 ,1,2, 李执2, 陈强2, 李阳2, 陈诗豪2, 裴英2, 周勇3, 程梦萍3, 唐豪3, 王际睿1,3, 魏育明1,3, 刘登才1,3, 陈黎4, 郑有良1,3, 蒲至恩,1,2
,1,2, 李执2, 陈强2, 李阳2, 陈诗豪2, 裴英2, 周勇3, 程梦萍3, 唐豪3, 王际睿1,3, 魏育明1,3, 刘登才1,3, 陈黎4, 郑有良1,3, 蒲至恩,1,2Development and Utilization of KASP Marker for Se Concentration in Synthetic Wheat SHW-L1
WEI GuangHui,1,2, LI Zhi2, CHEN Qiang2, LI Yang2, CHEN ShiHao2, PEI Ying2, ZHOU Yong3, CHENG MengPing3, TANG Hao3, WANG JiRui1,3, WEI YuMing1,3, LIU DengCai1,3, CHEN Li4, ZHENG YouLiang1,3, PU ZhiEn,1,2通讯作者:
责任编辑: 李莉
收稿日期:2019-12-2接受日期:2020-03-7网络出版日期:2020-10-16
| 基金资助: |
Received:2019-12-2Accepted:2020-03-7Online:2020-10-16
作者简介 About authors
魏广辉,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (2139KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
魏广辉, 李执, 陈强, 李阳, 陈诗豪, 裴英, 周勇, 程梦萍, 唐豪, 王际睿, 魏育明, 刘登才, 陈黎, 郑有良, 蒲至恩. 人工合成小麦SHW-L1高硒含量KASP分子标记开发及其应用[J]. 中国农业科学, 2020, 53(20): 4103-4112 doi:10.3864/j.issn.0578-1752.2020.20.001
WEI GuangHui, LI Zhi, CHEN Qiang, LI Yang, CHEN ShiHao, PEI Ying, ZHOU Yong, CHENG MengPing, TANG Hao, WANG JiRui, WEI YuMing, LIU DengCai, CHEN Li, ZHENG YouLiang, PU ZhiEn.
0 引言
【研究意义】人体需要22种元素来维持身体健康,其中硒是构成含硒蛋白与含硒酶的重要组分,含硒复合物具有清除体内自由基、抗衰老、增强人体免疫力、拮抗重金属毒性等生物功能,近年来已成为国内外研究的热点[1,2]。中国是世界上缺硒最严重的国家之一,约有一亿多人口膳食结构中硒含量不足。与动物性食品相比,植物性膳食中的硒相对含量较低[3,4],其中,小麦作为人类的主要植物性膳食来源之一,籽粒中的硒含量与健康息息相关。在小麦中,硒主要以硒代蛋氨酸和甲基硒代半胱氨酸形式存在[5],不同形态的硒对人体的作用不同[6],而这两种形式的有机硒对人体健康的有效性远大于无机硒。故利用好小麦中的硒源是一条人体补硒的高效、低廉、简单和易行的途径,开展高硒小麦选育是克服硒元素缺乏的最经济有效方法之一。由于对硒元素检测手段颇为复杂,限制了育种单位对这一性状的选育,因此,有必要将控制小麦籽粒硒含量的基因或者标记开发成简单、易操作的PCR标记来解决这一难题。【前人研究进展】随着生物技术的发展,越来越多的分子标记用于育种[7,8],目前,在基因组层面丰度最大的为单核苷酸多态性(single nucleotide polymorphisms,SNP)[9]。并且SNP已在很大程度上取代了已广泛测序的作物品种(如小麦)的简单序列重复序列(simple sequence repeats,SSR)用于分子标记辅助选择。竞争性等位基因特异性PCR(kompetitive allele specific PCR,KASP)可以针对指定的SNP和InDel(insertions and deletions,InDels)进行高精度双等位基因分型。相比于其他的验证技术,KASP具有更高的分析稳定性和准确度、反应成本更低、通量更大,是一项优质价廉的基因分型技术[10,11]。这项技术是基于引物末端碱基的特异匹配来对SNP分型以及检测InDels[12,13]。通过全基因组关联分析(genome-wide association study,GWAS)与SNP标记联合使用,可以挖掘出大量的重要农艺性状的相关位点,开发相关的KASP标记,可以快速方便地进行材料的选择,包括在育种过程中对抗逆性、株高、穗长等性状的直接选择[14]。YU等[15]利用GWAS鉴定了小麦品系中与Ug99茎锈病抗性相关的基因座,标记特征关联鉴定了12个与抗性显著相关的SNP标记并开发了KASP标记,这些标记物可用于小麦对Ug99茎锈病抗性的育种中的标记辅助选择。ZENG等[16]在小麦条锈病基因的发掘中,对重组自交系中利用BSA后的SNP阵列进行了单倍型分析,并开发了用于标记辅助选择的KASP标记用来进行条锈病的选择。可见在小麦中利用SNP标记开发的KASP标记用于分子标记辅助选择是可行的。分子标记的广泛应用同时推动了硒相关基因的定位研究,孙明茂[17]、张现伟[18]研究得到的水稻硒含量相关QTL位点将用以对杂种后代的选择改良;赵敏[19]通过筛选富硒基因型进行富硒大米及其农产品的研究与开发;陈大清等[20]通过对拟南芥耐硒突变体的基因定位,挖掘硒耐受基因与硒敏感基因;裴英[21]通过图谱构建得出小麦硒含量相关的QTL,利用高硒人工合成小麦资源SHW-L1与普通小麦品种川麦32杂交获得重组自交系群体(RILs),利用SSR标记与DArT标记技术对F10代的RILs群体进行基因型检测,构建了含有1 874个DArT标记和299个SSR标记、总长度3 985.6 cM的遗传图谱。并结合多年多点对籽粒硒含量进行了定位,得到10个硒相关QTL位点,主要分布于3D、5A染色体。【本研究切入点】利用SNP开发出的KASP标记筛选高硒小麦资源的研究仍鲜见报道。【拟解决的关键问题】本研究利用SHW-L1×川麦32重组自交系群体已有的660K小麦SNP图谱芯片对RILs群体进行了图谱密化,把位于5A染色体上的QTL区间进一步缩小,开发稳定基因内的SNP位点为KASP标记,用于高硒小麦材料的筛选。1 材料与方法
