Construction and Annotation of Ascosphaera apis Full-Length Transcriptome Utilizing Nanopore Third-Generation Long-Read Sequencing Technology
DU Yu,1, ZHU ZhiWei,1, WANG Jie1, WANG XiuNa3,4, JIANG HaiBin1, FAN YuanChan1, FAN XiaoXue1, CHEN HuaZhi1, LONG Qi1, CAI ZongBing1, XIONG CuiLing1,2, ZHENG YanZhen1, FU ZhongMin1,2, CHEN DaFu,1,2, GUO Rui,1,21 College of Animal Sciences (College of Bee Science), Fujian Agriculture and Forestry University, Fuzhou 350002 2Apitherapy Research Institution, Fujian Agriculture and Forestry University, Fuzhou 350002 3College of Life Sciences, Fujian Agriculture and Forestry University, Fuzhou 350002 4Key Laboratory of Pathogenic Fungi and Mycotoxins of Fujian Province (Fujian Agriculture and Forestry University), Fuzhou 350002
Abstract 【Objective】Purified mycelia sample (Aam) and spore sample (Aas) were sequenced using third-generation nanopore long-read sequencing technology, followed by construction and annotation of high-quality full-length transcriptome.【Method】Aam and Aas were respectively sequenced using Oxford Nanopore PromethION platform. Guppy software was used to conduct base calling of raw reads. Clean reads were obtained after filtering out short fragments and low-quality raw reads. Full-length transcripts were identified by recognizing primers at both ends of clean reads. Full-length transcripts were aligned to Nr, Swissprot, KOG, eggNOG, Pfam, GO and KEGG databases to gain corresponding annotations. Four approaches such as CPC, CNCI, CPAT, and Pfam were used to predict lncRNAs, and the intersection was deemed to be high-reliability lncRNAs.【Result】In total, 6 321 704 and 6 259 727 raw reads were yielded from nanopore sequencing of Aam and Aas, and after quality control, 5 669 436 and 6 233 159 clean reads were obtained, including 4 497 102 (79.32%) and 4 963 101 (79.62%) full-length clean reads. Additionally, 9 859 and 16 795 non-redundant full-length transcripts were identified, with a N50 of 1 482 and 1 658 bp, an average length of 1 187 and 1 303 bp, and a maximum length of 6 472 and 6 815 bp, respectively. Venn analysis showed that 6 512 non-redundant full-length transcripts were shared by Aam and Aas, while 3 347 and 10 283 ones were specific for Aam and Aas, respectively. Besides, a total of 20 142 full-length transcripts were identified in Aam and Aas, among them 20 809, 11 151, 17 723, 12 164, 11 340 and 9 833 full-length transcripts could be annotated to Nr, KOG, eggNOG, Pfam, GO and KEGG