,095 SNPs in more than 30,000 lncRNA transcripts in human and mouse, respectively. A large number of SNPs were predicted with the potential to impact on the miRNA-lncRNA interaction. The experimental evidence and conservation of miRNA-lncRNA interaction, as well as miRNA expressions from TCGA were also integrated to prioritize the miRNA-lncRNA interactions and SNPs on the binding sites. Furthermore, by mapping SNPs to GWAS results, we found that 142 human lncRNA SNPs are GWAS tagSNPs and 197,827 lncRNA SNPs are in the GWAS linkage disequilibrium regions. All these data for human and mouse lncRNAs were imported into lncRNASNP database (http://bioinfo.life.hust.edu.cn/lncRNASNP/), which includes two sub-databases lncRNASNP-human and lncRNASNP-mouse. The lncRNASNP database has a user-friendly interface for searching and browsing through the SNP, lncRNA and miRNA sections.
URLPMID:4834207
Despite the fact that a large quantity of noncoding RNAs (ncRNAs) have been identified, their functions remain unclear. To enable researchers to have a better understanding of ncRNAs鈥 functions, we updated the NPInter database to version 3.0, which contains experimentally verified interactions between ncRNAs (excluding tRNAs and rRNAs), especially long noncoding RNAs (lncRNAs) and other biomolecules (proteins, mRNAs, miRNAs and genomic DNAs). In NPInter v3.0, interactions pertaining to ncRNAs are not only manually curated from scientific literature but also curated from high-throughput technologies. In addition, we also curated lncRNA鈥搈iRNA interactions fromin silicopredictions supported by AGO CLIP-seq data. When compared with NPInter v2.0, the interactions are more informative (with additional information on tissues or cell lines, binding sites, conservation, co-expression values and other features) and more organized (with divisions on data sets by data sources, tissues or cell lines, experiments and other criteria). NPInter v3.0 expands the data set to 491,416 interactions in 188 tissues (or cell lines) from 68 kinds of experimental technologies. NPInter v3.0 also improves the user interface and adds new web services, including a local UCSC Genome Browser to visualize binding sites. Additionally, NPInter v3.0 defined a high-confidence set of interactions and predicted the functions of lncRNAs in human and mouse based on the interactions curated in the database. NPInter v3.0 is available athttp://www.bioinfo.org/NPInter/. Database URL:http://www.bioinfo.org/NPInter/
URLPMID:26818453
Motivation Recent studies have revealed that large numbers of non-coding RNAs are transcribed in humans, but only a few of them have been identified with their functions. Identification of the...
URLPMID:24297251
Although microRNAs (miRNAs), other non-coding RNAs (ncRNAs) (e.g. lncRNAs, pseudogenes and circRNAs) and competing endogenous RNAs (ceRNAs) have been implicated in cell-fate determination and in various human diseases, surprisingly little is known about the regulatory interaction networks among the multiple classes of RNAs. In this study, we developed starBase v2.0 (http://starbase.sysu.edu.cn/) to systematically identify the RNA-RNA and protein-RNA interaction networks from 108 CLIP-Seq (PAR-CLIP, HITS-CLIP, iCLIP, CLASH) data sets generated by 37 independent studies. By analyzing millions of RNA-binding protein binding sites, we identified 鈭9000 miRNA-circRNA, 16 000 miRNA-pseudogene and 285,000 protein-RNA regulatory relationships. Moreover, starBase v2.0 has been updated to provide the most comprehensive CLIP-Seq experimentally supported miRNA-mRNA and miRNA-lncRNA interaction networks to date. We identified 鈭10,000 ceRNA pairs from CLIP-supported miRNA target sites. By combining 13 functional genomic annotations, we developed miRFunction and ceRNAFunction web servers to predict the function of miRNAs and other ncRNAs from the miRNA-mediated regulatory networks. Finally, we developed interactive web implementations to provide visualization, analysis and downloading of the aforementioned large-scale data sets. This study will greatly expand our understanding of ncRNA functions and their coordinated regulatory networks.