1.1 试验材料
供试材料总共152份,均来自四川农业大学小麦研究所,其中人工合成小麦亲本SHW-L1和川麦32及其衍生的含138个株系的RILs用于KASP标记开发。SHW-L1在温江种植条件下的硒含量为0.1048 mg·kg-1,约3倍高于川麦32的硒含量0.0401 mg·kg-1[22,23]。通过前期利用SSR标记、DArT标记研究,发现与硒含量相关的QTL来源于SHW-L1[22,23]。为了验证该标记的可靠性,选用了以人工合成小麦SHW-L1作为亲本衍生出的如蜀麦969、蜀麦580、蜀麦980、蜀麦830、蜀麦114等14个品系用于验证KASP标记。所有材料于2015、2016年分别种植于四川农业大学崇州、温江试验生产基地,栽培措施同一般大田生产。其中用于SNP分型和硒含量表型匹配的重组自交系2015年种植于温江,表型选择标准为地方标准GB/T 22499-2008[24] 0.0400 mg·kg-1,籽粒硒含量大于0.0400 mg·kg-1的材料为高硒品系,籽粒硒含量低于0.0400 mg·kg-1的贫硒品系。后续用于KASP标记的验证材料于2016年种植于崇州试验基地,硒含量划分标准根据当年籽粒硒含量平均值0.0130 mg·kg-1进行,大于0.0130 mg·kg-1的材料为高硒。1.2 SNP图谱
参考杨剑[25]的方法构建重组自交系660K SNP图谱。1.3 小麦籽粒硒含量测定方法
利用原子荧光法[26]对材料进行硒含量测定,由宜宾市产品质量监督检验所检测。1.4 DNA提取方法
采用改良的CTAB法[27]方法提取幼苗新鲜叶片DNA,并且取1.5 μL DNA在0.8%琼脂糖凝胶上电泳检测纯度。1.5 引物合成与反应体系
利用Primer5.0软件(http://www.premierbiosoft. com/index.html),根据SNP标记侧翼序列信息进行KASP引物的设计,并通过生工生物工程(上海)股份有限公司进行引物合成。KASP验证条件:2条特异性正向引物3′末端碱基为SNP位点变异碱基(G或A),5′端分别加上FAM和HEX荧光序列标签序列。引物的混合比例为AL1:AL2:R:ddH2O=6:6:15:23,PCR反应体系为1 μL DNA、5 μL KASP Mix(LGC Genomics, Hoddeston, UK)、0.08 μL Μgcl2、2.52 μL ddH2O和1.4 μL混合引物。利用CFX96荧光定量仪进行检测,PCR反应程序为94℃ 15 min;94℃ 20 s,61—55℃ 60 s,10个循环(touch-down,每循环降低0.6℃);94℃ 20 s,55℃ 60 s,30个循环。
1.6 数据统计及分析
利用CFX96荧光定量仪配套软件BioRad CFX Manager的基因分型模块,读取各样品不同荧光信号值,并分析基因型;采用Microsoft Excel 2010进行数据的整理,SPSS 20.0进行方差分析,根据广义遗传力计算公式估算遗传力。遗传力计算公式[28]如下:h2=Vg/(Vg+Ve)=MSV/(MSV+MST/l+MSVT/rl+MSE)。
式中,Vg为品种的方差组分,Ve为残差的方差组分,MSV为品种的均方,MST为年份的均方,MSVT为品种与年份互作的均方,MSE为误差的均方,r为重复,l为年份。
2 结果
2.1 群体硒含量变异情况
通过对材料籽粒中的硒含量进行方差分析(表1),可以看出,群体品种间籽粒硒含量的差异达到极显著水平,不同年份间群体籽粒硒含量无显著差异,根据方差估算出小麦籽粒硒含量广义遗传力为66%。Table 1
表1
表1群体籽粒硒含量方差分析
Table 1
| 变异来源 Source of variation | 平方和 Type Ⅲ SS | 自由度 df | 均方 MS | F值 F value | Sig. |
|---|---|---|---|---|---|
| V | 0.02995031 | 114 | 0.00026272 | 13.410 | ** |
| T | 0.00009015 | 1 | 0.00009015 | 0.4890 | |
| V×T | 0.02100010 | 110 | 0.00019091 | ||
| 误差Error | 0.00415800 | 110 | 0.00003780 |
新窗口打开|下载CSV
2.2 SNP标记
通过对获得的多态性SNP标记进行偏分离筛选,去除偏分离后共获得142 430个SNP标记用于图谱构建。SNP标记和DArT标记以及SSR标记整合后的遗传图谱全长共17 889.62 cM,包含121 222个有效标记。基于前期定位结果,将控制硒含量的位点定位于3D、5A染色体上,由于3D染色体上标记密度不够,因此,本研究重点关注5A染色体上的信息。5A染色体上有7 207个标记,总长度为1 114.86 cM,密度为0.155 cM/Locus。2.3 KASP标记的开发
将重组自交系籽粒Se含量和整合后的5A染色体图谱进行计算,并将LOD阈值设为2,得到重组自交系在5A染色体上的QTL区域(表2)。Table 2
表2
表2筛选过LOD值后的QTL结果
Table 2
| 籽粒硒含量 Grain selenium content | 编号 Number | 染色体 Chromosome | 标记 Marker | 位置 Position (cM) | R2 | 加性效应 Additive | LOD |
|---|---|---|---|---|---|---|---|
| Grain se-1 | 1 | 5A | 22 | 43.62 | 0.0765 | 0.0003 | 2.5551 |
| Grain se-2 | 1 | 5A | 13 | 29.54 | 0.0654 | 0.0003 | 2.7476 |
| Grain se-2 | 2 | 5A | 16 | 32.66 | 0.0774 | 0.0003 | 3.4722 |
