0 引言
【研究意义】蜜蜂作为社会学模式昆虫,在发育学、行为学和神经生物学等方面具有重要价值[1]。同时,蜜蜂也是最重要的授粉昆虫,具有不可替代的经济价值和生态价值[2]。意大利蜜蜂(Apis mellifera ligustica,简称意蜂)具有优越的采集能力、造脾能力和分泌蜂王浆能力[3],在世界各地的养蜂生产中广泛使用。目前,有关蜜蜂中肠发育机理及调控机制的研究极为滞后,非编码RNA(non-coding RNA,ncRNA)在中肠发育过程中的作用的相关信息也十分有限。在转录组水平对长链非编码RNA(long non-coding RNA,lncRNA)在意蜂工蜂中肠发育中的作用进行研究,可为深入解析意蜂工蜂中肠发育的分子机理提供新的思路和线索。【前人研究进展】随着测序技术的发展,起初被认为基因转录“噪音”的ncRNA成为近几年的研究热点。在人类基因组中,编码蛋白序列所占比例不到2%,超过98%的序列是不编码蛋白质的[4]。ncRNA根据其长短和形状分为小RNA、lncRNA和环状RNA等。其中,lncRNA已被证明在剂量补偿效应[5]、表观遗传[6]、细胞周期[7]和细胞分化调控[8]等众多生命活动中发挥重要作用,因此成为生命科学领域的研究热点。lncRNA的作用方式多样,包括作为信号分子调控上下游基因转录[9],作为诱导分子充当分子阻断剂[10],作为引导分子与蛋白结合[11],作为支架分子发挥“脚手架”作用[12],作为小RNA的前体[13],以及作为竞争内源RNA结合miRNA对mRNA起剪接调控作用[14]。近年来,有关lncRNA在肿瘤发生机制方面作用的研究取得了显著进展,在脂肪代谢、肌肉发育及免疫抗病等机理方面作用的研究也取得了许多突破性进展[15,16]。然而,lncRNA在昆虫领域的相关研究较为滞后,仅在果蝇和家蚕等极少数模式昆虫中有较少的lncRNA相关研究报道[17]。此前,有研究报道lncRNA在蜜蜂的级型分化[18]、卵巢发育[19,20]和抵抗病毒入侵[21]中起重要的调控作用,但总体上有关蜜蜂lncRNA的研究仍非常有限。肠道是昆虫的重要消化器官和免疫器官。蜜蜂肠道的相关研究主要集中在肠道微生物方面[22,23,24,25,26,27],如ENGEL等[26]利用二代测序技术对意蜂工蜂后肠进行宏基因组测序,基于数据分析发现意蜂后肠内的少量细菌种类存在极大的遗传多样性,进一步的比较分析发现不同的细菌种类涉及不同的功能,包括宿主互作、菌膜形成及碳水化合物水解等。【本研究切入点】蜜蜂中肠发育机理及调控机制仍不明确,关于lncRNA在蜜蜂中肠发育过程中作用的研究未见报道。【拟解决的关键问题】结合RNA-seq技术和链特异性cDNA建库方法对意蜂7和10日龄的工蜂中肠进行测序,对中肠发育过程的lncRNA进行差异表达分析,进一步通过上下游基因分析和lncRNA-miRNA-mRNA调控网络分析DElncRNA在意蜂工蜂中肠发育过程中的作用。1 材料与方法
试验于2017年在福建农林大学蜂学学院蜜蜂保护实验室进行。1.1 生物材料
供试意大利蜜蜂工蜂取自福建农林大学蜂学学院教学蜂场。1.2 样品制备与Illumina测序
从群势较强的意蜂蜂群中选取有封盖子的巢脾,快速提至实验室,放入(34±0.5)℃培养箱中培养至工蜂出房,将刚出房的工蜂(记为0 d)放入干净的四周打孔以通风的塑料盒中,盒子上方插入一支装有50%(w/v)无菌糖水的饲喂器(图1),(34±0.5)℃条件下恒温培养,每日检查工蜂存活情况,及时清理死亡的工蜂。进行3次生物学重复。 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图1意蜂工蜂的人工饲养
-->Fig. 1Artificial rearing of A. m. ligustica worker
-->
选取意蜂7和10日龄工蜂中肠作为测序材料。分别快速拉取意蜂7日龄工蜂中肠(Am7)和10日龄工蜂中肠(Am10),液氮速冻后置于-80℃超低温冰箱保存备用,每只中肠的取样时间严格控制在15 s以内。首先用RNA抽提试剂盒(AxyPrepTM Multisource Total RNA Miniprep Kit)(TaKaRa公司,日本)抽提蜜蜂中肠样品的总RNA,为最大限度地保留所有非编码RNA(ncRNA),去除核糖体RNA后的mRNA和ncRNA用裂解缓冲液随机打断为小片段,作为模板用六碱基随机引物、缓冲液、dNTPs、RNase H和DNA polymerase I合成cDNA第二链。经过QiaQuick PCR试剂盒(Qiagen公司,德国)纯化并加EB缓冲液洗脱经末端修复、加碱基A,加测序接头,然后通过尿嘧啶-N-糖基化酶(UNG)降解cDNA第二链。消化产物经琼脂糖凝胶电泳和PCR扩增,PCR产物经高碘酸钠(Sigma 公司,美国)处理。委托广州基迪奥生物科技有限公司对上述cDNA文库进行双端(paired-end)测序,测序平台为Illumina HiSeq 4000。测序数据已上传NCBI SRA数据库,BioProject号:PRJNA406998。
1.3 高通量数据分析
1.3.1 LncRNA的生物信息学预测 对于下机数据,利用Perl脚本去除含有adaptor、未知核苷酸比例>10%和低质量reads,获得有效读段(clean reads)。使用短reads比对工具bowtie[28]分别将样品Am7-1、Am7-2、Am7-3、Am10-1、Am10-2和Am10-3的clean reads比对(mapping)到核糖体数据库(最多允许5个错配),去除mapping上核糖体的reads,利用TopHat2软件[29]将保留下来的数据进一步比对西方蜜蜂的参考基因组(Amel_4.5)[30]。利用FPKM(Fragments Per Kilobase of transcript per Million mapped reads)法计算基因表达量。利用R软件(version 2.16.2)计算各样品之间的相关性系数。lncRNA与mRNA相比在序列保守型、ORF长度等特点上具有一定差异,利用软件CPC[31]和CNCI[32]对新转录本的编码能力进行预测,选取二者的交集作为可靠的预测结果。
1.3.2 DElncRNA及其上下游基因分析 利用edgeR软件[33]进行lncRNA的差异分析,得到DElncRNA。LncRNA的功能与其编码基因位置临近的蛋白编码基因关系密切,位于上游的lncRNA可能与启动子或共表达基因的其他顺式作用元件存在交集,从而在转录或转录后水平进行基因表达调控;位于3′ UTR或基因下游的lncRNA可能参与其他调控作用。因此,对lncRNA进行注释,如果其位于一个基因的上游或下游,这些lncRNA有可能与顺式作用元件所在区域有交集,从而参与转录调控。利用Omicshare平台(http://www.omicshare.com/tools/index.php/)对DElncRNA的上下游基因进行GO(Gene Ontology)分类和KEGG pathway富集分析。
1.3.3 DElncRNA的调控网络构建 LncRNA可以竞争结合miRNA,从而减少miRNA结合mRNA,表明lncRNA可以通过miRNA调控mRNA的表达。利用RNAhybrid[34]、Miranda[35]和TargetScan[36]软件预测DElncRNA靶向结合的miRNA,以及miRNA结合的mRNA,根据上述靶向结合关系构建lncRNA-miRNA- mRNA的调控网络。利用Cytoscape软件[37]对上述调控网络进行可视化。