databases, respectively. Most of full-length transcripts were annotated to A. apis, Polytolypa hystricis and Histoplasma capsulatum. Moreover, GO database annotation demonstrated that the above-mentioned full-length transcripts could be annotated to 45 functional terms, involving in cell component-associated terms such as cell part, cell and organelle; molecular function-associated terms such as catalytic activity, binding and transporter activity; and biological process-associated terms such as cellular processes, metabolic processes and single-organism processes. KEGG database annotation indicated that these full-length transcripts could be annotated to 49 pathways, including biosynthesis of antibiotics, ribosome, biosynthesis of amino acid, carbon metabolism, spliceosome and so on. In addition, 648 lncRNAs were identified, including 480 long intergenic RNAs (lincRNAs), 119 anti-sense lncRNAs and 49 sense lncRNAs. 【Conclusion】The first high-quality full-length transcriptome was constructed and annotated in this work, which offers a key basis for exploration of the complexity of A. apis transcriptome, improvement of sequence and functional annotation of reference genome and further study on isoforms’ function of A. apis. Keywords:third-generation high-throughput sequencing technology;nanopore sequencing;full-length transcript;reference transcriptome;honeybee;Ascosphaera apis
PDF (3750KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文 本文引用格式 杜宇, 祝智威, 王杰, 王秀娜, 蒋海宾, 范元婵, 范小雪, 陈华枝, 隆琦, 蔡宗兵, 熊翠玲, 郑燕珍, 付中民, 陈大福, 郭睿. 利用第三代纳米孔长读段测序技术构建和注释蜜蜂球囊菌的全长转录组[J]. 中国农业科学, 2021, 54(4): 864-876 doi:10.3864/j.issn.0578-1752.2021.04.017 DU Yu, ZHU ZhiWei, WANG Jie, WANG XiuNa, JIANG HaiBin, FAN YuanChan, FAN XiaoXue, CHEN HuaZhi, LONG Qi, CAI ZongBing, XIONG CuiLing, ZHENG YanZhen, FU ZhongMin, CHEN DaFu, GUO Rui. Construction and Annotation of Ascosphaera apis Full-Length Transcriptome Utilizing Nanopore Third-Generation Long-Read Sequencing Technology[J]. Scientia Acricultura Sinica, 2021, 54(4): 864-876 doi:10.3864/j.issn.0578-1752.2021.04.017
(1)参照说明书步骤,利用TRizol试剂盒(Thermo Fisher公司,美国)分别提取Aam和Aas的总RNA;(2)引物退火,利用Maxima H Minus Reverse Transcriptase试剂盒(Thermo Fisher公司,美国)进行反转录,得到的cDNA添加switch oligo,再合成互补链;(3)对DNA进行损伤修复和末端修复,再利用磁珠对cDNA进行纯化;(4)委托北京百迈克生物科技有限公司对上述构建好的cDNA文库进行全长转录组测序,测序平台为PromethION(Oxford Nanopore Technologies公司,英国)。
利用Blast工具将上述所有全长转录本比对Nr[29]、Swissprot[30]、KOG[31]、eggNOG[32]、Pfam[33]、GO(Gene Ontology)[34]和KEGG(Kyoto Encyclopedia of Genes and Genomes)[35]数据库,获得相应的功能和通路注释信息。