| Grain se-2 | 3 | 5A | 21 | 35.58 | 0.0877 | 0.0004 | 3.9723 |
新窗口打开|下载CSV
将得到的QTL与图谱进行对照,以获得一个更大的QTL区间,区间内的SNP作为初筛选标记。根据SNP分型结果与籽粒硒含量数据进行比对再筛选(图1),总共分为3种,A型、B型和缺失。理想中的标记应该为A型和B型分离,并以某个籽粒硒含量的材料为界限,完全分布于两侧,但完全符合的标记基本不存在,因此,要从初筛选的标记中做二次筛选,只需选出基因型聚类较好的标记即可,最终选出5个标记用于开发籽粒硒含量相关的KASP分子标记(表3)。根据5个标记设计引物(表4),其中如AX-1引物包括2条等位基因特异性正向引物AL1和AL2,一条共同的反向引物R。
图1
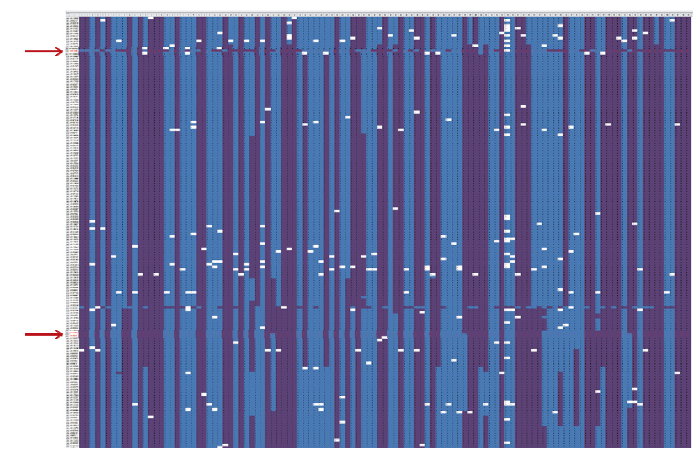 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1SNP分型与籽粒硒含量比对结果
X轴:RILs群体所有材料,按照籽粒硒含量从小到大顺序排列;Y轴:初筛选出的标记;箭头:2个SNP标记;蓝色:A型;紫色:B型;白色:缺失
Fig. 1SNP comparison of genotyping and grain selenium content
X-axis: All materials of the RILs population, arranged according to the selenium content of the grains in ascending order; Y-axis: Markers initially selected; Arrows: 2 SNP markers; Blue: Type A; Purple: Type B; White: Missing
Table 3
表3
表3所用SNP标记
Table 3
| 编号 Number | 染色体 Chromosome | 变异碱基 Base | 序列 Sequence (5′-3′) |
|---|---|---|---|
| AX-1 | 5A | A/G | CGTACCATGCGATCACACTTGGAGATGCGTGTGAG[A/G]GAAAAAGAGTACTCCATCCATCCAAATTCCATCCC |
| AX-2 | 5A | A/G | TGTAAATTAATTTGTTGTGGGTAGGGCATCTTGAG[A/G]AATGTTTACCTTCTTGCTGCTGCCTGGTTCGGTTT |
| AX-3 | 5A | A/C | GGGTGATAGCAAGGAAGCTGAAGAGCATGATGGAC[A/C]AGGGACAGACGGAAAAGAGTTGCAGGATGGACAAG |
| AX-4 | 5A | A/G | GATGACTTTGCGGCAATATTAAGAGATGTGCATCC[A/G]TTAGCAAGCAAGCTAGCGGACAAAACCAGCTTAAC |
| AX-5 | 5A | T/C | ACCCCTTGTTTAGTACCACCTCACCAAGTGAGAGT[C/T]ATTTCAACCGGTTTATAAGATTCAGCGTTCCAACC |
新窗口打开|下载CSV
Table 4
表4
表4KASP引物及序列
Table 4
 |
新窗口打开|下载CSV
2.4 KASP标记的初验证
针对5个标记分别进行KASP-SNP分型测试(图2),其中2个标记能够将2种基因型样本区分开,另外3个标记无法区分亲本基因型,因此剔除。通过测试的2个标记用于后续详细的测试。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2分子标记对不同籽粒硒含量样本的初次KASP-SNP分型检测
坐标轴内的每个点代表一个单株,数据点可能存在重叠;黑色点代表对照(ddH2O);橙色点代表高硒基因型;蓝色点代表低硒基因型。下同
Fig. 2Initial KASP-SNP typing of selenium content samples from different grains by molecular markers
These spots within the axis represent different single plant, and data spots might be overlapped; Black dots: Controls (ddH2O); Orange dots: High selenium gene genotypes; Blue dots: Low selenium genotypes. The same as below
2.5 标记AX-1、AX-2的KASP-SNP分型详细结果
在SNP荧光信号二维图(图3)中可以看到,2种基因型之间的夹角较大,且各信号点的荧光信号值也比较高,因此分型效果较好。其中,代表亲本人工合成小麦SHW-L1都是聚合在X轴的橙色信号点,亲本川麦32都是聚合在Y轴的蓝色信号点。利用基因型对材料进行分类,将对应的样本与其已经测出的硒含量进行比对(图4),其中AX-1标记所对应118个材料中鉴定为基因型低硒的为54个,平均籽粒硒含量为0.0145 mg·kg-1,相对应的表现型中,50个材料为低硒,4个材料为高硒(>0.0300 mg·kg-1),对低硒材料的选择效率为92.6%;群体中利用AX-1标记鉴定为基因型高硒的有64个,但其中47个材料表现型为低硒,17个为高硒,平均籽粒硒含量为0.0295 mg·kg-1,对高硒材料的选择效率为26.6%,2种基因型的材料籽粒硒含量存在显著差异(F=4.84,P<0.05)。AX-2标记所对应118个材料中鉴定为基因型低硒的为56个,平均籽粒硒含量为0.0141 mg·kg-1,其中52个材料表现为低硒,4个材料表现为高硒,对低硒材料的选择效率为92.9%;利用标记鉴定为基因型高硒的有62个,其中表现型中有45个为低硒,17个材料表现为高硒,平均籽粒硒含量为0.0303 mg·kg-1,对高硒材料的选择效率为28.4%,2种基因型的材料籽粒硒含量存在显著差异(F=5.43,P<0.05)。图3
