 ,1
,1Transcriptomic Analysis of Sclerotia Formation Induced by Low Temperature in Villosiclava virens
Lü ChuYang1, DENG PingChuan2, ZHANG XiaoLi1, SUN YuChao1, LIANG WuSheng1, HU DongWei,1通讯作者:
责任编辑: 岳梅
收稿日期:2020-03-23接受日期:2020-04-25网络出版日期:2020-11-16
| 基金资助: |
Received:2020-03-23Accepted:2020-04-25Online:2020-11-16
作者简介 About authors
吕楚阳,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (6888KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
吕楚阳, 邓平川, 张晓丽, 孙钰超, 梁五生, 胡东维. 低温诱导稻曲病菌菌核形成的转录组学分析[J]. 中国农业科学, 2020, 53(22): 4571-4583 doi:10.3864/j.issn.0578-1752.2020.22.005
Lü ChuYang, DENG PingChuan, ZHANG XiaoLi, SUN YuChao, LIANG WuSheng, HU DongWei.
开放科学(资源服务)标识码(OSID):

0 引言
【研究意义】近年来,我国稻曲病已从水稻次要病害上升为主要病害,成为全世界稻米主产区的主要真菌病害之一[1]。稻曲病是由子囊菌门麦角菌科的稻曲病菌(Villosiclava virens)侵染水稻颖花造成的。病原菌侵染后,整个颖花形成球状菌落,最外层有大量黄色和墨绿色的厚垣孢子层。在一定环境条件下,稻曲球表面可形成一个至数个菌核[2]。菌核及其有性生殖是稻曲病菌安全越冬和提供来年初侵染源的重要方式,菌核及其萌发过程的控制是稻曲病控制的中心环节和瓶颈问题。因此,深入研究菌核的分化与形成机制对探索稻曲病防控新策略具有重要意义。【前人研究进展】在稻曲病的侵染循环过程中,病菌菌核常被认为是造成病害初次侵染的重要来源。稻曲病菌菌核可以安全越冬,并在来年萌发产生大量的子囊孢子,且在时间上与稻曲病菌在水稻孕穗期侵染基本一致[3,4,5,6]。稻曲病菌菌核一般形成在高海拔或温带地区,且出现在稻曲球发育的后期[7,8],而在我国南方地区数量很少,在部分地区如广东尚未发现菌核[9]。但也有研究发现,位于长江中下游地区、属于亚热带地区的浙江省,稻曲病菌不但可产生菌核,且菌核的数量庞大[10]。笔者课题组近期研究表明,在稻曲球发育前期,低温是诱导稻曲病菌产生菌核的重要环境因子之一,室内夜间15℃处理3 d即足以诱导菌核形成[11];减少田间菌核数量可显著减轻稻曲病的发生[12]。为了验证低温是菌核产生的主导环境因子,笔者对低温诱导后的稻曲球进行了系统性解剖,结果发现稻曲球内部具有比以往的认识更多的隐含菌核。目前,转录组测序技术已经成功运用到病原菌形态发育研究中,YU等[13]利用转录组技术比较分析稻曲病菌子实体发育和产分生孢子阶段的基因表达差异,揭示了稻曲病菌的有性生殖相关候选基因;韩彦卿等[14]采用测序技术对接种稻曲病菌的抗、感病水稻进行了转录组学分析,初步探明参与水稻与稻曲病菌之间早期互作的调控网络。【本研究切入点】通过转录组测序技术,全面、快速地获取低温诱导菌核形成过程中的所有转录本信息,并系统分析相关的基因表达变化。【拟解决的关键问题】采用转录组测序技术对低温处理的稻曲病菌样品进行研究,探明稻曲病菌菌核形成初期的基因表达变化,阐释低温诱导稻曲病菌菌核形成的分子机制并了解其发育进程,为稻曲病防控提供理论依据。1 材料与方法
试验于2018—2019年在浙江大学完成。1.1 试验材料
对照组稻曲球样本采自浙江省杭州市桐庐县的稻曲病常发生地块,采集地区前10 d的日间平均气温为31.2℃。另采集浙江省宁波市奉化区前10 d的日间平均气温为23.2℃的稻曲球为处理组样本,本组样本的田间温度与对照组相差约10℃,视为低温处理(图1)。采集发育前期的稻曲球立即放入液氮中速冻,-80℃冰箱中保存。测序所用样品均设置3个生物学重复,每个重复3—5个稻曲球,分别记为对照组:TL911_1、TL911_2、TL911_3;低温处理组:FH1016_1、FH1016_2、FH1016_3。图1
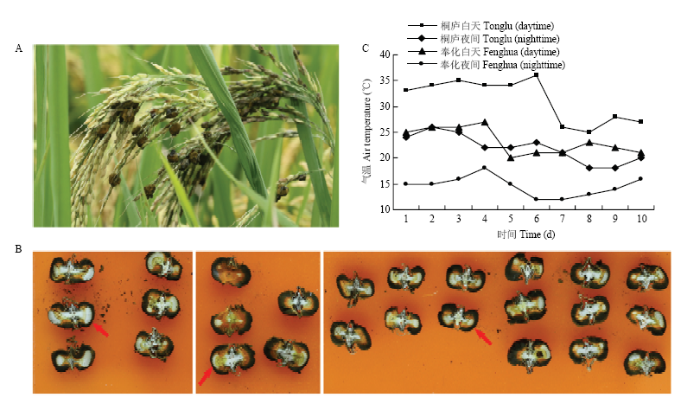 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1晚秋田间形成的稻曲球
A:田间稻曲病症状Symptoms of rice false smut in the field;B:稻曲球剖面观察到内生菌核着生于球体两侧There are some internal growth sclerotia from cross section observation of rice false smut ball;C:采集前10 d田间平均气温Field air temperature in 10 days before sampling
Fig. 1The rice false smut balls formed in late autumn in paddy field
1.2 试验方法
1.2.1 建库测序 将提取的稻曲病菌RNA利用琼脂糖凝胶电泳、Nanodrop、Qubit 2.0 Fluorometer、Agilent 2100等仪器进行降解程度、是否污染、纯度、浓度和完整性共5个方面的评估,质检合格后,以片段化的mRNA为模板,随机寡核苷酸为引物,在M-MuLV逆转录酶体系中合成cDNA第一条链,随后用RNaseH降解RNA链,并在DNA polymerase I体系下,以dNTPs为原料合成cDNA第二条链。纯化后的双链cDNA经过末端修复、加A尾并连接测序接头,用AMPure XP beads筛选250—300 bp左右的cDNA,进行PCR扩增并再次使用AMPure XP beads纯化PCR产物,构建cDNA文库,并委托北京诺禾致源生物信息科技有限公司利用Illumina HiSeqTM 4000测序平台进行高通量测序。1.2.2 转录组数据分析 利用Hisat2(v2.1.0)[15]将各样本数据(clean reads)分别与稻曲病菌“UV-8b”参考基因组序列(
1.2.3 差异表达基因的qRT-PCR验证 为验证转录组数据准确性,随机选取6个DEG进行qRT-PCR定量。利用Primer Premier 5.0设计qRT-PCR引物,内参基因选用α-tubulin,引物列表详见表1。随后采用仪器CFX96 Real-Time System(BIO-RAD)进行qPCR扩增,反应程序:95℃ 5 min;95℃ 10 s,60℃ 30 s,40个循环,每个样品设置3次重复。采用2-ΔΔCT法计算差异基因相对表达量[17]。
Table 1
表1
表1实时荧光定量PCR所用引物
Table 1
| 序号No. | 蛋白名称 Protein name | 蛋白ID Protein ID | 引物序列 Primer sequence (5′-3′) | 扩增长度 Amplicon size (bp) |
|---|---|---|---|---|
| 1 | Protein rds1 | KDB11131.1 | F: TCCAACGTGCGGGAATACA | 164 |
| R: CCAAACTGGCGGAAAATC | ||||
| 2 | Biotrophy-associated secreted protein 2 | KDB17523.1 | F: GCCTCAGCAACACCGACT | 220 |
| R: GCCTTTACCTCGTCCGCC | ||||
| 3 | Glycoside hydrolase | KDB17641.1 | F: TCCTCGCCACCATCTCGT | 178 |
| R: CGCCTCATCGCCCTCAAC | ||||
| 4 | Cytosolic phospholipase A2 zeta | KDB15918.1 | F: ATGGGCGTCTTTGGGAGCG | 165 |
| R: GGGAATTGTGGCGGGATCT | ||||
| 5 | CRE-TRX-1 protein | KDB18233.1 | F: GCCAAATCCCGACAAAAG | 142 |
| R: GGCGGCGTAGTCACCATA | ||||
| 6 | Methylenetetrahydrofolate reductase 1 | KDB13169.1 | F: TCCTCGCCACCATCTCGTC | 179 |
| R: CCGCCTCATCGCCCTCAAC | ||||
| 7 | α-tubulin | KDB12764.1 | F: GCTCTCGTGCTTGCTCTTGG | 144 |
| R: ATCACTTCGTCCTTGCGTTT |
新窗口打开|下载CSV
2 结果
2.1 测序数据质量分析