1.4 lncRNA的RT-PCR验证
随机选取9个lncRNA,根据它们的序列利用DNAMAN软件设计相应的特异性引物。委托上海生工生物工程有限公司合成引物。利用RNA抽提试剂盒(TaKaRa,中国)提取Am7和Am10的总RNA,作为模板进行反转录,得到的cDNA作为模板进行PCR。PCR反应体系(20 μL)包括cDNA模板1 μL,上游引物1 μL,下游引物1 μL,Mixture 10 μL,无菌水补至20 μL。PCR程序:94℃预变性5 min;94℃变性50 s,55℃退火30 s,72℃延伸50 s,共34个循环;72℃再延伸10 min。PCR产物经1.5%琼脂糖凝胶电泳和凝胶成像仪(上海培清,中国)检测。1.5 DElncRNA的实时荧光定量PCR(RT-qPCR)验证
为了验证RNA-seq数据的可靠性,随机选取5个DElncRNA进行RT-qPCR验证。利用DNAMAN软件设计相应的特异性引物。委托上海生工生物工程有限公司合成引物。利用RNA抽提试剂盒(TaKaRa,中国)提取Am7和Am10的总RNA,作为模板进行反转录,得到的cDNA作为模板进行PCR。RT-qPCR反应按照SYBR Green Dye试剂盒(Vazyme公司,中国)操作说明书进行,每个反应进行3次重复。反应体系(20 μL)包含正、反向引物(10.0 μmol·L-1)各1 μL,cDNA模板DNA 1 μL,SYBR Green Dye 10 μL,DEPC水7 μL。qRT-PCR反应在ABI 7500荧光定量PCR仪(ABI公司,美国)上进行,反应条件:95℃预变性1 min,95℃变性15 s,60℃延伸30 s,共40个循环,最后72℃延伸45 s。利用2-ΔΔCt法对上述基因的相对表达量进行计算。2 结果
2.1 数据质控与评估
在实验室条件下对意蜂工蜂进行人工饲养(图1),Am7和Am10中肠样品的测序得到raw reads平均分别为134 802 058和147 051 470条,过滤后得到clean reads平均分别为134 166 157和146 293 288条。各样品的平均Q20和Q30分别为97.34%和93.86%(表1)。Am7与Am10的组内各生物学重复之间的Pearson相关系数均在0.97以上,说明各样品的重复性较好(图2)。上述结果说明本研究的测序数据质量良好,可用于进一步分析。Table 1
表1
表1RT-seq 数据概览
Table 1Overview of RNA-seq datasets
| 样品 Sample | 原始读段 Raw reads | 有效读段 Clean reads | 99.9%的碱基正确率 Q20 (%) | 99.99%的碱基正确率 Q30 (%) |
|---|---|---|---|---|
| Am7-1 | 160844082 | 160049106 (99.51%) | 97.41 | 94.00 |
| Am7-2 | 129878194 | 129283918 (99.54%) | 97.56 | 94.19 |
| Am7-3 | 113683898 | 113165446 (99.54%) | 97.52 | 94.03 |
| Am10-1 | 160537248 | 159765346 (99.52%) | 97.27 | 93.84 |
| Am10-2 | 149230808 | 148494716 (99.51%) | 97.28 | 93.77 |
| Am10-3 | 131386354 | 130619802 (99.42%) | 96.98 | 93.34 |
新窗口打开
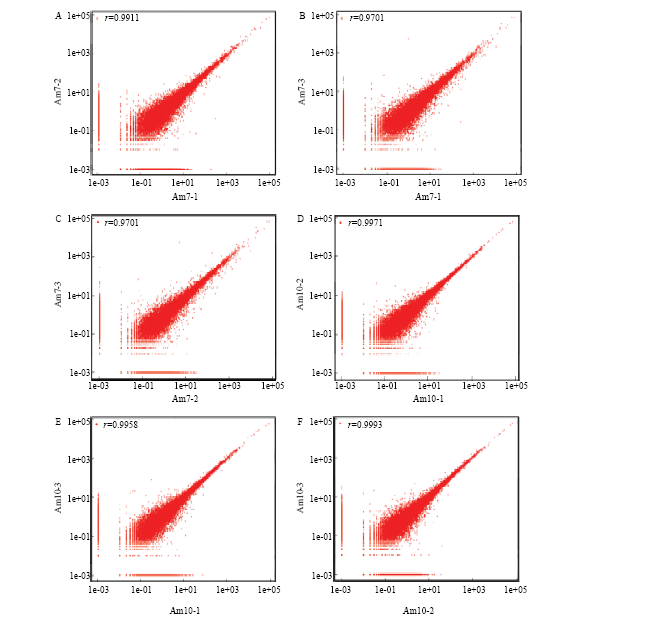 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图2各意蜂工蜂中肠样品生物学重复间的相关性
横纵坐标均表示基因表达量(FPKM)
-->Fig. 2Pearson correlation between every two biological repeats within each A. m. ligustica midgut sample
The horizontal and vertical coordinates represent gene expression level (FPKM)
-->
随机挑选9个lncRNA进行RT-PCR验证,电泳结果显示其中有8个lncRNA均能扩增出符合预期的目的片段,说明本研究预测出的绝大多数的lncRNA真实存在(图3)。相关引物信息详见表2。进一步对上述8个验证的lncRNA的上下游基因进行预测,其中TCONS_00020918可调控西方蜜蜂王浆主蛋白1编码基因,TCONS_00021005可调控西方蜜蜂未知蛋白LOC725PPP,TCONS_00025221可调控西方蜜蜂未知蛋白LOC726321、LOC100577788和LOC102656594。
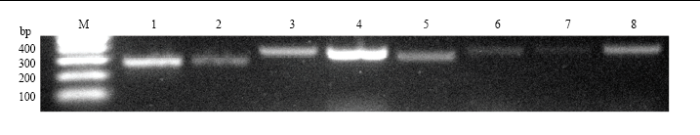 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图3测序数据的RT-PCR 验证
-->Fig. 3RT-PCR validation of RNA-seq data
M: DNA marker; 1: TCONS_00020918; 2: TCONS_00021005; 3: TCONS_00019675; 4: TCONS_00019678; 5: TCONS_00025221; 6: TCONS_00025232; 7: TCONS_00025235; 8: TCONS_00025236
-->
Table 2
表2
表2RT-PCR与RT-qPCR引物信息
Table 2Information of primers for RT-PCR and RT-qPCR
| 引物名称 Primer name | 引物序列 Primer sequence (5′-3′) |
|---|---|
| 1-F | GGCTGAAGATTTCGGATTC |
| 1-R | AGAAGGAGGCAAGGAGGAT |
| 2-F | GCAAAGACGGAAAGATGG |
| 2-R | CCGATGAGTGTGTTCAGTTT |
| 3-F | GCCTGTTAGCCATAGTAAGACG |
| 3-R | AGAGTGTTGAGCAGCGTTG |
| 4-F | CGAGGATGAGCAACTGACA |
| 4-R | GCTACGAGCCAGAAGTCTTT |
| 5-F | CGCAGTAATGAAAGCATAGG |
| 5-R | CGCATCGTGTAACCATAAGA |
| 6-F | CCTCTTGGAGATTCCGATACAG |
| 6-R | CGTTACCACCATTCAACACG |
| 7-F | CCTCTTGGAGATTCCGATACAG |
| 7-R | ACCATTCAACACGAGCACC |
| 8-F | CCTCTTGGAGATTCCGATACAG |
| 8-R | ACCACCATTCAACACGAGC |
| RE1-F | GTTGCTCAAACATCCGAGT |
| RE1-R | CGTTCCATCTTCCTCCAAG |
| RE2-F | TCGTATTCTACAGGGCTTGG |