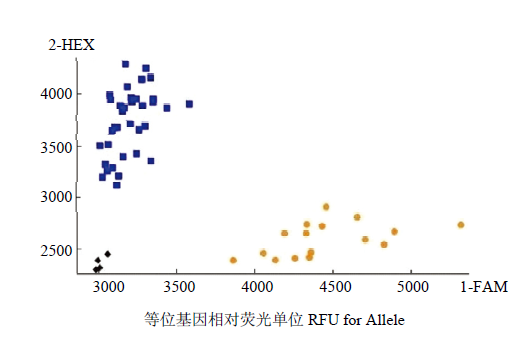 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3分子标记对Rils138群体籽粒硒含量的KASP-SNP分型
Fig. 3KASP-SNP typing of selenium content in grain of RILs 138 population by molecular markers
图4
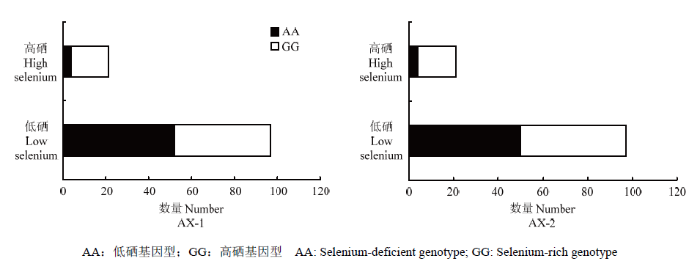 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4KASP标记AX-1和AX-2不同等位变异在118份材料中的分布
AA:低硒基因型;GG:高硒基因型
Fig. 4Distribution of KASP-labeled AX-1 and AX-2 alleles in 118 materials
AA: Selenium-deficient genotype; GG: Selenium-rich genotype
按照0.0400 mg·kg-1的标准对群体表现型进行划分,其中118个材料中仅有21份材料硒含量表现为高硒,表现型比例严重偏离1:1,也不符合数量性状的正态分布。其中不论AX-1标记还是AX-2标记对应高硒表现型材料均为17个,即表明高硒基因型和高硒表现型的匹配度达到81%。可见,在利用标记剔除低硒基因型后的剩余高硒基因型材料中,再利用标记进行筛选时效率会更高。
2.6 KASP标记的检验
为了验证AX-1和AX-2能否用于其他相关高硒小麦的筛选,选取14个由SHW-L1衍生育成的材料测试籽粒含量,按照群体当年籽粒硒含量平均值(0.0130 mg·kg-1)进行了KASP-SNP分型测试,图5为部分品种(系)硒含量结果。结果表明,标记能很好地区分高硒和低硒籽粒样品,通过与籽粒硒含量表型数据对比(表5),可以看出,各验证材料的基因型与表型结果基本一致,可以认为开发出的2个标记对于利用人工合成小麦SHW-L1衍生出的小麦品种(系)均有筛选籽粒硒含量相对高低的作用。图5
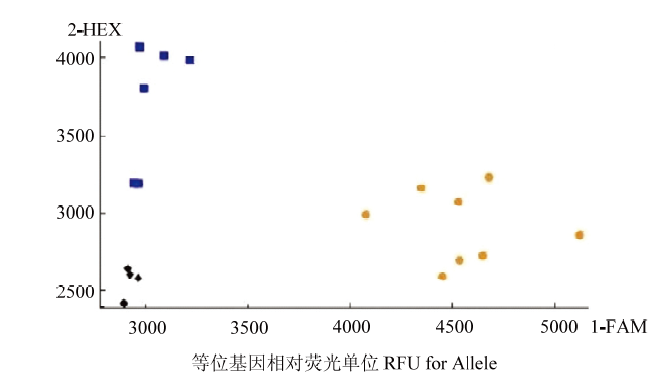 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5分子标记对不同验证样本籽粒硒含量的定性检测
Fig. 5Qualitative detection of selenium content in grain of different validation samples by molecular markers
Table 5
表5
表5部分供试材料分型检验结果与表型数据对比表
Table 5
| 品种或自交系 Cultivar or inbred lines | 籽粒硒含量 Selenium content in grains (mg·kg-1) | SNP基因分型 SNP genotyping |
|---|---|---|
| 蜀麦969 Shumai 969 | 0.020 | 高硒High Se |
| 蜀麦580 Shumai 580 | 0.011 | 低硒Low Se |
| 蜀麦980 Shumai 980 | 0.009 | 低硒Low Se |
| 蜀麦830 Shumai 830 | 0.017 | 高硒High Se |
| 蜀麦114 Shumai 114 | 0.013 | 高硒 High Se |
新窗口打开|下载CSV
3 讨论
3.1 高硒KASP标记的开发利用
硒是小麦籽粒中的微量元素,受基因型和环境的共同影响。在检测手段不能普及的前提下,培育高硒品种存在测试手段和技术上的瓶颈,开发简单、易操作分子标记用于快速筛选高硒小麦材料有助于高硒小麦品种的选育,本研究开发的标记将有助于育种人员开展高硒品种选育工作。分子标记辅助育种如DArT标记、SSR标记已经广泛应用于小麦育种上,苟璐璐等[29]利用231个小麦产量与品质相关性状的一致性QTL区段中的SSR标记,通过关联分析揭示四川地方品种产量和品质相关性状的遗传特征,这些SSR标记位点和区段为通过分子标记辅助选择手段利用和发掘四川小麦地方品种产量和品质优异相关基因或区段提供了理论指导。之后基于SNP芯片开发的KASP标记也被用于对抗逆性、种质资源和种子纯度等性状的直接选育,如GmSNAP11- 5149分子标记应用于辅助抗大豆胞囊线虫品种选育和抗病种质资源鉴定[30];张利莎等[31]研究表明SNP标记能够有效鉴定混杂度低至5%的麦芽样品,且基于KASP技术的SNP标记可以满足麦芽纯度的快速定量检测需要。可见SNP标记作为第三代分子标记,随着其标记水平的提升和成本的降低,应用也越来越广泛,用于构建遗传图谱和遗传多样性分析等,但在分子标记辅助选择育种中的应用还不够普遍化,在小麦品质育种上的应用更少,尤其是针对籽粒中含量很少的微量元素。
近年来,对于硒含量相关基因的定位还多停留在对QTL位点发掘阶段,性状的复杂性和分子标记实用性使高硒小麦育种进展缓慢。由于SNP技术的发展,本研究在前期研究的基础上进一步深入,将前期定位于5A上的新QTL位点的区间进一步缩小,开发出基于SNP位点的KASP标记。本研究开发的2个KASP标记均来源于人工合成小麦SHW-L1,不仅能在人工合成小麦群体中起到鉴定籽粒硒含量相关基因型的作用,在以人工合成小麦为亲本的其他后代材料中,也能有效区分高硒和贫硒材料。SHW-L1作为亲本具有很好的配合力,衍生了一大批材料,本研究选择的14个高代品种(系)验证材料中,其中5个材料(文中列出的仅为审定或者即将审定的高代材料)能够利用本标记进行有效区分高硒材料和低硒材料,且表型和基因型一致,可见该标记也可用于SHW-L1后代的筛选,快速鉴定其后代育种材料中籽粒硒含量的高低。随着对人工合成小麦的研究越来越深入[32,33,34],发现人工合成小麦的后代如川麦42,蜀麦969等具有氮素利用效率高等特点,人工合成小麦是否具有其他元素高效利用的特点还需要进一步关注,同时本研究也为元素高效利用的SNP标记开发提供了参考。