URL [本文引用: 1]
URLPMID:23051702 [本文引用: 1]
As a result of the development of high-throughput sequencing and efficient microarray analysis, global gene expression analysis has become an easy and readily available form of data collection. In many research and disease models however, steady state levels of target gene mRNA does not always directly correlate with steady state protein levels. Post-transcriptional gene regulation is a likely explanation of the divergence between the two. Driven by the binding of RNA Binding Proteins (RBP), post-transcriptional regulation affects mRNA localization, stability and translation by forming a Ribonucleoprotein (RNP) complex with target mRNAs. Identifying these unknown de novo mRNA targets from cellular extracts in the RNP complex is pivotal to understanding mechanisms and functions of the RBP and their resulting effect on protein output. This protocol outlines a method termed RNP immunoprecipitation-microarray (RIP-Chip), which allows for the identification of specific mRNAs associated in the ribonucleoprotein complex, under changing experimental conditions, along with options to further optimize an experiment for the individual researcher. With this important experimental tool, researchers can explore the intricate mechanisms associated with post-transcriptional gene regulation as well as other ribonucleoprotein interactions.
URL [本文引用: 1]
URLPMID:21963238 [本文引用: 1]
Abstract Long noncoding RNAs (lncRNAs) are key regulators of chromatin state, yet the nature and sites of RNA-chromatin interaction are mostly unknown. Here we introduce Chromatin Isolation by RNA Purification (ChIRP), where tiling oligonucleotides retrieve specific lncRNAs with bound protein and DNA sequences, which are enumerated by deep sequencing. ChIRP-seq of three lncRNAs reveal that RNA occupancy sites in the genome are focal, sequence-specific, and numerous. Drosophila roX2 RNA occupies male X-linked gene bodies with increasing tendency toward the 3' end, peaking at CES sites. Human telomerase RNA TERC occupies telomeres and Wnt pathway genes. HOTAIR lncRNA preferentially occupies a GA-rich DNA motif to nucleate broad domains of Polycomb occupancy and histone H3 lysine 27 trimethylation. HOTAIR occupancy occurs independently of EZH2, suggesting the order of RNA guidance of Polycomb occupancy. ChIRP-seq is generally applicable to illuminate the intersection of RNA and chromatin with newfound precision genome wide. Copyright 脗漏 2011 Elsevier Inc. All rights reserved.
In: Ørom U, ed.
URL [本文引用: 1]
Capture Hybridization Analysis of RNA Targets (CHART) has recently been developed to map the genome-wide binding profile of chromatin-associated RNAs. This protocol uses a small number of 22–28 nucleo
In: Dassi E, ed.
URL [本文引用: 1]
URLPMID:25366541 [本文引用: 1]
Abstract Post-transcriptional gene regulation is robustly regulated by RNA-binding proteins (RBPs). Here we describe the collection of RNAs regulated by AUF1 (AU-binding factor 1), an RBP linked to cancer, inflammation and aging. Photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) analysis reveals that AUF1 primarily recognizes U-/GU-rich sequences in mRNAs and noncoding RNAs and influences target transcript fate in three main directions. First, AUF1 lowers the steady-state levels of numerous target RNAs, including long noncoding RNA NEAT1, in turn affecting the organization of nuclear paraspeckles. Second, AUF1 does not change the abundance of many target RNAs, but ribosome profiling reveals that AUF1 promotes the translation of numerous mRNAs in this group. Third, AUF1 unexpectedly enhances the steady-state levels of several target mRNAs encoding DNA-maintenance proteins. Through its actions on target RNAs, AUF1 preserves genomic integrity, in agreement with the AUF1-elicited prevention of premature cellular senescence.