| RE2-R | TCGCTTCCTTCGTTTAGG |
| RE3-F | GGTTTACTATGCTCCGACGA |
| RE3-R | GGTGATACCGATGGACTCA |
| RE4-F | AGCCAACAGGTGAAATGTG |
| RE4-R | AGGTGTCAGACTGCGGTAA |
| RE5-F | CGTTTCTCGTGCTGCTCTCT |
| RE5-R | AGATGCCACACTTGGATGG |
| Actin-F | CACTCCTGCTATGTATGTCGC |
| Actin-R | GGCAAAGCGTATCCTTCA |
新窗口打开
2.2 DElncRNA及其上下游基因分析
LncRNA的差异表达分析结果显示,在Am7 vs Am10比较组中共有3 890个DElncRNA,包括2 005个上调lncRNA和1885个下调lncRNA。lncRNA可通过影响其编码基因的上下游基因而发挥作用。共预测出1 698个DElncRNA的上下游基因,其中上游基因、下游基因和重叠基因分别为485、535和593个。对DElncRNA编码基因的上下游基因进行GO分类,结果显示它们分布在生物学进程、细胞组分和分子功能3大类,涉及42个GO term,富集基因数最多的前10个GO term分别为结合(355 genes)、细胞进程(315 genes)、代谢进程(299 genes)、单细胞进程(262 genes)、催化活性(259 genes)、细胞(147 genes)、细胞组分(147 genes)、细胞膜(138 genes)、细胞膜组分(131 genes)和细胞器(117 genes)(图4)。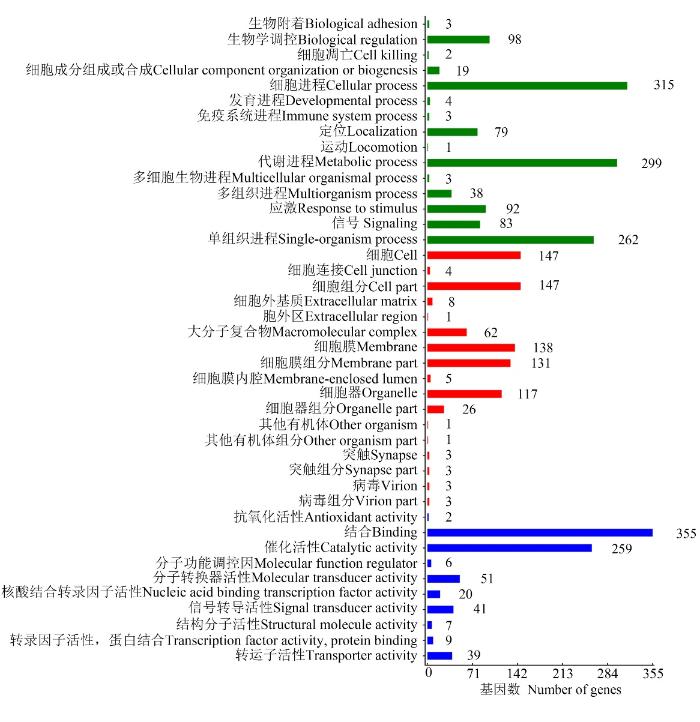 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图4DElncRNA上下游基因的GO分类
-->Fig. 4GO classification of DElncRNAs’ upstream and downstream genes
-->
进一步对DElncRNA编码基因的上下游基因进行KEGG pathway富集分析,结果显示它们富集在251个pathway,富集基因数最多的前10位pathway是Hippo信号通路(36 genes)、Wnt信号通路(19 genes)、癌症microRNA(16 genes)、癌症通路(16 genes)、剪接体(15 genes)、PI3K-AKt信号通路(14 genes)、碳代谢(12 genes)、单纯疱疹感染(12 genes)、癌症蛋白聚糖(12 genes)和神经营养因子信号通路(11 genes)(图5-A)。Hippo信号通路的概貌如图5-B所示。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图5DElncRNA的上下游基因的KEGG pathway富集分析==A:Pathway富集分析;B:Hippo信号通路概貌
-->Fig. 5KEGG pathway enrichment analysis of DElncRNA’s upstream and downstream genes
Enrichment analysis;General picture of Hippo signaling pathway
-->
2.3 DElncRNA的调控网络分析
LncRNA可以作为一种竞争性内源RNA,可通过结合miRNA减少靶向结合mRNA的miRNA数,从而减轻miRNA对mRNA的抑制作用[38]。利用软件预测DElncRNA靶向结合的miRNA,以及miRNA靶向结合的mRNA,其中靶向结合miRNA最多的DElncRNA为TCONS_00004891(36 miRNAs)、XR_001704571.1(28 miRNAs)和TCONS_00024939(27 miRNAs),靶向结合mRNA最多的miRNA为ame-miR-6001-3p(98 mRNAs)、mir-136-y(61 mRNAs)和mir-410-y(38 mRNAs)。进一步利用Cytoscape软件对DElncRNA-miRNA-mRNA调控网络进行可视化,结果显示上调、下调lncRNA与miRNA和mRNA间均形成复杂的调控网络(图6),共有125个lncRNA预测到靶向结合的miRNA,其中仅有6个lncRNA靶向结合1个miRNA,其余的119个lncRNA均靶向结合2个或更多个miRNA。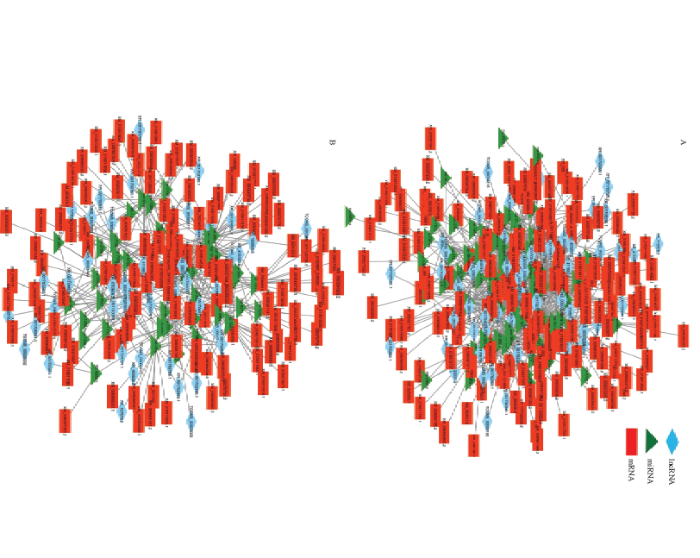 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图6意蜂工蜂中肠发育过程中差异表达lncRNA的调控网络
A:上调lncRNA的lncRNA-miRNA-mRNA网络lncRNA-miRNA-mRNA networks of up-regulated lncRNAs;B:下调lncRNA的lncRNA-miRNA-mRNA网络 lncRNA-miRNA-mRNA networks of down-regulated lncRNAs
-->Fig. 6Regulation network of DElncRNAs during the developmental process of A. m. ligustica worker’s midgut
-->
2.4 DElncRNA的qRT-PCR验证
随机挑取6个DElncRNA进行RT-qPCR验证,结果显示其中有5个DElncRNA表达水平的变化趋势和转录组数据中相应DElncRNA表达水平的变化趋势一致(图7),说明本研究中的测序数据真实可靠。相关引物序列信息详见表2。对验证的5个DElncRNA进行上下游基因分析,发现TCONS_00029692可调控肥大细胞脱粒肽编码基因,TCONS_00038012可调控未知蛋白LOC100576733编码基因。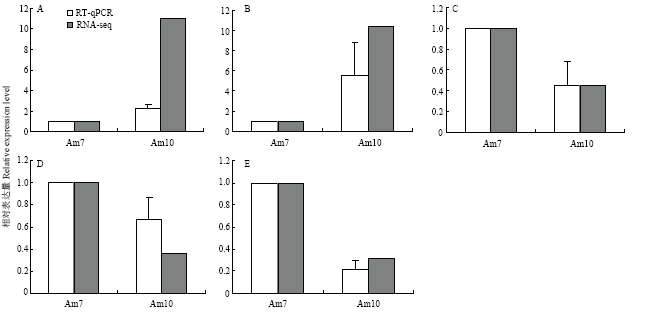 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图7转录组数据的RT-qPCR验证
A: XR_409562.2; B: TCONS_00011956; C: TCONS_00012589; D: TCONS_00038012; E: TCONS_00029692
-->Fig. 7RT-qPCR validation of transcriptome data
-->
3 讨论