值得注意的是,在本研究中,无论是AX-1还是AX-2标记,将供试材料可以按照基因型分为2类,即高硒和低硒材料,AX-1标记高硒基因型与贫硒基因型比例为50:54=1:1.18,AX-2标记基因型比例为52:56=1:1.10,等位基因符合1:1的分离比例。但按照表现型来划分,高硒材料:低硒材料=21:97,低硒材料的数目远远多于高硒材料数目,表型严重偏离了基因型,即通过KASP标记无法直接筛选出所有的贫硒材料,或者只筛选出高硒材料,需要根据标记不同等位变异在群体中的分布来考虑筛选方法以提高筛选效率。因此可通过反向操作,直接筛选去掉贫硒基因型材料,即可以筛选一半表现型为贫硒的材料,这样只会筛选掉极少部分高硒材料,以此来提升一倍的选择效率。对于剩下的高硒基因型,由于其对高硒材料选择效率不高,应当采取传统育种方法,结合表型筛选。但同时需要明确的是,具有高硒含量的表现型材料中,有81%的材料具有高硒的基因型,也表明了试验所得标记的可靠性。
高硒基因型的材料表现为低硒的表现型,即基因分型结果和表现型不匹配的现象在本研究中比较常见,可能有以下几种原因:一是植物籽粒从土壤中获取营养元素有3个步骤,吸收、转运以及物质在各个器官的再分配。但由于土壤中硒浓度太低(土壤含硒量为0.2600 μg·g-1),造成了材料吸收效率较低,进而引起籽粒硒积累量较低,但不排除这些具备高硒基因型的材料拥有高效吸收或者转运的潜力,在土壤硒充足时,具备高硒基因型的材料的籽粒硒积累量可能会更高。由于各个高硒基因型材料间也存在吸收转运能力的差异,因此高硒基因型中仍然会有高积累和低积累的材料存在;二是小麦籽粒硒含量是由多基因控制的数量性状,位于5A的位点差异并不能完全覆盖材料间硒含量的差异。本文推测这是基因型和表现型之间存在偏差的主要原因。
3.2 KASP标记的其他用途
现有研究表明,植物中硒元素不仅对植物生长有利,还能降低镉等重金属的吸收,因此开发高硒的KASP标记不仅在于小麦籽粒硒含量的提高,还可以起到“一因多效”的作用,即用于小麦低积累镉和汞品种和品系的筛选。现有研究表明单施Se可对Cd[35]、Hg[36]等重金属起到钝化作用,减少水稻对重金属的积累,能有效降低水稻吸收Cd,阻控Cd向地上部以及籽实中的转运。小麦中硒对镉的抑制主要原因是硒能促进茎和根的生长,降低茎中镉的浓度[37],从而降低小麦籽粒中的镉含量。另外在水稻中已有通过QTL聚合筛选高硒低镉材料的报道[38,39],这也为小麦富硒低镉材料的选育提供了思路和成功范例,希望本研究开发的高硒KASP标记能成为小麦富硒低镉品种选育的开端,为解决中国现存的部分土壤问题中镉含量较高现实情况前提下提供小麦安全生产的新思路。4 结论
开发的KASP标记AX-1和AX-2能够区分来源于人工合成小麦群体以及人工合成小麦衍生品种或自交系籽粒硒含量基因型,并有效鉴定其籽粒硒含量的相对高低,可用于高硒小麦材料的筛选。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1016/j.foodchem.2009.01.054URL [本文引用: 1]
DOI:10.7606/j.issn.1009-1041.2011.02.028URL [本文引用: 1]

为给富硒小麦生产提供理论依据,采用大田取样、盆栽试验和田间试验方法,同时结合室内分析,研究了河北省冬小麦硒的含量及小麦富硒施肥技术。结果表明:(1)河北省冬小麦含硒量变化范围为0.010~0.222 mg·kg-1,其中46.67%的冬小麦缺硒,35.00%的冬小麦轻度缺硒,正常含硒量的小麦仅占18.33%;(2)通过土壤施硒和叶面喷硒可有效地提高冬小麦籽粒的含硒量,达到富硒小麦的要求,而浸种只能作为一种辅助措施。以亚硒酸钠作硒肥,生产富硒小麦的土壤施硒量以0.10~0.25 mg Se·kg-1为宜;叶面喷硒的适宜浓度为40 mg Se·L-1,每公顷用液量750 kg,以小麦抽穗至灌浆初期进行喷洒效果较好。因此,建议在河北省的土壤和气候条件下,生产富硒小麦的土壤适宜施硒量为225~562.5 g·hm-2,叶面喷硒的适宜施硒量为30 g·hm-2。
DOI:10.7606/j.issn.1009-1041.2011.02.028URL [本文引用: 1]
为给富硒小麦生产提供理论依据,采用大田取样、盆栽试验和田间试验方法,同时结合室内分析,研究了河北省冬小麦硒的含量及小麦富硒施肥技术。结果表明:(1)河北省冬小麦含硒量变化范围为0.010~0.222 mg·kg-1,其中46.67%的冬小麦缺硒,35.00%的冬小麦轻度缺硒,正常含硒量的小麦仅占18.33%;(2)通过土壤施硒和叶面喷硒可有效地提高冬小麦籽粒的含硒量,达到富硒小麦的要求,而浸种只能作为一种辅助措施。以亚硒酸钠作硒肥,生产富硒小麦的土壤施硒量以0.10~0.25 mg Se·kg-1为宜;叶面喷硒的适宜浓度为40 mg Se·L-1,每公顷用液量750 kg,以小麦抽穗至灌浆初期进行喷洒效果较好。因此,建议在河北省的土壤和气候条件下,生产富硒小麦的土壤适宜施硒量为225~562.5 g·hm-2,叶面喷硒的适宜施硒量为30 g·hm-2。
[本文引用: 1]
[本文引用: 1]
DOI:10.1017/S0007114507812001URLPMID:17925056 [本文引用: 1]
The mean dietary exposure to the nutrient elements iodine, Fe, Se and Na by eight age-sex groups of the New Zealand population was estimated from foods purchased and prepared as for consumption. A total of 968 samples comprising 121 foods were collected and analysed. Mean daily exposures were calculated from mean concentration levels of the selected nutrients in each food combined with simulated diets for a 25+-year-old