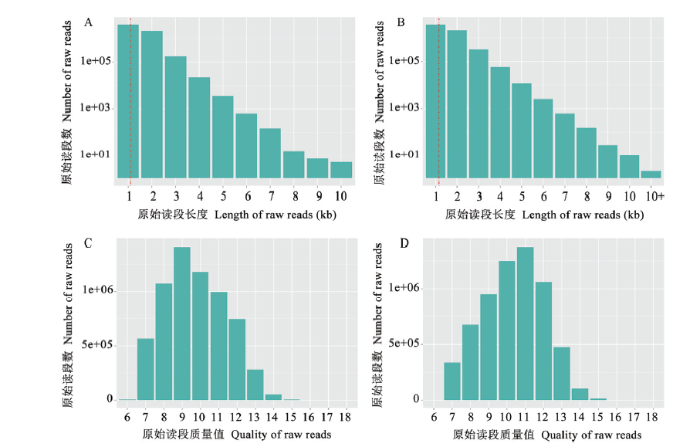
Fig. 1Length and quality distribution of raw reads generated from nanopore long-read sequencing of A. apis mycelium and spore
A:球囊菌菌丝测序产生的原始读段的长度分布Length distribution of raw reads produced from sequencing of Aam;B:球囊菌孢子测序产生的原始读段的长度分布Length distribution of raw reads produced from sequencing of Aas;C:球囊菌菌丝测序产生的原始读段的质量值分布Quality distribution of raw reads produced from sequencing of Aam;D:球囊菌孢子测序产生的原始读段的质量值分布Quality distribution of raw reads produced from sequencing of Aas
Fig. 2Length distribution of full-length clean reads and redundant clean reads-removed full-length transcripts
A:球囊菌菌丝测序产生的全长有效读段Full-length clean reads yielded from sequencing of Aam;B:球囊菌孢子测序产生的全长有效读段Full-length clean reads yielded from sequencing of Aas;C:球囊菌菌丝测序产生的全长转录本Full-length transcripts yielded from sequencing of Aam;D:球囊菌孢子测序产生的全长转录本 Full-length transcripts yielded from sequencing of Aas
ZHANG ZN, XIONG CL, XU XJ, HUANG ZJ, ZHENG YZ, LUOQ, LIUM, LI WD, TONG XY, ZHANGQ, LIANGQ, GUOR, CHEN DF. De novo assembly of a reference transcriptome and development of SSR markers for Ascosphaera apis. Acta Entomologica Sinica, 2017,60(1):34-44. (in Chinese) [本文引用: 5]
GUOR, LI WD, CHEN DF, XIONG CL, ZHENG YZ, FU ZM, XU XJ, HUANG ZJ, LUOQ. Highly-expressed gene differences between Ascosphaera apis stressing the larval gut of Apis mellifera ligustica and the pure culture of Ascosphaera apis Microbiology China, 2018,45(2):368-375. (in Chinese) [本文引用: 2]
CHEN DF, WANG HQ, LI WD, XIONG CL, ZHENG YZ, FU ZM, XU XJ, HUANG ZJ, GUOR. Analysis of highly expressed genes of Ascosphaera apis infecting the gut of Apis cerana cerana larvae and its in vitro culture Journal of Fujian Agriculture and Forestry University (Natural Science Edition), 2017,46(5):562-568. (in Chinese) [本文引用: 2]
GUOR, CHEN HZ, TONG XY, XIONG CL, ZHENG YZ, FU ZM, XIE YL, WANG HP, ZHAO HX, CHEN DF. Structural optimization of annotated genes and identification of novel genes in Ascosphaera apis Journal of China Agricultural University, 2019,24(1):61-68. (in Chinese) [本文引用: 2]
GUOR, WANG HP, CHEN HZ, XIONG CL, ZHENG YZ, FU ZM, ZHAO HX, CHEN DF. Identification of Ascosphaera apis microRNAs and investigation of their regulation networks Acta Microbiologica Sinica, 2018,58(6):1077-1089. (in Chinese) [本文引用: 2]
GUOR, CHEN DF, XIONG CL, HOU CS, ZHENG YZ, FU ZM, DIAO QY, ZHANGL, WANG HQ, HOU ZX, LI WD, KUMARD, LIANGQ. Identification of long non-coding RNAs in the chalkbrood disease pathogen Ascospheara apis Journal of Invertebrate Pathology, 2018,156:1-5. [本文引用: 3]