此前,蜜蜂肠道的相关研究集中在肠道微生物方面,有关蜜蜂肠道的发育机理、ncRNA在肠道发育过程中作用的研究未见报道。前人研究结果表明lncRNA与生物的神经发育、细胞组织发育、细胞周期调控、干细胞分化、免疫应答和癌症发生等过程关系密切。近年来,随着二代测序技术的迅速发展和广泛应用,越来越多的lncRNA在动物、植物和微生物中被鉴定出来,但蜜蜂lncRNA的研究较为滞后,相关信息极为有限。为探究意蜂工蜂中肠的发育机理及调控机制,本研究结合RNA-seq技术和链特异性建库方法对意蜂7和10日龄工蜂中肠进行测序,基于高质量的测序数据共预测出6 353个lncRNA,RT-PCR验证的扩增成功率高达88.9%,表明本研究预测出的绝大多数lncRNA真实存在。相对于非特异性建库,特异性建库的方法在合成cDNA第二链时,将dTTP替换为dUTP,加上接头后再用UNG酶降解第二链,因而该方法可以确定转录本来自正链或负链,故能更准确地获得基因的结构和基因表达信息。lncRNA的表达具有时间和组织特异性,本研究的测序对象是意蜂工蜂中肠组织,它们在其他组织中是否表达以及表达水平有待于进一步研究。本研究中人工饲养工蜂的过程中仅饲喂糖水,工蜂的肠道内会有少量流体且多聚集在后肠,为排除杂质对测序结果的影响,舍弃了后肠,将中肠组织用于测序。测得的原始数据包含工蜂本身的数据和少量的中肠内微生物的数据,为排除后者的干扰,对原始数据进行了严格的过滤和质控,先比对核糖体数据库以去除rRNA数据,再比对西方蜜蜂基因组去除未比对上的数据,保留下来的能比对上的数据用于后续的生物信息学分析,考虑到昆虫和微生物的物种亲缘关系较远,二者的基因保守性很低,因而比对上参考基因组的数据理论上皆为意蜂工蜂中肠本身的数据。LncRNA的功能与其编码基因坐标临近的蛋白编码基因相关,位于上游的lncRNA可能与启动子或共表达基因的其他顺式作用元件有交集,从而在转录或转录后水平对基因的表达进行调控[39]。本研究中,DElncRNA的上下游基因达到1 697个,GO富集分析结果显示分别有299个和4个上下游基因涉及代谢进程和发育进程,有259个上下游基因涉及氧化磷酸化,表明DElncRNA参与意蜂工蜂中肠的物质和能量代谢过程调控。本研究发现,315个上下游基因涉及细胞进程,表明DElncRNA广泛参与意蜂工蜂中肠的细胞生命活动。此外,还发现分别有92个和3个上下游基因涉及应激反应和免疫系统进程,暗示相应的DElncRNA在意蜂工蜂中肠发育中发挥调控作用。王浆主蛋白(MRJP)为幼虫发育提供必要的可利用氮元素,在蜜蜂行为中有潜在的调控作用,还参与决定蜂群中雌性蜂的级型分化等,其中MRJP1在蜂王浆主蛋白中含量最丰富,所占比例达31%,占蜂王浆中水溶性蛋白的48%[40]。本研究发现,TCONS_00020918可调控西方蜜蜂MRJP1编码基因,表明该lncRNA可能在意蜂的营养吸收、级型分化中发挥重要的调控功能。此外,多个DElncRNA可能调控未知蛋白编码基因,具体调控关系的明确有赖于Nr数据库蛋白功能注释信息的完善及进一步的实验验证。
蜜蜂肠道是物质和能量合成与代谢的主要场所。本研究中,DElncRNA上下游基因的KEGG富集分析结果显示,共有221个上下游基因富集在多达65个物质代谢通路上,包括甘油酯代谢(5 genes)和脂肪酸的生物合成(2 genes)等10条脂代谢相关通路,柠檬酸盐循环(6 genes)和丙酮酸盐代谢(2 genes)等13条碳水化合物代谢相关通路,赖氨酸降解(3 genes)和精氨酸生物合成(2 genes)等13条氨基酸代谢相关通路,以及嘌呤代谢(10 genes)和嘧啶代谢(1 gene),表明DElncRNA广泛参与意蜂工蜂中肠发育过程中的物质代谢调控。物质合成与代谢往往伴随着旺盛的能量代谢,本研究发现共有15个上下游基因富集在甲烷代谢(5 genes)、氧化磷酸化(5 genes)和硫代谢(2 genes)等能量代谢通路,表明相应的DElncRNA在意蜂工蜂中肠的能量代谢方面具有重要的调控功能。蜜蜂肠道是物质消化吸收的主要位置。本研究发现28个上下游基因富集在7个消化系统通路上,包括胰腺分泌(8 genes)、蛋白消化和吸收(2 genes)、胃酸分泌(7 genes)、脂肪体消化和吸收(4 genes)、唾液分泌(4 genes)、维生素消化和吸收(1 genes)和胆汁分泌(2 genes),表明DElncRNA参与调控意蜂工蜂中肠对物质的消化和吸收。Hippo信号通路可以调节器官的大小[41,42],也能与其他信号通路相互作用共同调节中肠组织的稳态[43]。Notch信号影响细胞正常形态发生的多个过程,包括多能细胞分化、细胞凋亡、细胞增殖及细胞边界的形成[44]。本研究发现,分别有36、19和8个上下游基因分别富集在Hippo、Wnt和Notch信号通路。此外,还发现7、3、3和7个上下游基因富集在TGF-beta、mTOR、Hedgehog和胰岛素信号通路。结果表明相应的DElncRNA可通过参与调控上述信号通路对意蜂工蜂中肠发育进行调控。
角质层和围食膜是昆虫免疫防御的第一道防线[45]。如果病原微生物突破第一道防线,包括蜜蜂在内的昆虫将会启动细胞和体液免疫抵抗病原的入侵。蜜蜂的细胞免疫包括内吞作用、黑化作用、吞噬作用、蛋白的酶促水解等,体液免疫主要为抗菌肽的合成与释放[46]。本研究中,DElncRNA上下游基因的KEGG pathway富集分析结果显示分别有11、9、7、5和4个上下游基因富集在溶酶体、内吞作用、泛素蛋白水解、吞噬体和黑化作用等细胞免疫通路,此外,分别有13、3、1和1个上下游基因富集在MAPK、Jak-STAT、NF-Kappa B和Toll-like受体等体液免疫通路。上述结果表明相应的DElncRNA同时参与了宿主的细胞和体液免疫的调控过程,推测其在宿主的免疫防御过程发挥关键作用。
SALMENA等提出内源性竞争RNA(ceRNA)假说[38],该假说认为lncRNA和mRNA等RNA可以通过miRNA应答原件(MRE)与miRNA竞争性结合,从而对基因表达进行调控。此后,ceRNA假说已被越来越多的研究[47]所证明。为进一步揭示DElncRNA的作用,本研究通过靶向关系预测DElncRNA结合的miRNA以及miRNA结合的mRNA,并构建三者间的调控网络,发现部分上调与下调lncRNA位于调控网络中心位置且结合较多的miRNA。其中,部分lncRNA可结合多个miRNA,如TCONS_00004891、XR_001704571.1和TCONS_00024939分别结合36、28和27个miRNA,表明这些lncRNA可能在意蜂工蜂中肠发育过程中发挥重要的调控功能。此外,也有部分lncRNA共同靶向结合一个miRNA,如有多达50个lncRNA能够靶向结合ame-miR-6001-3p。COLLINS等在研究真社会昆虫等级分化过程中发现,bte-miR-6001-3p在欧洲熊蜂(Bombus terrestris)等级分化的幼虫中差异表达显著,ame-miR-6001-3p在西方蜜蜂等级分化的幼虫中差异表达较弱,说明miR-6001-3p影响蜜蜂卵巢发育[48]。上述结果表明DElncRNA可作为ceRNA吸附结合miRNA,从而对意蜂工蜂中肠发育过程进行调控。目前,lncRNA的功能研究多集中在人类、哺乳动物和重要疾病领域[17],在果蝇中也有少量研究[49],但包括蜜蜂[18,19,20,21]在内的绝大多数昆虫的lncRNA研究仍处于初级阶段,相关的lncRNA信息十分有限。本研究预测出的多数lncRNA,其具体的调控关系尚不明确,需要进一步克隆出lncRNA全长序列并深入探究其功能,这是未来的研究方向。
本研究仅对意蜂7和10日龄工蜂中肠进行了测序和lncRNA相关分析,若要全面解析意蜂工蜂中肠的发育机理及调控机制,需要对更多日龄的工蜂中肠进行测序,进而在全局水平进行更加深入的分析,例如对所有DElncRNA进行基因权重共表达分析(WGCNA)。
4 结论
通过对意蜂工蜂中肠发育过程中的lncRNA及其功能进行深入分析,提供了意蜂工蜂中肠发育过程中的DElncRNA信息,揭示了DElncRNA广泛参与意蜂工蜂中肠的新陈代谢、细胞活动和免疫调控并作为竞争性内源RNA(ceRNA)发挥作用,为关键lncRNA的筛选和功能研究提供了必要的数据支持。The authors have declared that no competing interests exist.