URLPMID:25592151 [本文引用: 1]
HULC is a long noncoding RNA overexpressed in hepatocellular carcinoma (HCC), but its functional contributions in this setting have not been determined. In this study, we explored the hypothesis that HULC contributes to malignant development by supporting abnormal lipid metabolism in hepatoma cells. HULC modulated the deregulation of lipid metabolism in HCC by activating the acyl-CoA synthetase subunit ACSL1. Immunohistochemical analysis of tissue microarrays revealed that approximately 77% (180/233) of HCC tissues were positive for ACSL1. Moreover, HULC mRNA levels correlated positively with ACSL1 levels in 60 HCC cases according to real-time PCR analysis. Mechanistic investigations showed that HULC upregulated the transcriptional factor PPARA, which activated the ACSL1 promoter in hepatoma cells. HULC also suppressed miR-9 targeting of PPARA mRNA by eliciting methylation of CpG islands in the miR-9 promoter. We documented the ability of HULC to promote lipogenesis, thereby stimulating accumulation of intracellular triglycerides and cholesterol in vitro and in vivo. Strikingly, ACSL1 overexpression that generates cholesterol was sufficient to enhance the proliferation of hepatoma cells. Further, cholesterol addition was sufficient to upregulate HULC expression through a positive feedback loop involving the retinoid receptor RXRA, which activated the HULC promoter. Overall, we concluded that HULC functions as an oncogene in hepatoma cells, acting mechanistically by deregulating lipid metabolism through a signaling pathway involving miR-9, PPARA, and ACSL1 that is reinforced by a feed-forward pathway involving cholesterol and RXRA to drive HULC signaling.
URLPMID:29078316 [本文引用: 1]
Abstract Uncoupling protein 1 (UCP1) is localized on the inner mitochondrial membrane and generates heat by uncoupling ATP synthesis from proton transit across the inner membrane. UCP1 is a key element of nonshivering thermogenesis and is most likely important in the regulation of body adiposity. Pigs (Artiodactyl family Suidae ) lack a functional UCP1 gene, resulting in poor thermoregulation and susceptibility to cold, which is an economic and pig welfare issue owing to neonatal mortality. Pigs also have a tendency toward fat accumulation, which may be linked to their lack of UCP1, and thus influences the efficiency of pig production. Here, we report application of a CRISPR/Cas9-mediated, homologous recombination (HR)-independent approach to efficiently insert mouse adiponectin-UCP1 into the porcine endogenous UCP1 locus. The resultant UCP1 knock-in (KI) pigs showed an improved ability to maintain body temperature during acute cold exposure, but they did not have alterations in physical activity levels or total daily energy expenditure (DEE). Furthermore, ectopic UCP1 expression in white adipose tissue (WAT) dramatically decreased fat deposition by 4.89% ( P < 0.01), consequently increasing carcass lean percentage (CLP; P < 0.05). Mechanism studies indicated that the loss of fat upon UCP1 activation in WAT was linked to elevated lipolysis. UCP1 KI pigs are a potentially valuable resource for agricultural production through their combination of cold adaptation, which improves pig welfare and reduces economic losses, with reduced fat deposition and increased lean meat production. Published under the PNAS license.
URLPMID:19397524 [本文引用: 1]
Long non-coding RNA (long ncRNA) is a novel class of ncRNA that may be involved in critical cellular processes. A considerable number of mammalian long ncRNAs have now been isolated but only a small number of these nucleic acids have been functionally well characterized. In this study, to determine the structure, regulation and function of long ncRNA in pigs, TncRNA was isolated from this mammal and its potential function during pig foetus development was identified. We anticipated that this would provide new insights into functional genomic studies in the pig. Using LongSAGE libraries generated from Chinese indigenous Tongcheng and Landrace pigs at three prenatal stages, a novel porcine long ncRNA was identified, TncRNA , which was found to be differentially expressed during myogenesis. The full-length cDNA for this gene is 3409 bp, and it harbours a typical polyadenylation signal sequence located 18 bp upstream from the 3' poly (A) tail. Genomic sequence analysis showed that pig TncRNA is alternatively spliced and several transcripts were detected. Using the INRA芒聙聯University of Minnesota porcine radiation hybrid panel, TncRNA was assigned to SSC2 and found to be closely linked to the microsatellite marker SW256 . Porcine TncRNA was found to be expressed in all tissues examined but in variable amounts. Comparisons between the expression profiles of TncRNA at different development stages in Tongcheng and Landrace pigs revealed up-regulation of this molecule in prenatal skeletal muscle, and differential expression in 90-day-old foetal skeletal muscle between these two pig breeds. This is the first report to describe a long ncRNA in pig. Moreover, the distinct expression pattern and structure of porcine TncRNA suggest that it performs complex and critical functions during foetal development.