转录组分析共计6个样本,包括稻曲病菌对照组样本TL911_1、TL911_2、TL911_3和低温处理组样本FH1016_1、FH1016_2、FH1016_3。过滤带有接头(adapter)序列和低质量reads,共得到59.78 G的clean data,样本平均数据量约为9.96 G,其中,近93.17%的序列能够比对到参考基因组(表2)。以上结果表明测序产出数据质量良好,可进行后续的生物信息学分析。基于各基因表达量,计算样本间相关系数(R2),R2越接近1,表明样本之间表达模式的相似度越高。由图2-A可知,对照组、低温处理组3个生物学重复间任意两个样本R2均>0.99,表明生物学重复性好、数据可靠。此外,比较了不同处理下的基因表达相关性,发现对照组TL911与处理组FH1016之间的平均R2为0.93,低于组内生物学重复,说明低温处理组的基因表达与对照组之间存在一定差异,暗示低温诱导稻曲病菌菌核形成过程中确实发生了基因转录变化。样本间层级聚类也有着相近的结果,即相同样本、不同生物学重复优先聚类(图2-B)。
Table 2
表2
表2转录组测序数据以及与参考基因组比对结果
Table 2
| 样品 Sample | 总数据 Total reads | 比对数据 Mapped reads | 比对率 Mapped ratio (%) | 有效序列 Clean bases (G) | GC含量 GC content (%) | Q30 (%) |
|---|---|---|---|---|---|---|
| TL911_1 | 59349736 | 55846678 | 94.10 | 8.90 | 57.15 | 93.51 |
| TL911_2 | 59738802 | 55976056 | 93.70 | 8.96 | 56.95 | 93.09 |
| TL911_3 | 72656794 | 68033430 | 93.64 | 10.90 | 56.85 | 93.01 |
| FH1016_1 | 59387684 | 54809829 | 92.29 | 8.91 | 57.09 | 92.45 |
| FH1016_2 | 63542726 | 58890927 | 92.68 | 9.53 | 57.25 | 93.53 |
| FH1016_3 | 83852442 | 77646536 | 92.60 | 12.58 | 57.22 | 93.53 |
| 总计Total | 398528184 | 371203456 | - | 59.78 | - | - |
新窗口打开|下载CSV
图2
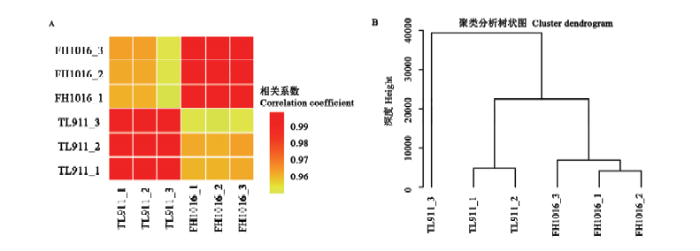 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2样本间关系的整体特征
A:任意两个样本间基因表达数据集的相关系数Correlation coefficients of gene expression data sets between any two samples;B:聚类树状图显示不同样本中基因表达的整体关系,分支长度表示变化程度Cluster dendrogram shows global relationships of gene expression in different samples. The branch length indicates the degree of variance
Fig. 2Overall characterization of sample relationship
综合TL911和FH1016样本基因表达分析,共检测到8 426个基因发生一定程度表达(图3-A),占稻曲病菌基因组基因的97.13%。按照DEG筛选标准,只有793个基因表达量发生了显著性变化,占总体基因的9.41%,其中,398个基因表达上调,395个基因表达下调(图3-B)。上、下调表达倍数最高的前6位基因及其相对表达量、FPKM表达量分别见图3-C、3-D。
图3
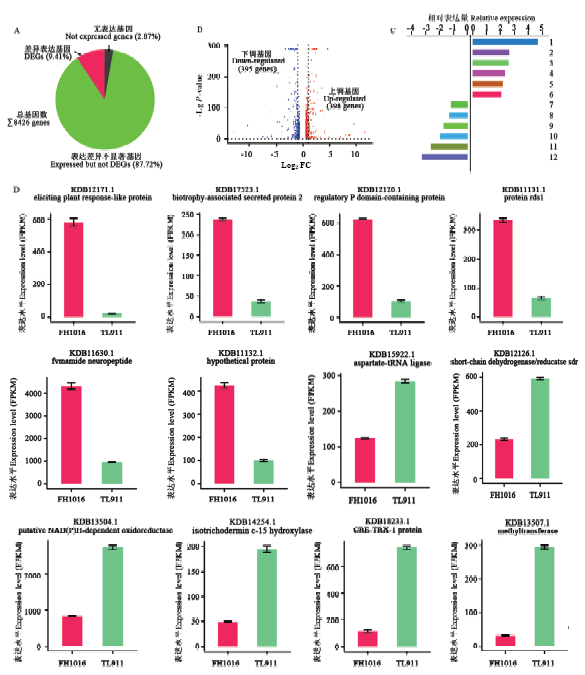 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3低温处理下稻曲病菌表达差异显著基因的比较分析
A:表达差异显著基因比例The proportion of DEGs;B:低温处理下稻曲病菌表达显著上调和下调基因分布The distribution of up- and down-regulated expression genes in V. virens under low temperature;C:前6位上、下调表达差异显著基因的相对表达量Relative expression of top-6 up- and down-regulated DEGs。1: Eliciting plant response-like protein; 2: Biotrophy-associated secreted protein 2; 3: Regulatory P domain-containing protein; 4: Protein rds1; 5: Fvmamide neuropeptide; 6: Hypothetical protein; 7: Aspartate-tRNA ligase; 8: Short-chain dehydrogenase/reductase sdr; 9: Putative NAD(P)H-dependent oxidoreductase; 10: Isotrichodermin c-15 hydroxylase; 11: CRE-TRX-1 protein; 12: Methyltransferase;D:前6位上、下调表达差异显著基因的FPKM表达量FPKM expression of top-6 up- and down-regulated DEGs
Fig. 3The comparative analysis of DEGs in V. virens under low temperature
2.2 GO富集分析
GO注释主要从3个角度描述基因的生物学功能,包括分子功能、生物过程和细胞成分。DEG在分子功能分类中,显著富集于氧化还原酶活性(GO:0016491),其上调基因142个,下调基因89个;阳离子结合(GO:0043169),其上调基因146个,下调基因83个;金属离子结合(GO:0046872),其上调基因145个,下调基因83个;过渡金属离子结合(GO:0046914),其上调基因113个,下调基因61个(图4-A);而在生物过程类别中,碳水化合物代谢过程(GO:0005975)和氧化还原进程(GO:0055114)等通路富集程度最高(图4-B);进一步对分子功能中氧化还原酶活性功能条目的DEG分析,发现NADPH氧化酶(KDB12731.1)、过氧化氢酶(KDB11363.1)、FAD依赖性氧化还原酶(KDB11634.1)、肌氨酸氧化酶(KDB12820.1)在低温处理稻曲病菌中显著上调,结合生物过程类别中氧化还原过程的相对活跃,推测这些真菌机体的氧化还原反应可能与稻曲病菌菌核形成有关。另外,在所有的GO分类中包含上调与下调基因的表达,但整体上并未发现有显著富集的下调功能条目。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4稻曲病菌低温诱导菌核形成相关基因的鉴定及富集分析
A:GO分子功能GO molecular function;B:GO生物过程GO biological process;C:KEGG途径KEGG pathway
Fig. 4Identification and enrichment analysis of genes involved in low temperature response in V. virens at the initial stage
2.3 KEGG代谢途径分析
DEG被注释到91条通路中,共获得显著富集通路8条,分别涉及次生代谢产物生物合成、淀粉和蔗糖代谢、糖酵解/糖异生、碳代谢、甲烷代谢、抗生素的生物合成、丙氨酸、天冬氨酸和谷氨酸代谢、氨基糖和核苷酸糖代谢(表3、图4-C)。这些结果进一步表明,稻曲病菌菌核在形成过程中具有活跃的营养物质代谢和能量代谢。Table 3
表3
表3差异表达基因在KEGG代谢通路中显著富集分析
Table 3
| 途径ID Pathway ID | 途径描述 Description of pathway | DEG数量 DEGs number | 上调数量 Up-regulated number | 下调数量 Down-regulated number | P值 P-value |
|---|---|---|---|---|---|
| fgr01110 | 次生代谢产物生物合成Biosynthesis of secondary metabolites | 174 | 119 | 55 | 0.000456851 |
| fgr00500 | 淀粉和蔗糖代谢Starch and sucrose metabolism | 25 | 20 | 5 | 0.001216228 |