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
| [1] | . Background The honey bee is an important model system for increasing understanding of molecular and neural mechanisms underlying social behaviors relevant to the agricultural industry and basic science. The western honey bee, Apis mellifera, has served as a model species, and its genome sequence has been published. In contrast, the genome of the Asian honey bee, Apis cerana, has not yet been sequenced. A. cerana has been raised in Asian countries for thousands of years and has brought considerable economic benefits to the apicultural industry. A cerana has divergent biological traits compared to A. mellifera and it has played a key role in maintaining biodiversity in eastern and southern Asia. Here we report the first whole genome sequence of A. cerana. Results Using de novo assembly methods, we produced a 238 Mbp draft of the A. cerana genome and generated 10,651 genes. A.cerana-specific genes were analyzed to better understand the novel characteristics of this honey bee species. Seventy-two percent of the A. cerana-specific genes had more than one GO term, and 1,696 enzymes were categorized into 125 pathways. Genes involved in chemoreception and immunity were carefully identified and compared to those from other sequenced insect models. These included 10 gustatory receptors, 119 odorant receptors, 10 ionotropic receptors, and 160 immune-related genes. Conclusions This first report of the whole genome sequence of A. cerana provides resources for comparative sociogenomics, especially in the field of social insect communication. These important tools will contribute to a better understanding of the complex behaviors and natural biology of the Asian honey bee and to anticipate its future evolutionary trajectory. |
| [2] | |
| [3] | |
| [4] | . Eukaryotic cells make many types of primary and processed RNAs that are found either in specific sub-cellular compartments or throughout the cells. A complete catalogue of these RNAs is not yet available and their characteristic sub-cellular localizations are also poorly understood. Since RNA represents the direct output of the genetic information encoded by genomes and a significant proportion of a cell鈥檚 regulatory capabilities are focused on its synthesis, processing, transport, modifications and translation, the generation of such a catalogue is crucial for understanding genome function. Here we report evidence that three quarters of the human genome is capable of being transcribed, as well as observations about the range and levels of expression, localization, processing fates, regulatory regions and modifications of almost all currently annotated and thousands of previously unannotated RNAs. These observations taken together prompt to a redefinition of the concept of a gene. |
| [5] | . Non-coding RNAs (ncRNAs) are receiving more and more attention not only as an abundant class of genes, but also as regulatory structural elements (some located in mRNAs). A key feature of RNA function is its structure. Computational methods were developed early for folding and prediction of RNA structure with the aim of assisting in functional analysis. With the discovery of more and more ncRNAs, it has become clear that a large fraction of these are highly structured. Interestingly, a large part of the structure is comprised of regular Watson-Crick and GU wobble base pairs. This and the increased amount of available genomes have made it possible to employ structure-based methods for genomic screens. The field has moved from folding prediction of single sequences to computational screens for ncRNAs in genomic sequence using the RNA structure as the main characteristic feature. Whereas early methods focused on energy-directed folding of single sequences, comparative analysis based on structure preserving changes of base pairs has been efficient in improving accuracy, and today this constitutes a key component in genomic screens. Here, we cover the basic principles of RNA folding and touch upon some of the concepts in current methods that have been applied in genomic screens forde novoRNA structures in searches for novel ncRNA genes and regulatory RNA structure on mRNAs. We discuss the strengths and weaknesses of the different strategies and how they can complement each other. |
| [6] | . There is a growing interest for noncoding RNA (ncRNA)-mediated epigenetic regulation of transcription in diverse biological functions. Recent evidence suggests that a subset of long ncRNA epigenetically regulate the transcription of multiple genes in chromosomal domains via interaction with chromatin. , , revealed that chromatin regulatory ncRNAs share common epigenetic pathways in the silencing of multiple genes. |
| [7] | . The long noncoding MALAT1 RNA is upregulated in cancer tissues and its elevated expression is associated with hyper-proliferation, but the underlying mechanism is poorly understood. We demonstrate that MALAT1 levels are regulated during normal cell cycle progression. Genome-wide transcriptome analyses in normal human diploid fibroblasts reveal that MALAT1 modulates the expression of cell cycle genes and is required for G1/S and mitotic progression. Depletion of MALAT1 leads to activation of p53 and its target genes. The cell cycle defects observed in MALAT1-depleted cells are sensitive to p53 levels, indicating that p53 is a major downstream mediator of MALAT1 activity. Furthermore, MALAT1-depleted cells display reduced expression of B-MYB (Mybl2), an oncogenic transcription factor involved in G2/M progression, due to altered binding of splicing factors on B-MYB pre-mRNA and aberrant alternative splicing. In human cells, MALAT1 promotes cellular proliferation by modulating the expression and/or pre-mRNA processing of cell cycle-regulated transcription factors. These findings provide mechanistic insights on the role of MALAT1 in regulating cellular proliferation. |
| [8] | . 61ADNCR is the most downregulated lncRNA during adipogenic differentiation.61ADNCR acts as a competing endogenous RNA for miR-204.61miR-204 promotes adipogenesis by targeting SIRT1.61ADNCR increases the expression of SIRT1 in a miR-204-dependent manner. |
| [9] | . Long intervening noncoding RNAs (lincRNAs) are transcribed from thousands of loci in mammalian genomes and might play widespread roles in gene regulation and other cellular processes. This Review outlines the emerging understanding of lincRNAs in vertebrate animals, with emphases on how they are being identified and current conclusions and questions regarding their genomics, evolution and mechanisms of action. |
| [10] | . Nat Genet. 2011 Jun 5;43(7):621-9. doi: 10.1038/ng.848. Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov't; Research Support, U.S. Gov't, Non-P.H.S. |
| [11] | . 78 RNA-binding protein HuR recruits let-7 to lincRNA-p21, lowers lincRNA-p21 stability 78 lincRNA-p21 selectively binds JUNB and CTNNB1 mRNAs, represses their translation 78 With low HuR, lincRNA-p21 levels rise, repressing JunB and β-catenin translation 78 With high HuR, lincRNA-p21 levels decline, rising JunB and β-catenin translation |
| [12] | . The steroid receptor RNA activator (SRA) has previously been characterized as belonging to the growing family of functional non-coding RNAs. However, we recently reported the Western blot detection of a putative endogenous SRA protein (SRAP) in breast cancer cells. Herein, we successfully suppressed the expression of this protein through specific RNA interference assay, unequivocally confirming its existence. Moreover, using database searches and Western blot analysis, we also showed that SRAP is highly conserved among chordata. Overall, our results suggest that SRA is the first example of a new class of functional RNAs also able to encode a protein. |
| [13] | . In mammals, dosage compensation is achieved by X-chromosome inactivation (XCI) in the female. The noncoding Xist gene initiates silencing of the X chromosome, whereas its antisense partner Tsix blocks silencing. The complementarity of Xist and Tsix RNAs has long suggested a role for RNA interference (RNAi). Here, we report that murine Xist and Tsix form duplexes in vivo. During XCI, the duplexes are processed to small RNAs (sRNAs), most likely on the active X (Xa) in a Dicer-dependent manner. Deleting Dicer compromises sRNA production and derepresses Xist. Furthermore, without Dicer, Xist RNA cannot accumulate and histone 3 lysine 27 trimethylation is blocked on the inactive X (Xi). The defects are partially rescued by truncating Tsix. Thus, XCI and RNAi intersect, down-regulating Xist on Xa and spreading silencing on Xi. |
| [14] | . |
| [15] | . Annotation on the reference genome of the C57BL6/J mouse has been an ongoing project ever since the draft genome was first published. Initially, the principle focus was on the identification of all protein-coding genes, although today the importance of describing long non-coding RNAs, small RNAs, and pseudogenes is recognized. Here, we describe the progress of the GENCODE mouse annotation project, which combines manual annotation from the HAVANA group with Ensembl computational annotation, alongside experimental and in silico validation pipelines from other members of the consortium. We discuss the more recent incorporation of next-generation sequencing datasets into this workflow, including the usage of mass-spectrometry data to potentially identify novel protein-coding genes. Finally, we will outline how the C57BL6/J genebuild can be used to gain insights into the variant sites that distinguish different mouse strains and species. The online version of this article (doi:10.1007/s00335-015-9583-x) contains supplementary material, which is available to authorized users. |