URLPMID:27589905 [本文引用: 1]
61We report here the first analysis of lncRNA landscape during muscle development.61lncMDis a muscle-specific lncRNA which is tightly regulated during myogenesis.61lncMDpromotes muscle differentiation by acting as a ceRNA for miR-125b.61IGF2is an important target of miR-125b in bovine myogenesis.61lncMDincreasesIGF2expression in a miR-125b-dependent manner.
URLPMID:28473774 [本文引用: 1]
Long non-coding RNAs (lncRNAs) play important roles in epigenetic regulation of skeletal muscle development. In our previous RNA-seq study (accession numberGSE58755), we found that lncRNA-Six1 is an lncRNA that is differentially expressed between White Recessive Rock (WRR) and Xinghua (XH) chicken. In this study, we have further demonstrated that lncRNA-Six1 is located 432 bp upstream of the gene encoding the protein Six homeobox 1 (Six1). A dual-luciferase reporter assay identified that lncRNA-Six1 overlaps theSix1proximal promoter. In lncRNA-Six1, a micropeptide of about 7.26 kDa was found to play an important role in the lncRNA-Six1in cisactivity. Overexpression of lncRNA-Six1 promoted the mRNA and protein expression level of theSix1gene, while knockdown of lncRNA-Six1 inhibitedSix1expression. Moreover, tissue expression profiles showed that both the lncRNA-Six1 and theSix1mRNA were highly expressed in chicken breast tissue. LncRNA-Six1 overexpression promoted cell proliferation and induced cell division. Conversely, its loss of function inhibited cell proliferation and reduced cell viability. Similar effects were observed after overexpression or knockdown of theSix1gene. In addition, overexpression or knockdown ofSix1promoted or inhibited, respectively, the expression levels of muscle-growth-related genes, such asMYOG, MYHC, MYOD, IGF1R, andINSR. Taken together, these data demonstrate that lncRNA-Six1 carries outcis-acting regulation of the protein-encodingSix1gene, and encodes a micropeptide to activateSix1gene, thus promoting cell proliferation and being involved in muscle growth.
URLPMID:28726188 [本文引用: 1]
Abstract Long noncoding RNAs (lncRNAs) are key regulatory factors for gene expression in a variety of biological processes; however, the role of lncRNAs in muscle formation and development is poorly understood, particularly in cattle. Here, we identified a highly expressed lncRNA in muscle, lncYYW, by high-throughput sequencing in bovine longissimus, scapular, intercostal, and gluteus muscles. The expression of lncYYW increased gradually during myoblast differentiation. Overexpression of lncYYW increased the number of cells in the DNA synthesis (S) stage of the cell cycle and upregulated the expression of two well-established myogenic markers, myogenin and myosin heavy chain. A microarray analysis showed that lncYYW positively regulates the expression of growth hormone 1 and its downstream genes, AKT1 and PIK3CD, in bovine myoblasts. This discovery provides a good foundation for further study of the mechanism of action of lncYYW during bovine myoblast development. Taken together, our results reveal a novel lncRNA associated with bovine myoblast proliferation and differentiation. This lncRNA will play a crucial and critical role in future studies of bovine muscle development.