GUOR, CHEN DF, CHEN HZ, FU ZM, XIONG CL, HOU CS, ZHENG YZ, GUO YL, WANG HP, DUY, DIAO QY. Systematic investigation of circular RNAs in Ascosphaera apis, a fungal pathogen of honeybee larvae Gene, 2018,678:17-22. [本文引用: 2]
LU HY, GIORDANOF, NING ZM. Oxford Nanopore MinION sequencing and genome assembly Genomics Proteomics and Bioinformatics, 2016,14(5):265-279. [本文引用: 2]
WORKMAN RE, TANG AD, TANG PS, JAINM, TYSON JR, RAZAGHIR, ZUZARTE PC, GILPATRICKT, PAYNEA, QUICKJ, et al. Nanopore native RNA sequencing of a human poly (A) transcriptome Nature Methods, 2019,16(12):1297-1305. [本文引用: 2]
LEA WA, PARNELL SC, WALLACE DP, CALVET JP, ZELENCHUK LV, ALVAREZ NS, WARD CJ. Human-specific abnormal alternative splicing of wild-type PKD1 induces premature termination of polycystin-1 Journal of the American Society of Nephrology, 2018,29(10):2482-2492. [本文引用: 1]
CHEN SY, DENG FL, JIA XB, LIC, LAI SJ. A transcriptome atlas of rabbit revealed by PacBio single-molecule long-read sequencing Scientific Reports, 2017,7:7648. [本文引用: 1]
BAYEGAA, OIKONOMOPOULOSS, ZORBASE, WANG YC, GREGORIOU ME, TSOUMANI KT, MATHIOPOULOS KD, RAGOUSSISJ. Transcriptome landscape of the developing olive fruit fly embryo delineated by Oxford Nanopore long-read RNA-Seq bioRxiv, 2018. doi: https://doi.org/10.1101/478172. [本文引用: 2]
CHAOQ, GAO ZF, ZHANGD, ZHAO BG, DONG FQ, FU CX, LIU LJ, WANG BC. The developmental dynamics of the Populus stem transcriptome Plant Biotechnology Journal, 2019,17(1):206-219. [本文引用: 1]
ZHU CH, LI XF, ZHENG JY. Transcriptome profiling using Illumina- and SMRT-based RNA-seq of hot pepper for in-depth understanding of genes involved in CMV infection Gene, 2018,666:123-133. [本文引用: 1]
TOMBáCZD, BALáZSZ, CSABAIZ, MOLDOVáNN, SZ?CSA, SHAROND, SNYDERM, BOLDOGK?IZ. Characterization of the dynamic transcriptome of a herpesvirus with long-read single molecule real-time sequencing Scientific Reports, 2017,7:43751. [本文引用: 1]
TOMBáCZD, BALáZSZ, CSABAIZ, SNYDERM, BOLDOGKOIZ. Long-read sequencing revealed an extensive transcript complexity in herpesviruses Frontiers in Genetics, 2018,9:259. [本文引用: 1]
CHEN HZ, ZHU ZW, JIANG HB, WANGJ, FAN YC, FAN XX, WAN JQ, LU JX, XIONG CL, ZHENG YZ, FU ZM, CHEN DF, GUOR. Comparative analysis of microRNAs and corresponding target mRNAs in Ascospheara apis mycelium and spore Scientia Agricultura Sinica, 2020,53(17):3606-3619. (in Chinese) [本文引用: 1]