| [16] | . Abstract Long non-coding RNAs (lncRNAs) are largely heterogeneous and functionally uncharacterized. Here, using FANTOM5 cap analysis of gene expression (CAGE) data, we integrate multiple transcript collections to generate a comprehensive atlas of 27,919 human lncRNA genes with high-confidence 5' ends and expression profiles across 1,829 samples from the major human primary cell types and tissues. Genomic and epigenomic classification of these lncRNAs reveals that most intergenic lncRNAs originate from enhancers rather than from promoters. Incorporating genetic and expression data, we show that lncRNAs overlapping trait-associated single nucleotide polymorphisms are specifically expressed in cell types relevant to the traits, implicating these lncRNAs in multiple diseases. We further demonstrate that lncRNAs overlapping expression quantitative trait loci (eQTL)-associated single nucleotide polymorphisms of messenger RNAs are co-expressed with the corresponding messenger RNAs, suggesting their potential roles in transcriptional regulation. Combining these findings with conservation data, we identify 19,175 potentially functional lncRNAs in the human genome. |
| [17] | . 长链非编码RNA(long noncoding RNA,lncRNA)是一类转录本长度超过200 nt的非编码RNA,主要通过转录调控和转录后调控调节基因的表达,也可通过影响蛋白质定位和端粒复制发挥其强大的生物学功能。本文在介绍lncRNA的特征、分类及其主要作用机制的基础上,综述了有关昆虫lncRNA的鉴定及功能研究等方面的最新进展。近5年来已经从黑腹果蝇Drosophila melanogaster、小菜蛾Plutella xylostella和褐飞虱Nilaparvata lugens等8种昆虫中鉴定出了大量lncRNA,为进一步研究lncRNA在昆虫生长发育过程中的功能奠定了重要基础。非编码RNA参与调控害虫抗药性的分子机制已经成为昆虫毒理学研究的一个新兴领域,因此本文对有关lncRNA与害虫抗药性关系的最新研究进展也做了介绍。 . 长链非编码RNA(long noncoding RNA,lncRNA)是一类转录本长度超过200 nt的非编码RNA,主要通过转录调控和转录后调控调节基因的表达,也可通过影响蛋白质定位和端粒复制发挥其强大的生物学功能。本文在介绍lncRNA的特征、分类及其主要作用机制的基础上,综述了有关昆虫lncRNA的鉴定及功能研究等方面的最新进展。近5年来已经从黑腹果蝇Drosophila melanogaster、小菜蛾Plutella xylostella和褐飞虱Nilaparvata lugens等8种昆虫中鉴定出了大量lncRNA,为进一步研究lncRNA在昆虫生长发育过程中的功能奠定了重要基础。非编码RNA参与调控害虫抗药性的分子机制已经成为昆虫毒理学研究的一个新兴领域,因此本文对有关lncRNA与害虫抗药性关系的最新研究进展也做了介绍。 |
| [18] | . 蜜蜂的级型分化被证实是由蜂王浆中的Royalactin决定,工蜂和蜂王幼虫在级型分化时编码基因的表达差异也被广泛研究.我们发现,在蜜蜂幼虫的级型分化过程中,lncRNA也有着显著的表达差异,因此认为,lncRNA也参与了蜜蜂的级型分化过程.进一步的分析显示,lncRNA可能通过影响上下游基因的转录和功能执行的方式,在蜜蜂早期发育的多细胞组织发育、神经系统发育和转录调控的过程中起到重要的调控作用. . 蜜蜂的级型分化被证实是由蜂王浆中的Royalactin决定,工蜂和蜂王幼虫在级型分化时编码基因的表达差异也被广泛研究.我们发现,在蜜蜂幼虫的级型分化过程中,lncRNA也有着显著的表达差异,因此认为,lncRNA也参与了蜜蜂的级型分化过程.进一步的分析显示,lncRNA可能通过影响上下游基因的转录和功能执行的方式,在蜜蜂早期发育的多细胞组织发育、神经系统发育和转录调控的过程中起到重要的调控作用. |
| [19] | . Division of labor in social insect colonies relies on a strong reproductive bias that favors queens. Although the ecological and evolutionary success attained through caste systems is well sketched out in terms of ultimate causes, the molecular and cellular underpinnings driving the development of caste phenotypes are still far from understood. Recent genomics approaches on honey bee developmental biology revealed a set of genes that are differentially expressed genes in larval ovaries and associated with transgressive ovary size in queens and massive cell death in workers. Amongst these, two contigs called special attention, both being over 200 bp in size and lacking apparent coding potential. Herein, we obtained their full cDNA sequences. These and their secondary structure characteristics placed in evidence that they are bona fide long noncoding RNAs (lncRNA) differentially expressed in larval ovaries, thus named lncov1 and lncov2. Genomically, both map within a previously identified QTL on chromosome 11, associated with transgressive ovary size in honey bee workers. As lncov1 was over-expressed in worker ovaries we focused on this gene. Real-time qPCR analysis on larval worker ovaries evidenced an expression peak coinciding with the onset of autophagic cell death. Cellular localization analysis through fluorescence in situ hybridization revealed perinuclear spots resembling omega speckles known to regulate trafficking of RNA-binding proteins. With only four lncRNAs known so far in honey bees, two expressed in the ovaries, these findings open a novel perspective on regulatory factors acting in the fine tuning of developmental processes underlying phenotypic plasticity related to social life histories. |
| [20] | . The honey bee (Apis mellifera) is a highly diverse species commonly used for honey production and pollination services. The oviposition of the honey bee queen affects the development and overall performance of the colony. To investigate the ovary activation and oviposition processes on a molecular level, a genome-wide analysis of lncRNAs, miRNAs and mRNA expression in the ovaries of the queens was performed to screen for differentially expressed coding and noncoding RNAs. Further analysis identified relevant candidate genes or RNAs. The analysis of the RNA profiles in different oviposition phase of the queens revealed that 740 lncRNAs, 81 miRNAs and 5,481 mRNAs were differently expressed during the ovary activation; 88 lncRNAs, 13 miRNAs and 338 mRNAs were differently expressed during the oviposition inhibition process; and finally, 100 lncRNAs, four miRNAs and 497 mRNAs were differently expressed during the oviposition recovery process. In addition, functional annotation of differentially expressed RNAs revealed several pathways that are closely related to oviposition, including hippo, MAPK, notch, Wnt, mTOR, TGF-beta and FoxO signaling pathways. Furthermore, in the QTL region for ovary size, 73 differentially expressed genes and 14 differentially expressed lncRNAs were located, which are considered as candidate genes affecting ovary size and oviposition. Moreover, a core set of genes served as bridges among different miRNAs were identified through the integrated analysis of lncRNA-miRNA-mRNA network. The observed dramatic expression changes of coding and noncoding RNAs suggest that they may play a critical role in honey bee queens oviposition. The identified candidate genes for oviposition activation and regulation could serve as a resource for further studies of genetic markers of oviposition in honey bees. |
| [21] | . Long non-coding RNAs (lncRNAs) are a class of RNAs that do not encode proteins. Recently, lncRNAs have gained special attention for their roles in various biological process and diseases. In an attempt to identify long intergenic non-coding RNAs (lincRNAs) and their possible involvement in honey bee development and diseases, we analyzed RNA-seq datasets generated from Asian honey bee (Apis cerana) and western honey bee (Apis mellifera). We identified 2470 lincRNAs with an average length of 1011 bp from A. cerana and 1514 lincRNAs with an average length of 790 bp in A. mellifera. Comparative analysis revealed that 5 % of the total lincRNAs derived from both species are unique in each species. Our comparative digital gene expression analysis revealed a high degree of tissue-specific expression among the seven major tissues of honey bee, different from mRNA expression patterns. A total of 863 (57 %) and 464 (18 %) lincRNAs showed tissue-dependent expression in A. mellifera and A. cerana, respectively, most preferentially in ovary and fat body tissues. Importantly, we identified 11 lincRNAs that are specifically regulated upon viral infection in honey bees, and 10 of them appear to play roles during infection with various viruses. This study provides the first comprehensive set of lincRNAs for honey bees and opens the door to discover lincRNAs associated with biological and hormone signaling pathways as well as various diseases of honey bee. The online version of this article (doi:10.1186/s12864-015-1868-7) contains supplementary material, which is available to authorized users. |
| [22] | . |
| [23] | . The animal gut is a habitat for diverse communities of microorganisms (microbiota). Honeybees and bumblebees have recently been shown to harbour a distinct and species poor microbiota, which may confer protection against parasites. Here, we investigate diversity, host specificity and transmission mode of two of the most common, yet poorly known, gut bacteria of honeybees and bumblebees: Snodgrassella alvi (Betaproteobacteria) and Gilliamella apicola (Gammaproteobacteria). We analysed 16S rRNA gene sequences of these bacteria from diverse bee host species across most of the honeybee and bumblebee phylogenetic diversity from North America, Europe and Asia. These focal bacteria were present in 92% of bumblebee species and all honeybee species but were found to be absent in the two related corbiculate bee tribes, the stingless bees (Meliponini) and orchid bees (Euglossini). Both Snodgrassella alvi and Gilliamella apicola phylogenies show significant topological congruence with the phylogeny of their bee hosts, albeit with a considerable degree of putative host switches. Furthermore, we found that phylogenetic distances between Gilliamella apicola samples correlated with the geographical distance between sampling locations. This tentatively suggests that the environmental transmission rate, as set by geographical distance, affects the distribution of G. apicola infections. We show experimentally that both bacterial taxa can be vertically transmitted from the mother colony to daughter queens, and social contact with nest mates after emergence from the pupa greatly facilitates this transmission. Therefore, sociality may play an important role in vertical transmission and opens up the potential for co-evolution or at least a close association of gut bacteria with their hosts. |