URLPMID:25753296 [本文引用: 1]
Long intergenic non-coding RNAs (lincRNAs) play important roles in many cellular processes. Here, we present the first systematic identification and characterization of lincRNAs in fetal porcine skeletal muscle. We obtained a total of 55.02 million 90-bp paired-end reads and assembled 54,550 transcripts using cufflinks. We developed a pipeline to identify 570 multi-exon lincRNAs by integrating a set of previous approaches. These putative porcine lincRNAs share many characteristics with mammalian lincRNAs, such as a relatively short length, small number of exons and low level of sequence conservation. We found that the porcine lincRNAs were preferentially located near genes mediating transcriptional regulation rather than those with developmental functions. We further experimentally analyzed the features of a conserved mouse lincRNA gene and found that isoforms 1 and 4 of this lincRNA were enriched in the cell nucleus and were associated with polycomb repressive complex 2 (PRC2). Our results provide a catalog of fetal porcine lincRNAs for further experimental investigation of the functions of these genes in the skeletal muscle developmental process.
URLPMID:28183149 [本文引用: 1]
Abstract Long non-coding RNAs (lncRNAs) participated in growth and development of skeletal muscle; however, little is known about their response to testosterone deficiency in porcine skeletal muscle. We compared lean mass related carcass traits and lncRNAs expression files in Longissimus dorsi (LD) muscle between intact and castrated Huainan male pigs. The results showed that castration significantly reduced eye muscle area and lean meat percentage (P02<020.05), but increased the fat mass weight (P02<020.05). Meanwhile, 8946 lncRNAs, including 6743 intergenic lncRNAs (lincRNAs), 498 anti-sense lncRNAs, and 1705 intronic lncRNAs, were identified in porcine LD, among which, 385 lncRNAs were considered as the differentially expressed candidates between intact groups and castrated groups (q-value02<020.05). Functional analysis indicated that these differently expressed lncRNAs and their target genes were involved in the estrogen receptor signaling pathway and skeletal and muscular system development and function. We first detect porcine muscular lncRNA response to castration, and the results suggested that lncRNAs and their target genes participated in the regulation of testosterone deficiency-related skeletal muscle growth. 08 2017 Japanese Society of Animal Science.
URLPMID:22374175 [本文引用: 1]
Vertebrate genomes encode thousands of non-coding RNAs including short non-coding RNAs (such as microRNAs) and long non-coding RNAs (lncRNAs). Chicken ( Gallus gallus ) is an important model organism for developmental biology, and the recently assembled genome sequences for chicken will facilitate the understanding of the functional roles of non-coding RNA genes during development. The present study concerns the first systematic identification of lncRNAs using RNA-Seq to sample the transcriptome during chicken muscle development. A computational approach was used to identify 281 new intergenic lncRNAs in the chicken genome. Novel lncRNAs in general are less conserved than protein-coding genes and slightly more conserved than random non-coding sequences. The present study has provided an initial chicken lncRNA catalog and greatly increased the number of chicken ncRNAs in the non-protein coding RNA database. Furthermore, the computational pipeline presented in the current work will be useful for characterizing lncRNAs obtained from deep sequencing data.
URLPMID:5420592 [本文引用: 1]
Embryonic growth and development of skeletal muscle is a major determinant of muscle mass, and has a significant effect on meat production in chicken. To assess the protein expression profiles during embryonic skeletal muscle development, we performed a proteomics analysis using isobaric tags for relative and absolute quantification (iTRAQ) in leg muscle tissues of female Xinghua chicken at embryonic age (E) 11, E16, and 1-day post hatch (D1). We identified 3,240 proteins in chicken embryonic muscle and 491 of them were differentially expressed (fold change 鈮 1.5 or 鈮 0.666 andp< 0.05). There were 19 up- and 32 down-regulated proteins in E11 vs. E16 group, 238 up- and 227 down-regulated proteins in E11 vs. D1 group, and 13 up- and 5 down-regulated proteins in E16 vs. D1 group. Protein interaction network analyses indicated that these differentially expressed proteins were mainly involved in the pathway of protein synthesis, muscle contraction, and oxidative phosphorylation. Integrative analysis of proteome and our previous transcriptome data found 189 differentially expressed proteins that correlated with their mRNA level. The interactions between these proteins were also involved in muscle contraction and oxidative phosphorylation pathways. The lncRNA-protein interaction network found four proteins DMD, MYL3, TNNI2, and TNNT3 that are all involved in muscle contraction and may be lncRNA regulated. These results provide several candidate genes for further investigation into the molecular mechanisms of chicken embryonic muscle development, and enable us to better understanding their regulation networks and biochemical pathways.