male and female, a 19-24-year-old male, a 11-14-year-old boy and girl, a 5-6-year-old child, a 1-3-year-old toddler and a 6-12-month-old infant. Food concentrations and dietary exposures are reported and compared with nutrient reference values (for example, recommended daily intakes, adequate intakes or upper limits). Dietary iodine exposures for all age-sex groups were well below recommended levels and have steadily decreased since 1982, raising concern especially for the physical and mental development of infants and young children. Fe exposures meet the recommended daily intake for the average male and 11-14 year olds but are only about half that recommended for adult females. Se exposure is about 20 % less than optimal for females. Na exposures, excluding discretionary salt, are above the acceptable exposure level for all age-sex groups, and exceed the upper intake limits for 25+-year-old males, 19-24-year-old young males, and 11-14-year-old boys and girls by up to 125 % for an average consumer.
DOI:10.1016/j.foodchem.2011.07.102URL [本文引用: 1]
DOI:10.1016/j.aca.2009.08.013URL [本文引用: 1]
[本文引用: 1]
.
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
DOI:10.1111/pbi.12113URLPMID:24034357 [本文引用: 1]
The advances in genotyping technology provide an opportunity to use genomic tools in crop breeding. As compared to field selections performed in conventional breeding programmes, genomics-based genotype screen can potentially reduce number of breeding cycles and more precisely integrate target genes for particular traits into an ideal genetic background. We developed a whole-genome single nucleotide polymorphism (SNP) array, RICE6K, based on Infinium technology, using representative SNPs selected from more than four million SNPs identified from resequencing data of more than 500 rice landraces. RICE6K contains 5102 SNP and insertion-deletion (InDel) markers, about 4500 of which were of high quality in the tested rice lines producing highly repeatable results. Forty-five functional markers that are located inside 28 characterized genes of important traits can be detected using RICE6K. The SNP markers are evenly distributed on the 12 chromosomes of rice with the average density of 12 SNPs per 1 Mb and can provide information for polymorphisms between indica and japonica subspecies as well as varieties within indica and japonica groups. Application tests of RICE6K showed that the array is suitable for rice germplasm fingerprinting, genotyping bulked segregating pools, seed authenticity check and genetic background selection. These results suggest that RICE6K provides an efficient and reliable genotyping tool for rice genomic breeding.