| fgr00010 | 糖酵解/糖异生Glycolysis/Gluconeogenesis | 23 | 17 | 6 | 0.002826092 |
| fgr01200 | 碳代谢Carbon metabolism | 64 | 42 | 22 | 0.003358729 |
| fgr00680 | 甲烷代谢Methane metabolism | 15 | 10 | 5 | 0.004476103 |
| fgr01130 | 抗生素的生物合成Biosynthesis of antibiotics | 128 | 86 | 42 | 0.004815518 |
| fgr00250 | 丙氨酸、天冬氨酸和谷氨酸代谢Alanine, aspartate and glutamate metabolism | 19 | 13 | 6 | 0.006041910 |
| fgr00520 | 氨基糖和核苷酸糖代谢Amino sugar and nucleotide sugar metabolism | 27 | 15 | 12 | 0.006396843 |
新窗口打开|下载CSV
2.4 差异表达的基因家族分析
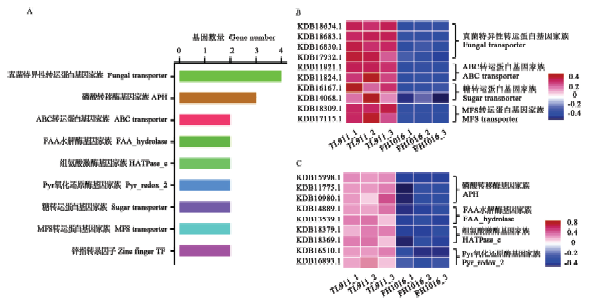
基因家族是指在进化过程中,由一个祖先基因通过基因复制产生两个或更多的拷贝,进而发生分化形成的一类具有相似结构和功能的基因,能够编码相似的蛋白质产物。本研究中DEG共注释到180个基因家族中,占DEG总数的22.7%(图5-A),其中111个基因家族表现为显著上调,占总数的61.67%;11个上调基因被注释到MFS转运蛋白基因家族中,富集程度最高,其他显著上调表达基因家族如糖转运蛋白(8个)、锌指转录因子(6个)、磷酸激酶(4个)等也有着较多成员受低温诱导(图5-B—E);显著下调基因家族占比38.33%,主要包括真菌特异性转运蛋白基因家族(4个)、磷酸转移酶基因家族(3个)等(图6)。推测显著差异数目较多的基因家族与低温诱导稻曲病菌菌核形成有关,可能参与低温胁迫的形态学发育和生理代谢响应。图5
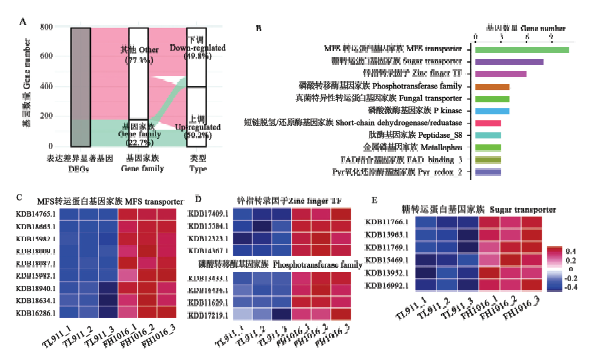 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5稻曲病菌低温诱导相关基因家族的表达分析
A:低温下稻曲病菌显著差异表达的基因家族Diagram showing differentially expressed gene family in V. virens under low temperature;B:含有2个以上上调表达成员的基因家族The distribution of gene family with more than 2 up-regulated members;C—E:基因家族成员表达模式The expression patterns of gene family
Fig. 5Expression analysis of gene family involved in low temperature response in V. virens at the initial stage
图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6低温诱导稻曲病菌下调表达的基因家族分析
A:具有2个以上下调表达成员的基因家族分布The distribution of gene family with more than 2 down-regulated members;B:转运蛋白基因家族表达模式The expression patterns of gene family for transporter;C:其他基因家族表达模式The expression patterns of other gene family
Fig. 6Expression analysis of down-regulated gene family in V. virens under low temperature
2.5 与低温诱导菌核形成相关的差异表达基因
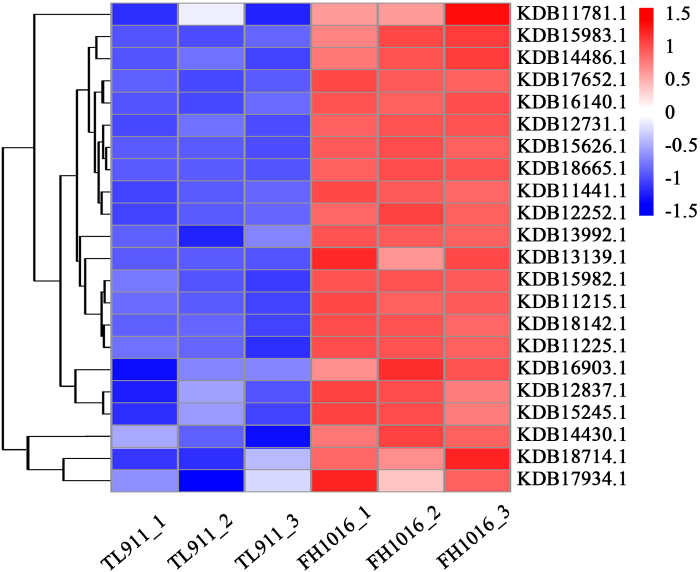
为研究稻曲病菌在低温处理后,菌核形成过程中基因表达差异,根据稻曲病菌基因组搜索获得基因编号,主要对氧化应激反应、信号转导、跨膜运输、细胞形态、生物合成等相关基因进行分析。并根据转录组数据的FPKM值绘制热图表示基因的相对表达量(图7)。这些基因在不同样本间的表达量具有显著差异。2.5.1 氧化应激反应相关基因分析 氧化应激反应能够诱导机体在遭受胁迫时产生的ROS(O2-和H2O2等),影响丝状真菌齐整小核菌(Sclerotium rolfsii)[18]和核盘菌(Sclerotinia sclerotiorum)[19]菌核的分化过程。而NADPH氧化酶(NADPH oxidase,Nox)、超氧化物歧化酶(superoxide dismutase,SOD)、过氧化氢酶(catalase,CAT)是影响真核生物细胞ROS水平和H2O2积累的关键酶类。对低温处理后的稻曲病菌中Nox、SOD和CAT基因表达变化进行研究,发现NADPH氧化酶基因(KDB12731.1、KDB18142.1)、SOD基因(KDB13992.1、KDB18714.1)、CAT基因(KDB11441.1、KDB15626.1)在低温处理的稻曲病菌中表达量明显增加(图7);结合转录组数据GO途径中差异基因在氧化还原酶活性和过氧化物酶的富集,推测由于低温胁迫,稻曲病菌的氧化应激反应强烈,内部活性氧水平升高,进而诱导了菌核的分化。
图7
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7样品TL911和FH1016中差异显著表达基因热图
Fig. 7Heatmap of DEGs in TL911 and FH1016
2.5.2 信号转导相关基因分析 丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)是真核类生物信号传递通路(MAPK通路)中的关键酶,能够调控真核细胞形态和生长。转录组数据中,该类基因(KDB16903.1)在低温诱导菌核的过程中显著差异表达(图7),表明信号传递通路被激活,能够将感知到的外界或胞内信号向下游传递,引发一系列的级联反应。
2.5.3 跨膜运输相关基因分析 主要协助转运蛋白超家族(MFS),是目前真菌主要的跨膜运输蛋白家族之一,在真菌营养吸收中起到重要作用。通过对DEG分析,发现有22个基因表达被注释到MFS蛋白家族中,结合图5-B、5-C看出MFS蛋白家族DEG在上调表达的基因家族分类中富集程度最高;其中3个MFS基因KDB15983.1、KDB18665.1、KDB15982.1上调表达量分别达到了2.46、2.14和2.11倍(图7)。另外,糖转运蛋白家族中绝大部分基因也是表现为显著上调富集(图5)。磷脂是微生物细胞膜的重要组成部分,而磷脂酰丝氨酸合成酶(phosphatidylserine synthase,Pss)能够催化磷脂酰丝氨酸的合成,是合成细胞膜成分的关键,该基因KDB14430.1在低温处理时表达量较高(图7)。
2.5.4 细胞形态相关基因分析 Ras和cytochrome P450(CYP450)蛋白家族能够调控菌丝的极性生长和形态发育[20,21],稻曲病菌转录组中注释到Ras蛋白家族中的3个基因KDB12837.1、KDB17934.1、KDB11781.1被诱导上调表达;此外,有22个基因表达被注释到CYP450蛋白中,其中18个家族基因表达被显著诱导,以KDB17652.1表达量上调幅度最大,为7.13倍(图7)。
2.5.5 生物合成基因相关分析 真菌菌核的发育形成往往伴随黑色素的沉积,聚酮合酶是真菌黑色素合成途径中的关键酶之一,通过对DEG进行分析,发现聚酮合酶基因(KDB13139.1)上调表达了1.65倍;此外几丁质和1,3-β-葡聚糖作为真菌细胞壁的组成成分,其几丁质合成酶(KDB15245.1、KDB11225.1、KDB11215.1、KDB14486.1)表达量也显著增高。分析富集在淀粉和蔗糖代谢途径中的DEG,发现参与合成1,3-β-葡聚糖的2个关键基因(KDB12252.1、KDB16140.1)在低温处理的稻曲球中显著上调(图7)。说明代谢产物黑色素、几丁质和多糖随菌核发育而被大量合成。
2.5.6 生殖相关基因分析 菌核作为无性繁殖体是稻曲病菌分化形成的一团紧密交织的菌丝体,越冬后的菌核可进行有性生殖,萌发形成子实体并产生子囊孢子。而Velvet家族蛋白被认为是真菌特有的无性和有性发育的重要调控因子,同时也影响着次级产物的合成[22,23]。通过与模式真菌比对,找出稻曲转录组中关于Velvet family的同源基因VEA(KDB14625.1),并发现其表达水平上调了1.31倍;其中,低温处理组FPKM为36.97、对照组FPKM为28.19,明显低于整个转录组数据的平均数值130,说明VEA在稻曲病菌的基因组内属于低表达的组成型基因。另外,稻曲病菌的有性配合主要受基因MAT1-1-1和MAT1-2-1调控,发现MAT1-2-1表达量极低(FPKM value<2),而交配型基因MAT1-1-1在低温处理组和对照组中均未检测到该基因的表达。