| [24] | . |
| [25] | . The gut of insects may harbour one of the largest reservoirs of a yet unexplored microbial diversity. To understand how specific insects select for their own bacterial communities, the structural diversity and variability of bacteria found in the gut of different bee species was analysed. For three successive years, adults and larvae of Apis mellifera ssp. carnica (honey bee), and Bombus terrestris (bumble bee), as well as larvae of Osmia bicornis (red mason bee) were collected at a flowering oilseed rape field. Total DNA was extracted from gut material and the bacterial diversity was analysed, independent of cultivation, by genetic profiling with single-strand conformation polymorphism (SSCP) of polymerase chain reaction (PCR)-amplified partial 16S rRNA genes. The SSCP profiles were specific for all bee species and for larvae and adults. Qualitative and quantitative differences were found in the bacterial community structure of larvae and adults of A. mellifera , but differences in B. terrestris were mainly quantitative. Sequencing of the PCR products revealed a dominance of Alpha -, Beta -, and Gammaproteobacteria , Bacteroidetes , and Firmicutes in all bee species. Single-strand conformation polymorphism profiles suggested a higher abundance and diversity of lactobacilli in adults of A. mellifera than in larvae. Further phylogenetic analyses indicated common bacterial phylotypes for all three bee species, e.g. those related to Simonsiella , Serratia , and Lactobacillus . Clades related to Delftia acidovorans , Pseudomonas aeruginosa or Lactobacillus intestinalis only contained sequences from larvae. Several of the bee-specific clusters also contained identical or highly similar sequences from bacteria detected in other A. mellifera subspecies from South Africa, suggesting the existence of cosmopolitan gut bacteria in bees. |
| [26] | . The honey bee, Apis mellifera, harbors a characteristic gut microbiota composed of only a few species which seem to be specific to social bees. The maintenance of this stable and distinct microbial community depends on the social lifestyle of these insects. As in other animals, the bacteria in the gut of honey bees probably govern important functions critical to host health. We recently sequenced a metagenome of the gut microbiota of A. mellifera, assigned gene contents to bins corresponding to the major species present in the honey bee gut, and compared functional gene categories between these species, and between the complete metagenome and those of other animals. Gene contents could be linked to different symbiotic functions with the host. Further, we found a high degree of genetic diversity within each of these species. In the case of the gammaproteobacterial species Gilliamella apicola, we experimentally showed a link between genetic variation of isolates and functional differences suggesting that niche partitioning within this species has emerged during evolution with its bee hosts. The consistent presence of only a few species, combined with strain variation within each of these species, makes the gut microbiota of social bees an ideal model for studying functional, structural, and evolutionary aspects of host-associated microbial communities: many characteristics resemble the gut microbiota of humans and other mammals, but the complexity is considerably reduced. In this addendum, we summarize and discuss our major findings and provide a detailed perspective on future research. |
| [27] | . |
| [28] | . Bowtie is an ultrafast, memory-efficient alignment program for aligning short DNA sequence reads to large genomes. For the human genome, Burrows-Wheeler indexing allows Bowtie to align more than 25 million reads per CPU hour with a memory footprint of approximately 1.3 gigabytes. Bowtie extends previous Burrows-Wheeler techniques with a novel quality-aware backtracking algorithm that permits mismatches. Multiple processor cores can be used simultaneously to achieve even greater alignment speeds. Bowtie is open source http://bowtie.cbcb.umd.edu . |
| [29] | . |
| [30] | . |
| [31] | . Recent transcriptome studies have revealed that a large number of transcripts in mammals and other organisms do not encode proteins but function as noncoding RNAs (ncRNAs) instead. As millions of transcripts are generated by large-scale cDNA and EST sequencing projects every year, there is a need for automatic methods to distinguish protein-coding RNAs from noncoding RNAs accurately and quickly. We developed a support vector machine-based classifier, named Coding Potential Calculator (CPC), to assess the protein-coding potential of a transcript based on six biologically meaningful sequence features. Tenfold cross-validation on the training dataset and further testing on several large datasets showed that CPC can discriminate coding from noncoding transcripts with high accuracy. Furthermore, CPC also runs an order-of-magnitude faster than a previous state-of-the-art tool and has higher accuracy. We developed a user-friendly web-based interface of CPC at http://cpc.cbi.pku.edu.cn. In addition to predicting the coding potential of the input transcripts, the CPC web server also graphically displays detailed sequence features and additional annotations of the transcript that may facilitate users' further investigation. |
| [32] | . |
| [33] | . |
| [34] | . MicroRNAs (miRNAs) are short RNAs that post-transcriptionally regulate the expression of target genes by binding to the target mRNAs. Although a large number of animal miRNAs has been defined, only a few targets are known. In contrast to plant miRNAs, which usually bind nearly perfectly to their targets, animal miRNAs bind less tightly, with a few nucleotides being unbound, thus producing more complex secondary structures of miRNA/target duplexes. Here, we present a program, RNA-hybrid, that predicts multiple potential binding sites of miRNAs in large target RNAs. In general, the program finds the energetically most favorable hybridization sites of a small RNA in a large RNA. Intramolecular hybridizations, that is, base pairings between target nucleotides or between miRNA nucleotides are not allowed. For large targets, the time complexity of the algorithm is linear in the target length, allowing many long targets to be searched in a short time. Statistical significance of predicted targets is assessed with an extreme value statistics of length normalized minimum free energies, a Poisson approximation of multiple binding sites, and the calculation of effective numbers of orthologous targets in comparative studies of multiple organisms. We applied our method to the prediction of miRNA targets in 3 TRs and coding sequence. RNAhybrid, with its accompanying programs RNAcalibrate and RNAeffective, is available for download and as a Web tool on the Bielefeld Bioinformatics Server (). |
| [35] | . MicroRNA.org (http://www.microrna.org) is a comprehensive resource of microRNA target predictions and expression profiles. Target predictions are based on a development of the miRanda algorithm which incorporates current biological knowledge on target rules and on the use of an up-to-date compendium of mammalian microRNAs. MicroRNA expression profiles are derived from a comprehensive sequencing project of a large set of mammalian tissues and cell lines of normal and disease origin. Using an improved graphical interface, a user can explore (i) the set of genes that are potentially regulated by a particular microRNA, (ii) the implied cooperativity of multiple microRNAs on a particular mRNA and (iii) microRNA expression profiles in various tissues. To facilitate future updates and development, the microRNA.org database structure and software architecture is flexibly designed to incorporate new expression and target discoveries. The web resource provides users with functional information about the growing number of microRNAs and their interaction with target genes in many species and facilitates novel discoveries in microRNA gene regulation. |
| [36] | . Plants and animals use small RNAs (microRNAs [miRNAs] and siRNAs) as guides for posttranscriptional and epigenetic regulation. In plants, miRNAs and trans-acting (ta) siRNAs form through distinct biogenesis pathways, although they both interact with target transcripts and guide cleavage. An integrated approach to identify targets of Arabidopsis thaliana miRNAs and ta-siRNAs revealed several new classes of small RNA-regulated genes, including conventional genes such as Argonaute2 and an E2-ubiquitin conjugating enzyme. Surprisingly, five ta-siRNA-generating transcripts were identified as targets of miR173 or miR390. Rather than functioning as negative regulators, miR173- and miR390-guided cleavage was shown to set the 21-nucleotide phase for ta-siRNA precursor processing. These data support a model in which miRNA-guided formation of a 5′ or 3′ terminus within pre-ta-siRNA transcripts, followed by RDR6-dependent formation of dsRNA and Dicer-like processing, yields phased ta-siRNAs that negatively regulate other genes. |
| [37] | . |
| [38] | Here, we present a unifying hypothesis about how messenger RNAs, transcribed pseudogenes, and long noncoding RNAs “talk” to each other using microRNA response elements (MREs) as letters of a new language. We propose that this “competing endogenous RNA” (ceRNA) activity forms a large-scale regulatory network across the transcriptome, greatly expanding the functional genetic information in the human genome and playing important roles in pathological conditions, such as cancer. |