CHEN HZ, WANGJ, ZHU ZW, JIANG HB, FAN YC, FAN XX, WAN JQ, LU JX, ZHENG YZ, FU ZM, XU GJ, CHEN DF, GUOR. Comparison and potential functional analysis of long non-coding RNAs between Ascosphaera apis mycelium and spore Scientia Agricultura Sinica, 2021,54(2):435-448. (in Chinese) [本文引用: 1]
CHEN HZ, FAN XX, DUY, FAN YC, WANGJ, JIANG HB, XIONG CL, ZHENG YZ, CHEN DF, GUOR. Nanopore-based long-read transcriptome data of Nosema ceranae-infected and un-infected western honeybee workers’ midguts bioRxiv, 2020. doi: https://doi.org/10.1101/2020.03.21.001958. [本文引用: 1]
DUY, FAN YC, CHEN HZ, WANGJ, XIONG CL, ZHENG YZ, CHEN DF, GUOR. A full-length transcriptome dataset of normal and Nosema ceranae-challenged midgut tissues of eastern honeybee workers bioRxiv, 2020. doi: https://doi.org/10.1101/2020.03.18. 997981. [本文引用: 1]
JENJAROENPUNP, WONGSURAWATT, PEREIRAR, PATUMCHAROENPOLP, USSERY DW, NIELSENJ, NOOKAEWI. Complete genomic and transcriptional landscape analysis using third-generation sequencing: A case study of Saccharomyces cerevisiae CEN.PK113-7D Nucleic Acids Research, 2018,46(7):e38. [本文引用: 1]
BOLDOGKOIZ, MOLDOVANN, BALAZSZ, SNYDERM, TOMBACZD. Long-read sequencing-A powerful tool in viral transcriptome research Trends in Microbiology, 2019,27(7):578-592. [本文引用: 1]
DENG YY, LI JQ, WU SF, ZHU YP, CHEN YW, HE FC. Integrated nr database in protein annotation system and its localization Computer Engineering, 2006,32(5):71-73, 76. (in Chinese) [本文引用: 1]
The UniProtConsortium. UniProt: The universal protein knowledgebase Nucleic Acids Research, 2017,45(D1):D158-D169. [本文引用: 1]
KOONIN EV, FEDOROVA ND, JACKSON JD, JACOBS AR, KRYLOV DM, MAKAROVA KS, MAZUMDERR, MEKHEDOV SL, NIKOLSKAYA AN, RAO BS, et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes Genome Biology, 2004,5(2):R7. [本文引用: 1]
POWELLS, FORSLUNDK, SZKLARCZYKD, TRACHANAK, ROTHA, HUERTA-CEPASJ, GABALDóN T, RATTEI T, CREEVEY C, KUHN M, JENSEN L J, VON MERING C, BORK P. eggNOG v4.0: Nested orthology inference across 3686 organisms Nucleic Acids Research, 2014,42(Database issue):D231-D239. [本文引用: 1]
FINN RD, BATEMANA, CLEMENTSJ, COGGILLP, EBERHARDT RY, EDDY SR, HEGERA, HETHERINGTONK, HOLML, MISTRYJ, SONNHAMMER E LL, TATEJ, PUNTAM. Pfam: The protein families database Nucleic Acids Research, 2014,42(Database issue):D222-D230. [本文引用: 2]
ASHBURNERM, BALL CA, BLAKE JA, BOTSTEIND, BUTLERH, CHERRY JM, DAVIS AP, DOLINSKIK, DWIGHT SS, EPPIG JT, et al. Gene ontology: Tool for the unification of biology Nature Genetics, 2000,25(1):25-29. [本文引用: 1]