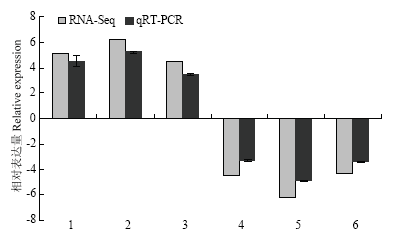
2.6 qRT-PCR验证基因表达
为验证转录组数据是否准确、可信,随机选取6个DEG进行qRT-PCR检测。qRT-PCR测定结果与转录组测序输出数据分析得到的变化趋势一致(图8),表明本研究中基于转录组数据得到的DEG是可信的。图8
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8随机挑选6个差异基因的实时荧光定量分析
Fig. 8qRT-PCR analysis of 6 randomly selected DEGs
1: Protein rds1; 2: Biotrophy-associated secreted protein 2; 3: Glycoside hydrolase; 4: Cytosolic phospholipase A2 zeta; 5: CRE-TRX-1 protein; 6: Methylenetetrahydrofolate reductase 1
3 讨论
氧化应激反应特别是ROS的形成,是诱发菌核分化和调节真菌发育的重要因素之一,高氧化状态可以驱动真菌菌核、有性结构的形成[24,25,26]。Nox能在真核生物中产生活性氧,是超氧化物的一个重要来源[27]。有研究表明,bcnoxa和bcnoxb参与了灰霉病菌(Botrytic cinerea)菌核的形成过程[28]。另一方面,在高氧化应激状态下,各种抗氧化酶和其他小分子(如SOD、CAT等)可能在保护真菌细胞免受ROS的有害影响方面起重要作用。SOD是细胞内ROS的第一道防线,是氧化应激过程中清除自由基最重要的酶之一,在各种生物系统中都会遇到ROS,其作用是消除O2-。CAT能够催化H2O2产生H2O和O2-,发挥抗氧化作用[29]。在本研究转录组数据中发现,CAT和SOD基因表达量升高,这可能归因于氧化还原反应的平衡被破坏和ROS含量相应地急剧增加。因此,SOD和CAT能在应激时主动清除Nox产生的活性氧,最终导致抗氧化酶浓度升高,这些结果间接证明了菌核形成与氧化应激密切相关。GRINTZALIS等研究表明,黄曲霉(Aspergillus flavus)能够在氧化应激反应中合成次级代谢产物黄曲霉素B1[30]。推测氧化应激反应不仅是菌核形成所需要的,也与菌核形成过程中次级代谢产物的合成有关联,这与本研究KEGG通路中差异基因在次级代谢产物合成途径中显著富集相吻合。此外,MAPK信号转导通路中的丝裂霉素活化蛋白激酶基因在低温处理组中上调表达,使得ROS能够通过MAPK途径将信号传递给下游,进一步影响稻曲病菌生理过程基因的表达。例如Pss基因表达量升高,稻曲病菌通过加速合成磷脂来保护细胞膜系统并确保膜上的功能酶能够正常行使功能,结合基因家族分析,MFS转运蛋白家族和糖转运蛋白家族的DEG数目较多且表达量变化明显,说明稻曲病菌在菌核形成过程中细胞营养代谢旺盛,自身能够在膜转运蛋白的协助下实现物质的快速转运,使机体应答低温胁迫。在细胞形态发育方面,Ras是一类调控细胞极性、周期和功能分化的信号分子,而CYP450蛋白在植物木质素中间体、甾醇、萜烯、类黄酮等多种次生代谢物的合成中起关键作用[31],也能够通过对激素的响应进而影响形态发育。Ras和CYP450的表达上调,推测其参与调控稻曲菌丝的细胞极性生长与形态发育。而锌指蛋白是一类具有手指状结构域的转录因子,在转录和翻译水平上调控基因的表达和细胞分化,如构巢曲霉(Aspergillus nidulans)中,nsdC蛋白是一种C2H2型锌指蛋白,基因敲除突变体的生长速率降低,并且完全不能形成子实体[32]。本研究锌指转录因子中6个显著上调,2个显著下调,锌指转录因子家族中的不同TF表达上调或下调,推测这些TF的种类及表达水平的变化可能与稻曲病菌对低温胁迫响应的形态发育有关。此外,代谢产物黑色素、几丁质和多糖也随菌核的发育而被大量合成。综上,稻曲病菌菌丝形成菌核无疑是对外界刺激的一种反应,通过自身形态学及生理代谢等改变对低温胁迫作出响应。
另外,在大部分丝状真菌中有一类velvet,VEA是Velvet蛋白复合物的核心蛋白,具有广泛存在性和功能保守性,能够调节菌核的形成。在寄生曲霉(Aspergillus parasiticus)和黄曲霉中VEA不仅能够调控菌核的发育、诱导黄曲霉毒素的产生,还进一步证实是机体发育和次级代谢之间分子联系的桥梁[33,34]。在烟曲霉(Aspergillus fumigatus)中VEA可以激活有性生殖,减少分生孢子的产生。但是其组成型的基因表达,使得VEA几乎不因无性或有性生殖的发生而有很大的表达变化[35],这与本研究在DEG分析中发现VEA较低的上调倍数相一致,故推测稻曲病菌的VEA可能以其他独特的方式调控菌核的形成。进一步挖掘稻曲病菌有性生殖基因的表达,发现在低温处理组与对照组中不仅MAT1-2-1表达量极低而且还未检测到MAT1-1-1的表达,推测是由于在低温环境下促进的是稻曲病菌菌核的快速形成,而菌核越冬后需要在适合的温度以及充足的光照下才能进行有性生殖形成子实体,所以导致在低温诱导菌核形成的过程中并未发现有性生殖基因的大量表达。由于低温诱导稻曲病菌菌核形成所涉及的分子机制尚未确定,因此通过转录组测序分析的大量基因将作为进一步研究的候选者,为今后稻曲病的防治提供数据和理论基础。
4 结论
通过对低温诱导稻曲病菌菌核形成的转录组数据进行深度挖掘,发现稻曲菌丝低温胁迫下,致使菌丝机体内部处于氧化应激状态,并通过信号转导途径放大,该过程由多个基因参与并调控基因家族成员,最终促使跨膜运输、细胞形态、生物合成等基因的上调表达,使得在形成菌核过程中蛋白表达活跃,达到合成细胞及物质的高峰期,进而促进菌核形成。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1111/mpp.12362URLPMID:26720072 [本文引用: 1]

Villosiclava virens (Vv) is an ascomycete fungal pathogen that causes false smut disease in rice. Recent reports have revealed some interesting aspects of the enigmatic pathogen to address the question of why it specifically infects rice flowers and converts a grain into a false smut ball. Comparative and functional genomics have suggested specific adaptation of Vv in the colonization of rice flowers. Anatomical studies have disclosed that Vv specifically infects rice stamen filaments before heading and intercepts seed formation. In addition, Vv can occupy the whole inner space of a spikelet embracing all floral organs and activate the rice grain-filling network, presumably for nutrient acquisition to support the development of the false smut ball. This profile provides a general overview of the rice false smut pathogen, and summarizes advances in the Vv life cycle, genomics and genetics, and the molecular Vv-rice interaction. Current understandings of the Vv-rice pathosystem indicate that it is a unique and interesting system which can enrich the study of plant-pathogen interactions. Taxonomy: Ustilaginoidea virens is the anamorph form of the pathogen (Kingdom Fungi; Phylum Ascomycota; Class Ascomycetes; Subclass Incertae sedis; Order Incertae sedis; Family Incertae sedis; Genus Ustilaginoidea). The teleomorph form is Villosiclava virens (Kingdom Fungi; Phylum Ascomycota; Class Ascomycetes; Subclass Sordariomycetes; Order