URLPMID:5220077 [本文引用: 1]
Long non-coding RNAs (lncRNAs) play important roles in transcriptional and post-transcriptional regulation. However, little is currently known about the mechanisms by which they regulate skeletal muscle development in the chicken. In this study, we used RNA sequencing to profile the leg muscle transcriptome (lncRNA and mRNA) at three stages of skeletal muscle development in the chicken: embryonic day 11 (E11), embryonic day 16 (E16), and 1 day after hatching (D1). In total, 129, 132, and 45 differentially expressed lncRNAs, and 1798, 3072, and 1211 differentially expressed mRNAs were identified in comparisons of E11 vs. E16, E11 vs. D1, and E16 vs. D1, respectively. Moreover, we identified thecis- andtrans-regulatory target genes of differentially expressed lncRNAs, and constructed lncRNA-gene interaction networks. In total, 126 and 200cis-targets, and two and threetrans-targets were involved in lncRNA-gene interaction networks that were constructed based on the E11 vs. E16, and E11 vs. D1 comparisons, respectively. The comparison of the E16 vs. D1 lncRNA-gene network comprised 25cis-targets. We determined that lncRNA target genes are potentially involved in cellular development, and cellular growth and proliferation using Ingenuity Pathway Analysis. The gene networks identified for the E11 vs. D1 comparison were involved in embryonic development, organismal development and tissue development. The present study provides an RNA sequencing based evaluation of lncRNA function during skeletal muscle development in the chicken. Comprehensive analysis facilitated the identification of lncRNAs and target genes that might contribute to the regulation of different stages of skeletal muscle development.
URLPMID:27550073 [本文引用: 1]
Long non-coding RNAs (lncRNAs) have been studied extensively over the past few years. Large numbers of lncRNAs have been identified in mouse, rat, and human, and some of them have been shown to play important roles in muscle development and myogenesis. However, there are few reports on the characterization of lncRNAs covering all the development stages of skeletal muscle in livestock. RNA libraries constructed from developinglongissimus dorsimuscle of fetal (45, 60, and 10502days of gestation) and postnatal (302days after birth) goat (Capra hircus) were sequenced. A total of 1,034,049,894 clean reads were generated. Among them, 3981 lncRNA transcripts corresponding to 2739 lncRNA genes were identified, including 3515 intergenic lncRNAs and 466 anti-sense lncRNAs. Notably, in pairwise comparisons between the libraries of skeletal muscle at the different development stages, a total of 577 transcripts were differentially expressed (P<650.05) which were validated by qPCR using randomly selected six lncRNA genes. The identified goat lncRNAs shared some characteristics, such as fewer exons and shorter length, with the lncRNAs in other mammals. We also found 1153 lncRNAs genes were neighbored 1455 protein-coding genes (<1002kb upstream and downstream) and functionally enriched in transcriptional regulation and development-related processes, indicating they may be incis-regulatory relationships. Additionally, Pearson’s correlation coefficients of co-expression levels suggested 1737 lncRNAs and 19,422 mRNAs were possibly intrans-regulatory relationships (r>650.95 orr<65610.95). These co-expressed mRNAs were enriched in development-related biological processes such as muscle system processes, regulation of cell growth, muscle cell development, regulation of transcription, and embryonic morphogenesis. This study provides a catalog of goat muscle-related lncRNAs, and will contribute to a fuller understanding of the molecular mechanism underpinning muscle development in mammals. The online version of this article (doi:10.1186/s12864-016-3009-3) contains supplementary material, which is available to authorized users.