URLPMID:12746733 [本文引用: 1]
DOI:10.1038/35103535URLPMID:11733746 [本文引用: 1]
Understanding the relationship between genetic variation and biological function on a genomic scale is expected to provide fundamental new insights into the biology, evolution and pathophysiology of humans and other species. The hope that single nucleotide polymorphisms (SNPs) will allow genes that underlie complex disease to be identified, together with progress in identifying large sets of SNPs, are the driving forces behind intense efforts to establish the technology for large-scale analysis of SNPs. New genotyping methods that are high throughput, accurate and cheap are urgently needed for gaining full access to the abundant genetic variation of organisms.
DOI:10.1007/s11032-013-9917-xURL [本文引用: 1]
Single nucleotide polymorphism (SNP) data can be obtained using one of the numerous uniplex or multiplex SNP genotyping platforms that combine a variety of chemistries, detection methods, and reaction formats. Kompetitive Allele Specific PCR (KASP) is one of the uniplex SNP genotyping platforms, and has evolved to be a global benchmark technology. However, there are no publications relating either to the technology itself or to its application in crop improvement programs. In this review, we provide an overview of the different aspects of the KASP genotyping platform, discuss its application in crop improvement, and compare it with the chip-based Illumina GoldenGate platform. The International Maize and Wheat Improvement Center routinely uses KASP, generating in excess of a million data points annually for crop improvement purposes. We found that (1) 81 % of the SNPs used in a custom-designed GoldenGate assay were transferable to KASP; (2) using KASP, negative controls (no template) consistently clustered together and rarely produced signals exceeding the threshold values for allele calling, in contrast to the situation observed using GoldenGate assays; (3) KASP's average genotyping error in positive control DNA samples was 0.7-1.6 %, which is lower than that observed using GoldenGate (2.0-2.4 %); (4) KASP genotyping costs for marker-assisted recurrent selection were 7.9-46.1 % cheaper than those of the BeadXpress and GoldenGate platforms; and (5) KASP offers cost-effective and scalable flexibility in applications that require small to moderate numbers of markers, such as quality control analysis, quantitative trait loci (QTL) mapping in bi-parental populations, marker-assisted recurrent selection, marker-assisted backcrossing, and QTL fine mapping.
URLPMID:27228161 [本文引用: 1]
DOI:10.1371/journal.pone.0171963URLPMID:28241006 [本文引用: 1]
Wheat stem rust (Puccinia graminis f. sp. tritici Eriks. and E. Henn.) is one of the most destructive diseases world-wide. Races belonging to Ug99 (or TTKSK) continue to cause crop losses in East Africa and threaten global wheat production. Developing and deploying wheat varieties with multiple race-specific genes or complex adult plant resistance is necessary to achieve durability. In the present study, we applied genome-wide association studies (GWAS) for identifying loci associated with the Ug99 stem rust resistance (SR) in a panel of wheat lines developed at the International Maize and Wheat Improvement Center (CIMMYT). Genotyping was carried out using the wheat 9K iSelect single nucleotide polymorphism (SNP) chip. Phenotyping was done in the field in Kenya by infection of Puccinia graminis f. sp. tritici race TTKST, the Sr24-virulent variant of Ug99. Marker-trait association identified 12 SNP markers significantly associated with resistance. Among them, 7 were mapped on five chromosomes. Markers located on chromosomes 4A and 4B overlapped with the location of the Ug99 resistance genes SrND643 and Sr37, respectively. Markers identified on 7DL were collocated with Sr25. Additional significant markers were located in the regions where no Sr gene has been reported. The chromosome location for five of the SNP markers was unknown. A BLASTN search of the NCBI database using the flanking sequences of the SNPs associated with Ug99 resistance revealed that several markers were linked to plant disease resistance analogues, while others were linked to regulatory factors or metabolic enzymes. A KASP (Kompetitive Allele Specific PCR) assay was used for validating six marker loci linked to genes with resistance to Ug99. Of those, four co-segregated with the Sr25-pathotypes while the rest identified unknown resistance genes. With further investigation, these markers can be used for marker-assisted selection in breeding for Ug99 stem rust resistance in wheat.
.
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
DOI:10.1016/S2095-3119(13)60640-1URL [本文引用: 2]
Micronutrient malnutrition affects over three billion people worldwide, especially women and children in developing countries. Increasing the bioavailable concentrations of essential elements in the edible portions of crops is an effective resolution to address this issue. To determine the genetic factors controlling micronutrient concentration in wheat, the quantitative trait locus (QTL) analysis for iron, zinc, copper, manganese, and selenium concentrations in two recombinant inbred line populations was performed. In all, 39 QTLs for five micronutrient concentrations were identified in this study. Of these, 22 alleles from synthetic wheat SHW-L1 and seven alleles from the progeny line of the synthetic wheat Chuanmai 42 showed an increase in micronutrient concentrations. Five QTLs on chromosomes 2A, 3D, 4D, and 5B found in both the populations showed significant phenotypic variation for 2-3 micronutrient concentrations. Our results might help understand the genetic control of micronutrient concentration and allow the utilization of genetic resources of synthetic hexaploid wheat for improving micronutrient efficiency of cultivated wheat by using molecular marker-assisted selection.