Hypocreales; Family Clavicipitaceae; Genus Villosiclava). Disease symptoms: The only visible symptom is the replacement of rice grains by ball-shaped fungal mycelia, namely false smut balls. When maturing, the false smut ball is covered with powdery chlamydospores, and the colour changes to yellowish, yellowish orange, green, olive green and, finally, to greenish black. Sclerotia are often formed on the false smut balls in autumn. Identification and detection: Vv conidia are round to elliptical, measuring 3-5 mum in diameter. Chlamydospores are ornamented with prominent irregularly curved spines, which are 200-500 nm in length. The sclerotia are black, horseshoe-shaped and irregular oblong or flat, ranging from 2 to 20 mm. Nested polymerase chain reaction (PCR) and quantitative PCR have been developed to specifically detect Vv presence in rice tissues and other biotic and abiotic samples in fields. Host range: Rice is the primary host for Vv. Natural infection by Vv has been found on several paddy field weeds, including Digitaria marginata, Panicum trypheron, Echinochloa crusgalli and Imperata cylindrica. However, the occurrence of infection in these potential alternative hosts is very rare. Life cycle: Vv infects rice spikelets at the late rice booting stage, and produces false smut balls covered with dark-green chlamydospores. Occasionally, sclerotia form on the surface of false smut balls in late autumn when the temperature fluctuates greatly between day and night. Both chlamydospores and sclerotia may serve as primary infection sources. Rainfall at the rice booting stage is a major environmental factor resulting in epidemics of rice false smut disease. Disease control: The use of fungicides is the major approach for the control of Vv. Several fungicides, such as cuproxat SC, copper oxychloride, tebuconazole, propiconazole, difenoconazole and validamycin, are often applied. However, the employment of resistant rice cultivars and genes has been limited, because of the poor understanding of rice resistance to Vv. Useful websites: Villosiclava virens genome sequence: http://www.ncbi.nlm.nih.gov/Traces/wgs/?val=JHTR01#contigs.
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
Villosiclava virens
DOI:10.1016/S2095-3119(16)61400-4URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s00294-015-0563-1URLPMID:26905382 [本文引用: 1]
Sexual reproduction of heterothallic clavicipitaceous fungus Villosiclava virens (anamorph: Ustilaginoidea virens) generates ascospores, which is considered as primary infection source of rice false smut disease. However, little is known about the molecular underpinnings of sexual reproduction in V. virens. In this study, transcriptomes of V. virens in fruiting body (FB) and sporulating mycelia (SM) were compared using Illumina paired-end sequencing technology. A total of 33,384,588 and 23,765,275 clean reads of FB and SM transcriptome profiles could be used to map cDNA of V. virens, respectively. We evaluated the gene expression variations between FB and SM, a total of 488 genes therein were significantly higher expressed in FB than SM, and 342 genes were significantly higher expressed genes in SM than FB. These differentially expressed genes were annotated using Kyoto Encyclopedia of Genes and Genomes and Gene Ontology databases. Several genes were found to specifically function in sexual reproduction, involving in mating type, pheromone synthesis, signaling transduction, transcription factors, and meiosis; additionally, a few of genes were presumed to function in conidia sporulation and infection. Comparative transcriptome analysis of V. virens during FB and SM provided an overview of gene expression profiles at the transcriptional level and provided hints to better understand the molecular mechanisms of sexual development. Additionally, the data presented here also proved benefit for mining of essential genes contributing to sexual conidiation and infection.
[本文引用: 1]
[本文引用: 1]
.