URLPMID:28763026 [本文引用: 1]
Abstract As an important type of noncoding RNA molecules, long non-coding RNAs (lncRNAs) act as versatile players in various biological processes. However, little is known about lncRNA regulators during sheep muscle growth. To explore functional lncRNAs during sheep muscle growth, we systematically investigated lncRNAs using strand-specific Ribo-Zero RNA sequencing at three key developmental stages in Hu sheep. A total of 6924 lncRNAs were obtained, and the differentially expressed lncRNAs and genes were screened from (control vs. experiment) fetus vs. lamb, lamb vs. adult, and fetus vs. adult comparisons, respectively. The quantitative real-time polymerase chain reaction (qRT-PCR) analysis results correlated well with the sequencing data. Moreover, functional annotation analysis based on the Gene Ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) databases showed that the target genes of the differentially expressed lncRNAs were significantly enriched in organ morphogenesis, skeletal system development as well as response to stimulus and some other terms related to muscle. Furthermore, a co-expression network of the differentially expressed target genes and lncRNAs was constructed and well-known muscle growth regulators such as retrotransposon-like 1 and Junctophilin-2 were included. Finally, we investigated the expression profiles of seven lncRNAs and their target genes, and found that they played vital roles in muscle growth. This study extends the sheep muscle lncRNA database and provides novel candidate regulators for future genetic and molecular studies on sheep muscle growth, which is helpful for optimizing the production of mutton.
URLPMID:24948191 [本文引用: 1]
Background The advent of large-scale gene expression technologies has helped to reveal in eukaryotic cells, the existence of thousands of non-coding transcripts, whose function and significance remain mostly poorly understood. Among these non-coding transcripts, long non-coding RNAs (lncRNAs) are the least well-studied but are emerging as key regulators of diverse cellular processes. In the present study, we performed a survey in bovine Longissimus thoraci of lincRNAs (long intergenic non-coding RNAs not overlapping protein-coding transcripts). To our knowledge, this represents the first such study in bovine muscle. Results To identify lincRNAs, we used paired-end RNA sequencing (RNA-Seq) to explore the transcriptomes of Longissimus thoraci from nine Limousin bull calves. Approximately 14鈥45 million paired-end reads were obtained per library. A total of 30,548 different transcripts were identified. Using a computational pipeline, we defined a stringent set of 584 different lincRNAs with 418 lincRNAs found in all nine muscle samples. Bovine lincRNAs share characteristics seen in their mammalian counterparts: relatively short transcript and gene lengths, low exon number and significantly lower expression, compared to protein-encoding genes. As for the first time, our study identified lincRNAs from nine different samples from the same tissue, it is possible to analyse the inter-individual variability of the gene expression level of the identified lincRNAs. Interestingly, there was a significant difference when we compared the expression variation of the 418 lincRNAs with the 10,775 known selected protein-encoding genes found in all muscle samples. In addition, we found 2,083 pairs of lincRNA/protein-encoding genes showing a highly significant correlated expression. Fourteen lincRNAs were selected and 13 were validated by RT-PCR . Some of the lincRNAs expressed in muscle are located within quantitative trait loci for meat quality traits. Conclusions Our study provides a glimpse into the lincRNA content of bovine muscle and will facilitate future experimental studies to unravel the function of these molecules. It may prove useful to elucidate their effect on mechanisms underlying the genetic variability of meat quality traits. This catalog will complement the list of lincRNAs already discovered in cattle and therefore will help to better annotate the bovine genome.

{kind=link}
{kind=link}
{kind=link}
{kind=link}