KANEHISAM, GOTOS, KAWASHIMAS, OKUNOY, HATTORIM. The KEGG resource for deciphering the genome Nucleic Acids Research, 2004,32(Database issue):D277-D280. [本文引用: 1]
XIONG CL, GENG SH, WANG XR, LIU SY, CHEN DF, ZHENG YZ, FU ZM, DUY, WANG HP, CHEN HZ, ZHOU DD, GUOR. Prediction, analysis and identification of long non-coding RNA in the midguts of Apis mellifera ligustica workers Chinese Journal of Applied Entomology, 2018,55(6):1034-1044. (in Chinese) [本文引用: 1]
KONGL, ZHANGY, YE ZQ, LIU XQ, ZHAO SQ, WEIL, GAOG. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine Nucleic Acids Research, 2007,35(Web Server issue):W345-W349. [本文引用: 1]
SUNL, LUO HT, BU DC, ZHAO GG, YU KT, ZHANG CH, LIU YN, CHEN RS, ZHAOY. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts Nucleic Acids Research, 2013,41(17):e166. [本文引用: 1]
WANGL, PARK HJ, DASARIS, WANGS, KOCHER JP, LIW. CPAT: Coding-potential assessment tool using an alignment-free logistic regression model Nucleic Acids Research, 2013,41(6):e74. [本文引用: 1]
CHEN DF, DUY, FAN XX, ZHU ZW, JIANG HB, WANGJ, FAN YC, CHEN HZ, ZHOU DD, XIONG CL, ZHENG YZ, XU XJ, LUOQ, GUOR. Reconstruction and functional annotation of Ascosphaera apis full-length transcriptome via PacBio single-molecule long-read sequencing bioRxiv, 2019. doi: https://doi.org/10.1101/770040. [本文引用: 1]
MAGIA, SEMERAROR, MINGRINOA, GIUSTIB, D’AURIZIO R. Nanopore sequencing data analysis: State of the art, applications and challenges Briefings in Bioinformatics, 2018,19(6):1256-1272. [本文引用: 1]
ARONSTEIN KA, MURRAY KD. Chalkbrood disease in honey bees Journal of Invertebrate Pathology, 2010,103(Suppl.1):S20-S29. [本文引用: 1]
LI JH, ZHENG ZY, CHEN DF, LIANGQ. Factors influencing Ascosphaera apis infection on honeybee larvae and observation on the infection process Acta Entomologica Sinica, 2012,55(7):790-797. (in Chinese) [本文引用: 1]
TAUBER JP, EINSPANIERR, EVANS JD, MCMAHON DP. Co-incubation of dsRNA reduces proportion of viable spores of Ascosphaera apis, a honey bee fungal pathogen Journal of Apicultural Research, 2020,59(5):791-799. [本文引用: 1]
 ,1, 祝智威
,1, 祝智威

 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT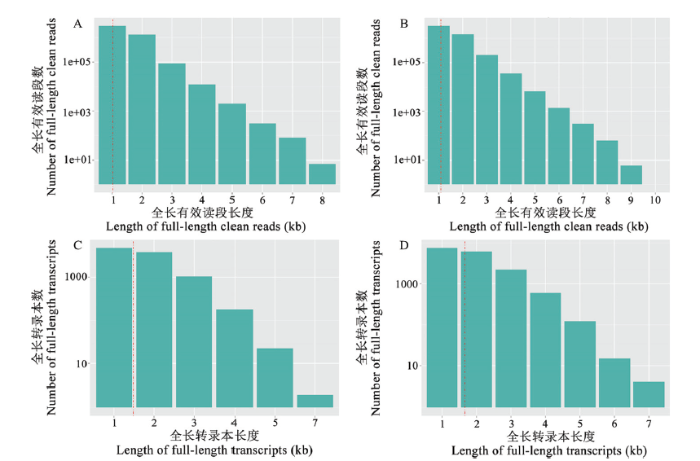 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT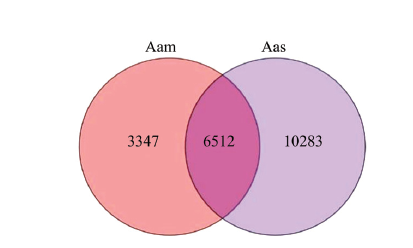 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT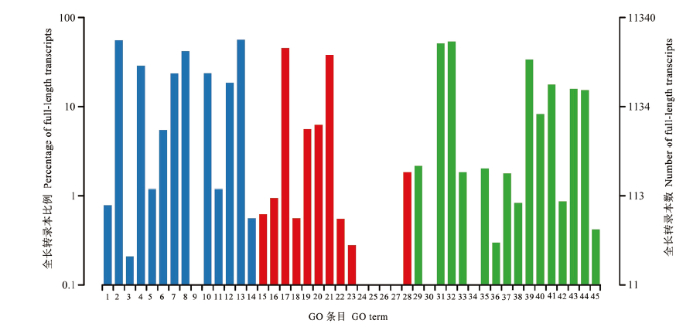 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT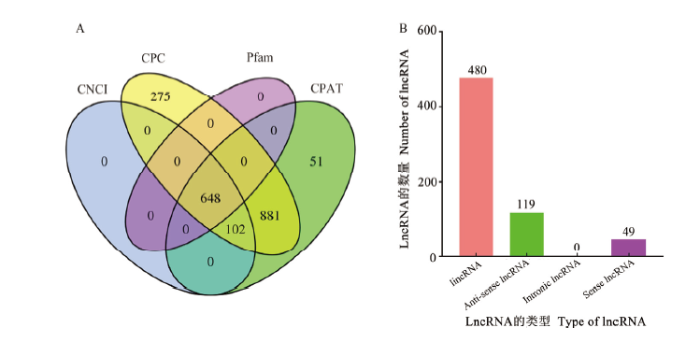 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}