DOI:10.1038/nprot.2016.095URLPMID:27560171 [本文引用: 1]
High-throughput sequencing of mRNA (RNA-seq) has become the standard method for measuring and comparing the levels of gene expression in a wide variety of species and conditions. RNA-seq experiments generate very large, complex data sets that demand fast, accurate and flexible software to reduce the raw read data to comprehensible results. HISAT (hierarchical indexing for spliced alignment of transcripts), StringTie and Ballgown are free, open-source software tools for comprehensive analysis of RNA-seq experiments. Together, they allow scientists to align reads to a genome, assemble transcripts including novel splice variants, compute the abundance of these transcripts in each sample and compare experiments to identify differentially expressed genes and transcripts. This protocol describes all the steps necessary to process a large set of raw sequencing reads and create lists of gene transcripts, expression levels, and differentially expressed genes and transcripts. The protocol's execution time depends on the computing resources, but it typically takes under 45 min of computer time. HISAT, StringTie and Ballgown are available from http://ccb.jhu.edu/software.shtml.
.
DOI:10.1038/nbt.1621URLPMID:20436464 [本文引用: 1]
High-throughput mRNA sequencing (RNA-Seq) promises simultaneous transcript discovery and abundance estimation. However, this would require algorithms that are not restricted by prior gene annotations and that account for alternative transcription and splicing. Here we introduce such algorithms in an open-source software program called Cufflinks. To test Cufflinks, we sequenced and analyzed >430 million paired 75-bp RNA-Seq reads from a mouse myoblast cell line over a differentiation time series. We detected 13,692 known transcripts and 3,724 previously unannotated ones, 62% of which are supported by independent expression data or by homologous genes in other species. Over the time series, 330 genes showed complete switches in the dominant transcription start site (TSS) or splice isoform, and we observed more subtle shifts in 1,304 other genes. These results suggest that Cufflinks can illuminate the substantial regulatory flexibility and complexity in even this well-studied model of muscle development and that it can improve transcriptome-based genome annotation.
.
DOI:10.1006/meth.2001.1262URLPMID:11846609 [本文引用: 1]
The two most commonly used methods to analyze data from real-time, quantitative PCR experiments are absolute quantification and relative quantification. Absolute quantification determines the input copy number, usually by relating the PCR signal to a standard curve. Relative quantification relates the PCR signal of the target transcript in a treatment group to that of another sample such as an untreated control. The 2(-Delta Delta C(T)) method is a convenient way to analyze the relative changes in gene expression from real-time quantitative PCR experiments. The purpose of this report is to present the derivation, assumptions, and applications of the 2(-Delta Delta C(T)) method. In addition, we present the derivation and applications of two variations of the 2(-Delta Delta C(T)) method that may be useful in the analysis of real-time, quantitative PCR data.
.
DOI:10.1016/j.funbio.2010.01.010URL [本文引用: 1]
DOI:10.1016/j.micres.2013.12.002URL [本文引用: 1]
.
DOI:10.1111/j.1365-2958.2010.07525.xURL [本文引用: 1]
P>Cdc42/Rho GTPases are universally important regulators of cellular morphogenesis. Whereas their functions are well characterized in budding yeast, they are only beginning to be understood in filamentous fungi. The recent systematic analysis of Cdc42, Rac1 and Rho function in Aspergillus niger provides the first global perspective on their respective roles in hyphal morphogenesis. Surprisingly, the partitioning of these roles between Cdc42 and Rac1 seems to vary even among related fungi. These observations highlight the variable use of a common signalling module in filamentous fungi.