| [39] | . Over the last decade, it has been increasingly demonstrated that the genomes of many species are pervasively transcribed, resulting in the production of numerous long noncoding RNAs (lncRNAs). At the same time, it is now appreciated that many types of DNA regulatory elements, such as enhancers and promoters, regularly initiate bi-directional transcription. Thus, discerning functional noncoding transcripts from a vast transcriptome is a paramount priority, and challenge, for the lncRNA field. In this review, we aim to provide a conceptual and experimental framework for classifying and elucidating lncRNA function. We categorize lncRNA loci into those that regulate gene expression in cis versus those that perform functions in trans and propose an experimental approach to dissect lncRNA activity based on these classifications. These strategies to further understand lncRNAs promise to reveal new and unanticipated biology with great potential to advance our understanding of normal physiology and disease. |
| [40] | . 蜜蜂的蜂王浆主蛋白具有为蜂王 和幼虫提供营养、影响蜂群社会行为及调节个体生理机能等作用,作为蜂王浆的主要成分对其他机体也可产生多方面的生物学功能。因此,近年来蜂王浆主蛋白的相 关研究备受关注。本文针对蜂王浆主蛋白的发现、种类、功能、系统进化及其基因表达情况进行了系统综述。 . 蜜蜂的蜂王浆主蛋白具有为蜂王 和幼虫提供营养、影响蜂群社会行为及调节个体生理机能等作用,作为蜂王浆的主要成分对其他机体也可产生多方面的生物学功能。因此,近年来蜂王浆主蛋白的相 关研究备受关注。本文针对蜂王浆主蛋白的发现、种类、功能、系统进化及其基因表达情况进行了系统综述。 |
| [41] | . Abstract The Hippo pathway has emerged as a conserved signaling pathway that is essential for the proper regulation of organ growth in Drosophila and vertebrates. Although the mechanisms of signal transduction of the core kinases Hippo/Mst and Warts/Lats are relatively well understood, less is known about the upstream inputs of the pathway and about the downstream cellular and developmental outputs. Here, we review recently discovered mechanisms that contribute to the dynamic regulation of Hippo signaling during Drosophila and vertebrate development. We also discuss the expanding diversity of Hippo signaling functions during development, discoveries that shed light on a complex regulatory system and provide exciting new insights into the elusive mechanisms that regulate organ growth and regeneration. |
| [42] | . First discovered in Drosophila, the Hippo signaling pathway is a conserved regulator of organ size. Central to this pathway is a kinase cascade leading from the tumor suppressor Hippo (Mst1 and Mst2 in mammals) to the oncoprotein Yki (YAP and TAZ in mammals), a transcriptional coactivator of target genes involved in cell proliferation and survival. Here, I review recent progress in elucidating the molecular mechanism and physiological function of Hippo signaling in Drosophila and mammals. These studies suggest that the core Hippo kinase cascade integrates multiple upstream inputs, enabling dynamic regulation of tissue homeostasis in animal development and physiology. |
| [43] | . The mechanisms that regulate mammalian organ size are poorly understood. It is unclear whether the pathways that control organ size also impinge on stem/progenitor cells. A highly expressed gene in stem cells is YAP1[1], the ortholog of Drosophila Yorkie, a downstream component of the Hippo pathway [2]. Mutations in components of this pathway produce tissue overgrowth phenotypes in the fly whereas mammalian orthologs, like salvador [3], merlin [4], LATS[5], and YAP1[6, 7], have been implicated in tumorigenesis. We report here that YAP1 increases organ size and causes aberrant tissue expansion in mice. YAP1 activation reversibly increases liver size more than 4-fold. In the intestine, expression of endogenous YAP1 is restricted to the progenitor/stem cell compartment, and activation of YAP1 expands multipotent undifferentiated progenitor cells, which differentiate upon cessation of YAP1 expression. YAP1 stimulates Notch signaling, and administration of -secretase inhibitors suppressed the intestinal dysplasia caused by YAP1. Human colorectal cancers expressing higher levels of YAP1 share molecular aspects with YAP1-induced dysplastic growth in the mouse. Our data show that the Hippo signaling pathway regulates organ size in mammals and can act on stem cell compartments, indicating a potential link between stem/progenitor cells, organ size, and cancer. |
| [44] | . Metazoans employ cytoprotective and regenerative strategies to maintain tissue homeostasis. Understanding the coordination of these strategies is critical to developing accurate models for aging and associated diseases. Here we show that cytoprotective Jun N-terminal kinase (JNK) signaling influences regeneration in the Drosophila gut by directing proliferation of intestinal stem cells (ISCs). Interestingly, this function of JNK contributes to the loss of tissue homeostasis in old and stressed intestines by promoting the accumulation of misdifferentiated ISC daughter cells. Ectopic Delta/Notch signaling in these cells causes their abnormal differentiation but also limits JNK-induced proliferation. Protective JNK signaling and control of cell proliferation and differentiation by Delta/Notch signaling thus have to be carefully balanced to ensure tissue homeostasis. Our findings suggest that this balance is lost in old animals, increasing the potential for neoplastic transformation. |
| [45] | |
| [46] | . Chalkbrood is a fungal disease of honey bee brood caused by Ascosphaera apis. This disease is now found throughout the world, and there are indications that chalkbrood incidence may be on the rise. In this review we consolidate both historic knowledge and recent scientific findings. We document the worldwide spread of the fungus, which is aided by increased global travel and the migratory nature of many beekeeping operations. We discuss the current taxonomic classification in light of the recent complete reworking of fungal systematics brought on by application of molecular methods. In addition, we discuss epidemiology and pathogenesis of the disease, as well as pathogen biology, morphology and reproduction. New attempts at disease control methods and management tactics are reviewed. We report on research tools developed for identification and monitoring, and also include recent findings on genomic and molecular studies not covered by previous reviews, including sequencing of the A. apis genome and identification of the mating type locus. |
| [47] | . A transposon-based mutagenesis screen uncovers a striking enrichment of putative PTEN ceRNAs among mutated genes that accelerate tumorigenesis in a mouse model of melanoma. One candidate gene, ZEB2, regulates PI3K/AKT signaling and tumor-suppressive properties of PTEN. |
| [48] | . In eusocial Hymenoptera (ants, bees and wasps), queen and worker adult castes typically arise via environmental influences. A fundamental challenge is to understand how a single genome can thereby produce alternative phenotypes. A powerful approach is to compare the molecular basis of caste determination and differentiation along the evolutionary trajectory between primitively and advanced eusocial species, which have, respectively, relatively undifferentiated and strongly differentiated adult castes. In the advanced eusocial honeybee, Apis mellifera, studies suggest that microRNAs (miRNAs) play an important role in the molecular basis of caste determination and differentiation. To investigate how miRNAs affect caste in eusocial evolution, we used deep sequencing and Northern blots to isolate caste-associated miRNAs in the primitively eusocial bumblebee Bombus terrestris. We found that the miRNAs Bte-miR-6001-5p and -3p are more highly expressed in queen- than in worker-destined late-instar larvae. These are the first caste-associated miRNAs from outside advanced eusocial Hymenoptera, so providing evidence for caste-associated miRNAs occurring relatively early in eusocial evolution. Moreover, we found little evidence that miRNAs previously shown to be associated with caste in A. mellifera were differentially expressed across caste pathways in B. terrestris, suggesting that, in eusocial evolution, the caste-associated role of individual miRNAs is not conserved. |
| [49] | . Abstract Fragile X syndrome is one of the most common forms of inherited intellectual disability. It is caused by mutations of the Fragile X mental retardation 1(FMR1) gene, resulting in either the loss or abnormal expression of the Fragile X mental retardation protein (FMRP). Recent research showed that FMRP participates in non-coding RNA pathways and plays various important roles in physiology, thereby extending our knowledge of the pathogenesis of the Fragile X syndrome. Initial studies showed that the Drosophila FMRP participates in siRNA and miRNA pathways by interacting with Dicer, Ago1 and Ago2, involved in neural activity and the fate determination of the germline stem cells. Subsequent studies showed that the Drosophila FMRP participates in piRNA pathway by interacting with Aub, Ago1 and Piwi in the maintenance of normal chromatin structures and genomic stability. More recent studies showed that FMRP is associated with lncRNA pathway, suggesting a potential role for the involvement in the clinical manifestations. In this review, we summarize the novel findings and explore the relationship between FMRP and non-coding RNA pathways, particularly the piRNA pathway, thereby providing critical insights on the molecular pathogenesis of Fragile X syndrome, and potential translational applications in clinical management of the disease. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}