DOI:10.1073/pnas.1306373110URL [本文引用: 1]
Head blight, which is caused by mycotoxin-producing fungi of the genus Fusarium, is an economically important crop disease. We assessed the potential of host-induced gene silencing targeting the fungal cytochrome P450 lanosterol C-14 alpha-demethylase (CYP51) genes, which are essential for ergosterol biosynthesis, to restrict fungal infection. In axenic cultures of Fusarium graminearum, in vitro feeding of CYP3RNA, a 791-nt double-stranded (ds)RNA complementary to CYP51A, CYP51B, and CYP51C, resulted in growth inhibition [half-maximum growth inhibition (IC50) = 1.2 nM] as well as altered fungal morphology, similar to that observed after treatment with the azole fungicide tebuconazole, for which the CYP51 enzyme is a target. Expression of the same dsRNA in Arabidopsis and barley rendered susceptible plants highly resistant to fungal infection. Microscopic analysis revealed that mycelium formation on CYP3RNA-expressing leaves was restricted to the inoculation sites, and that inoculated barley caryopses were virtually free of fungal hyphae. This inhibition of fungal growth correlated with in planta production of siRNAs corresponding to the targeted CYP51 sequences, as well as highly efficient silencing of the fungal CYP51 genes. The high efficiency of fungal inhibition suggests that host-induced gene-silencing targeting of the CYP51 genes is an alternative to chemical treatments for the control of devastating fungal diseases.
.
DOI:10.1007/s12275-018-8417-4URL [本文引用: 1]
.
DOI:10.1126/science.1155888URLPMID:18556559 [本文引用: 1]
Differentiation and secondary metabolism are correlated processes in fungi that respond to light. In Aspergillus nidulans, light inhibits sexual reproduction as well as secondary metabolism. We identified the heterotrimeric velvet complex VelB/VeA/LaeA connecting light-responding developmental regulation and control of secondary metabolism. VeA, which is primarily expressed in the dark, physically interacts with VelB, which is expressed during sexual development. VeA bridges VelB to the nuclear master regulator of secondary metabolism, LaeA. Deletion of either velB or veA results in defects in both sexual fruiting-body formation and the production of secondary metabolites.
DOI:10.1017/S0953756296002882URL [本文引用: 1]
.
DOI:10.1093/icb/icj034URLPMID:21672779 [本文引用: 1]
Sclerotium-forming filamentous fungi are of great agricultural and biological interest because they can be viewed as models of simple metamorphosis. They differentiate by asexually producing sclerotia but the processes involved in sclerotial metamorphosis were poorly understood. In 1997, it was shown for the first time that the sclerotial differentiation state in Sclerotium rolfsii concurred with increasing levels of lipid peroxides. This finding prompted the development of a theory supporting that sclerotial metamorphosis is induced by oxidative stress. Growth factors that reduce or increase oxidative stress are expected to inhibit or promote sclerotium metamorphosis, respectively. This theory has been verified by a series of published data on the effect of certain hydroxyl radical scavengers on sclerotial metamorphosis, on the identification and quantification of certain endogenous antioxidants (such as ascorbic acid, beta-carotene) in relation to the fungal undifferentiated and differentiated states, and on their inhibiting effect on sclerotial metamorphosis as growth nutrients. In 2004-2005, we developed assays for the measurement of certain redox markers of oxidative stress, such as the thiol redox state, the small-sized fragmented DNA, and the superoxide radical. These new advances allowed us to initiate studies on the exact role of glutathione, hydrogen peroxide, and superoxide radical on sclerotial metamorphosis. The emerging data, combined with similar data from other better-studied fungi, allowed us to make some preliminary postulations on the ROS-dependent biochemical signal transduction pathways in sclerotiogenic filamentous fungi.
.
DOI:10.1111/j.1365-2672.2010.04822.xURLPMID:20681971 [本文引用: 1]
AIMS: The purpose of this study was to investigate the role of H(2) O(2) and the related oxidative stress markers catalase (CAT) and lipid peroxidation in the sclerotial differentiation of the phytopathogenic filamentous fungi Sclerotium rolfsii, Sclerotinia minor, Sclerotinia sclerotiorum and Rhizoctonia solani. METHODS AND RESULTS: Using the H(2) O(2) -specific scopoletin fluorometric assay and the CAT-dependent H(2) O(2) consumption assays, it was found that the production rate of intra/extracellular H(2) O(2) and CAT levels in the sclerotiogenic fungi were significantly higher and lower, respectively, than those of their nondifferentiating counterpart strains. They peaked in the transition between the undifferentiated and the differentiated state of the sclerotiogenic strains, suggesting both a cell proliferative and differentiative role. In addition, the indirect indicator of oxidative stress, lipid peroxidation, was substantially decreased in the nondifferentiating strains. CONCLUSIONS: These findings suggest that the differentiative role of H(2) O(2) is expressed via induction of higher oxidative stress in the sclerotiogenic filamentous phytopathogenic fungi. SIGNIFICANCE AND IMPACT OF THE STUDY: This study shows that the direct marker of oxidative stress H(2) O(2) is involved in the sclerotial differentiation of the phytopathogenic filamentous fungi S. rolfsii, S. minor, S. sclerotiorum and R. solani, which could have potential biotechnological implications in terms of developing antifungal strategies by regulating intracellular H(2) O(2) levels.
DOI:10.1128/AEM.05472-11URL [本文引用: 1]
Numerous studies have shown both the detrimental and beneficial effects of reactive oxygen species (ROS) in animals, plants, and fungi. These organisms utilize controlled generation of ROS for signaling, pathogenicity, and development. Here, we show that ROS are essential for the pathogenic development of Sclerotinia sclerotiorum, an economically important fungal pathogen with a broad host range. Based on the organism's completed genome sequence, we identified two S. sclerotiorum NADPH oxidases (SsNox1 and SsNox2), which presumably are involved in ROS generation. RNA interference (RNAi) was used to examine the function of SsNox1 and SsNox2. Silencing of SsNox1 expression indicated a central role for this enzyme in both virulence and pathogenic (sclerotial) development, while inactivation of the SsNox2 gene resulted in limited sclerotial development, but the organism remained fully pathogenic. Delta Ssnox1 strains had reduced ROS levels, were unable to develop sclerotia, and unexpectedly correlated with significantly reduced oxalate production. These results are in accordance with previous observations indicating that fungal NADPH oxidases are required for pathogenic development and are consistent with the importance of ROS regulation in the successful pathogenesis of S. sclerotiorum.
DOI:10.1094/MPMI-21-6-0808URLPMID:18624644 [本文引用: 1]
Nicotinamide adenine dinucleotide (NADPH) oxidases have been shown to be involved in various differentiation processes in fungi. We investigated the role of two NADPH oxidases in the necrotrophic phytopathogenic fungus, Botrytis cinerea. The genes bcnoxA and bcnoxB were cloned and characterized; their deduced amino acid sequences show high homology to fungal NADPH oxidases. Analyses of single and double knock-out mutants of both NADPH oxidase genes showed that both bcnoxA and bcnoxB are involved in formation of sclerotia. Both genes have a great impact on pathogenicity: whereas bcnoxB mutants showed a retarded formation of primary lesions, probably due to an impaired formation of penetration structures, bcnoxA mutants were able to penetrate host tissue in the same way as the wild type but were much slower in colonizing the host tissue. Double mutants showed an additive effect: they were aberrant in penetration and colonization of plant tissue and, therefore, almost nonpathogenic. To study the structure of the fungal Nox complex in more detail, bcnoxR (encoding a homolog of the mammalian p67(phox), a regulatory subunit of the Nox complex) was functionally characterized. The phenotype of DeltabcnoxR mutants is identical to that of DeltabcnoxAB double mutants, providing evidence that BcnoxR is involved in activation of both Bcnox enzymes.
.
DOI:10.1017/S0953756204001352URL [本文引用: 1]
DOI:10.1128/AEM.01282-14URL [本文引用: 1]
We show here that oxidative stress is involved in both sclerotial differentiation (SD) and aflatoxin B1 biosynthesis in Aspergillus flavus. Specifically, we observed that (i) oxidative stress regulates SD, as implied by its inhibition by antioxidant modulators of reactive oxygen species and thiol redox state, and that (ii) aflatoxin B1 biosynthesis and SD are comodulated by oxidative stress. However, aflatoxin B1 biosynthesis is inhibited by lower stress levels compared to SD, as shown by comparison to undifferentiated A. flavus. These same oxidative stress levels also characterize a mutant A. flavus strain, lacking the global regulatory gene veA. This mutant is unable to produce sclerotia and aflatoxin B1. (iii) Further, we show that hydrogen peroxide is the main modulator of A. flavus SD, as shown by its inhibition by both an irreversible inhibitor of catalase activity and a mimetic of superoxide dismutase activity. On the other hand, aflatoxin B1 biosynthesis is controlled by a wider array of oxidative stress factors, such as lipid hydroperoxide, superoxide, and hydroxyl and thiyl radicals.
DOI:10.1371/journal.pone.0143549URLPMID:26606395 [本文引用: 1]
Lignosus rhinocerotis (Cooke) Ryvarden (tiger milk mushroom) has long been known for its nutritional and medicinal benefits among the local communities in Southeast Asia. However, the molecular and genetic basis of its medicinal and nutraceutical properties at transcriptional level have not been investigated. In this study, the transcriptome of L. rhinocerotis sclerotium, the part with medicinal value, was analyzed using high-throughput Illumina HiSeqTM platform with good sequencing quality and alignment results. A total of 3,673, 117, and 59,649 events of alternative splicing, novel transcripts, and SNP variation were found to enrich its current genome database. A large number of transcripts were expressed and involved in the processing of gene information and carbohydrate metabolism. A few highly expressed genes encoding the cysteine-rich cerato-platanin, hydrophobins, and sugar-binding lectins were identified and their possible roles in L. rhinocerotis were discussed. Genes encoding enzymes involved in the biosynthesis of glucans, six gene clusters encoding four terpene synthases and one each of non-ribosomal peptide synthetase and polyketide synthase, and 109 transcribed cytochrome P450 sequences were also identified in the transcriptome. The data from this study forms a valuable foundation for future research in the exploitation of this mushroom in pharmacological and industrial applications.
DOI:10.1534/genetics.109.101667URLPMID:19416940 [本文引用: 1]
The formation of the Aspergillus nidulans fruiting body is affected by a number of genetic and environmental factors. Here, the nsdC (never in sexual development) gene-encoding a putative transcription factor carrying a novel type of zinc-finger DNA-binding domain consisting of two C(2)H(2)'s and a C(2)HC motif that are highly conserved in most fungi but not in plants or animals-was investigated. Two distinct transcripts of 2.6 and 3.0 kb were generated from nsdC. The 2.6-kb mRNA accumulated differentially in various stages of growth and development, while the level of the 3.0-kb mRNA remained relatively constant throughout the life cycle. While the deletion of nsdC resulted in the complete loss of fruiting body formation under all conditions favoring sexual development, overexpression of nsdC not only enhanced formation of fruiting bodies (cleistothecia) but also overcame inhibitory effects of certain stresses on cleistothecial development, implying that NsdC is a key positive regulator of sexual development. Deletion of nsdC also retarded vegetative growth and hyperactive asexual sporulation, suggesting that NsdC is necessary not only for sexual development but also for regulating asexual sporulation negatively. Overexpression of veA or nsdD does not rescue the failure of fruiting body formation caused by nsdC deletion. Furthermore, nsdC expression is not affected by either VeA or NsdD, and vice versa, indicating that NsdC regulates sexual development independently of VeA or NsdD.
DOI:10.1007/s00253-006-0581-5URL [本文引用: 1]
The plant pathogenic fungus Aspergillus flavus produces several types of mycotoxins. The most well known are the carcinogenic compounds called aflatoxins. In addition, A. flavus produces cyclopiazonic acid and aflatrem mycotoxins, contributing to the toxicity of A. flavus infected crops. Cyclopiazonic acid is a specific inhibitor of calcium-dependent ATPase in the sarcoplasmic reticulum that results in altered cellular Ca++ levels. Aflatrem is a potent tremorgenic mycotoxin known to lead to neurological disorders. Previously we showed that a gene called veA controls aflatoxin and sclerotial production in A. parasiticus. In this study in A. flavus, we show that the veA homolog in A. flavus not only is necessary for the production of aflatoxins B1 and B2 and sclerotia, but also regulates the synthesis of the mycotoxins cyclopiazonic acid and aflatrem. The A. flavus ΔveA mutant was completely blocked in the production of aflatrem and showed greater than twofold decrease in cyclopiazonic acid production. The genes involved in the synthesis of cyclopiazonic acid are unknown; however, the aflatrem gene cluster has been characterized. Northern hybridization analysis showed that veA is required for expression of the A. flavus aflatrem genes atmC, atmG, and atmM. This is the first report of a regulatory gene governing the production of cyclopiazonic acid and aflatrem mycotoxins.
DOI:10.1128/EC.00088-09URLPMID:19411623 [本文引用: 1]
Aspergillus flavus, a mycotoxigenic filamentous fungus, colonizes several important agricultural crops, such as maize and peanuts. Two proteins, VeA and LaeA, known to form a nuclear complex in Aspergillus nidulans have been found to positively regulate developmental processes in several Aspergillus species. Here, an examination of near-isogenic A. flavus mutants differing in copy number of veA and laeA alleles (0, 1, or at least 2 each) revealed critical roles for VeA and LaeA in A. flavus development and seed colonization. In contrast to the wild type, both null mutants were unable to metabolize host cell lipid reserves and were inhibited by oleic acid in growth assays. The copy number of LaeA but not VeA appeared critical for a density-dependent sclerotial-to-conidial shift, since the multicopy laeA (MClaeA) strain produced relatively constant sclerotial numbers with increasing population size rather than showing the decrease in sclerotia seen in both the wild-type and MCveA strains. The MCveA-laeA strain yielded an intermediate phenotype. This study revealed unique roles of VeA and LaeA in seed pathogenesis and fungal biology, distinct from their cooperative regulatory functions in aflatoxin and sclerotial development.
DOI:10.1016/S1087-1845(02)00029-4URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}