DOI:10.1007/s11104-017-3556-7URL [本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[本文引用: 1]
(in Chinese)
[本文引用: 1]
DOI:10.1007/BF00303966URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
普通小麦(Triticum aestivum)地方品种具备了对当地自然生态条件的较强适应性和与之相对应的生产潜力。因此,从地方小麦品种中挖掘产量、品质、抗病虫及耐逆等优良基因资源,扩大当前小麦育种亲本的遗传基础,历来受到小麦遗传育种学家的高度重视。本研究通过对64个四川小麦地方品种进行了2年3个环境的表型精准鉴定,并利用231个小麦产量与品质相关性状的一致性QTL区段中的SSR标记,通过关联分析揭示四川地方品种产量和品质相关性状的遗传特征。表型鉴定结果显示,这些地方小麦品种具有多花多粒、分蘖能力强、成穗率高等特性,总体上属于中筋或弱筋小麦,且有效分蘖数、株高、小穗密度、穗长、可育小穗数、沉降值等性状遗传力较高,达50%以上。关联分析鉴定出18个多环境下稳定表达的SSR位点,与产量和品质性状极显著关联,其中1个(Xgwm372)同时与产量性状和品质性状关联,4个(Xwmc112, Xcfd5, Xwmc317和Xgwm372)最为稳定,3个(Xcfd5, Xgwm328和Xbarc181)关联到新的产量性状。相关分析表明,穗长与小穗密度呈极显著负相关,且有2个共同关联标记(Xgwm328和Xcfd5)。还鉴定出2A染色体上的Xgwm448-Xgwm328-Xgwm372区段(8.0 cM)在多环境下与穗长显著关联。上述SSR标记位点和区段为通过分子标记辅助选择手段利用和发掘四川小麦地方品种产量和品质优异相关基因或区段提供理论指导。
URL [本文引用: 1]
普通小麦(Triticum aestivum)地方品种具备了对当地自然生态条件的较强适应性和与之相对应的生产潜力。因此,从地方小麦品种中挖掘产量、品质、抗病虫及耐逆等优良基因资源,扩大当前小麦育种亲本的遗传基础,历来受到小麦遗传育种学家的高度重视。本研究通过对64个四川小麦地方品种进行了2年3个环境的表型精准鉴定,并利用231个小麦产量与品质相关性状的一致性QTL区段中的SSR标记,通过关联分析揭示四川地方品种产量和品质相关性状的遗传特征。表型鉴定结果显示,这些地方小麦品种具有多花多粒、分蘖能力强、成穗率高等特性,总体上属于中筋或弱筋小麦,且有效分蘖数、株高、小穗密度、穗长、可育小穗数、沉降值等性状遗传力较高,达50%以上。关联分析鉴定出18个多环境下稳定表达的SSR位点,与产量和品质性状极显著关联,其中1个(Xgwm372)同时与产量性状和品质性状关联,4个(Xwmc112, Xcfd5, Xwmc317和Xgwm372)最为稳定,3个(Xcfd5, Xgwm328和Xbarc181)关联到新的产量性状。相关分析表明,穗长与小穗密度呈极显著负相关,且有2个共同关联标记(Xgwm328和Xcfd5)。还鉴定出2A染色体上的Xgwm448-Xgwm328-Xgwm372区段(8.0 cM)在多环境下与穗长显著关联。上述SSR标记位点和区段为通过分子标记辅助选择手段利用和发掘四川小麦地方品种产量和品质优异相关基因或区段提供理论指导。
DOI:10.3724/SP.J.1006.2018.01600URL [本文引用: 1]
大豆胞囊线虫(Heterodera glycines Ichinohe)是严重危害世界范围大豆生产的害虫, 采用合理轮作和种植抗病品种可有效控制损失。为了开展分子标记辅助选择以加速抗病品种培育, 本研究针对前期发现的与大豆胞囊线虫3号小种(SCN3)抗性显著关联的非同义变异SNP位点Map-5149, 开发高通量、低成本的新型分子标记—竞争性等位基因特异PCR标记(kompetitive allele specific PCR, KASP), GmSNAP11-5149。利用GmSNAP11-5149鉴定了来自8个国家的202份代表性大豆抗感资源, 发现141份材料携带抗病基因型GmSNAP11-5149-AA, 平均雌虫指数为6.2%, 极显著低于58份携带感病基因型GmSNAP11-5149-GG材料的雌虫指数(61.1%), 方差分析表明, GmSNAP11-5149与胞囊线虫的抗性显著相关(F=44.18, P<0.0001), 对抗病材料的选择效率达到92%, GmSNAP11-5149可作为一个实用的分子标记应用于辅助抗大豆胞囊线虫品种选育和抗病种质资源鉴定。
DOI:10.3724/SP.J.1006.2018.01600URL [本文引用: 1]
大豆胞囊线虫(Heterodera glycines Ichinohe)是严重危害世界范围大豆生产的害虫, 采用合理轮作和种植抗病品种可有效控制损失。为了开展分子标记辅助选择以加速抗病品种培育, 本研究针对前期发现的与大豆胞囊线虫3号小种(SCN3)抗性显著关联的非同义变异SNP位点Map-5149, 开发高通量、低成本的新型分子标记—竞争性等位基因特异PCR标记(kompetitive allele specific PCR, KASP), GmSNAP11-5149。利用GmSNAP11-5149鉴定了来自8个国家的202份代表性大豆抗感资源, 发现141份材料携带抗病基因型GmSNAP11-5149-AA, 平均雌虫指数为6.2%, 极显著低于58份携带感病基因型GmSNAP11-5149-GG材料的雌虫指数(61.1%), 方差分析表明, GmSNAP11-5149与胞囊线虫的抗性显著相关(F=44.18, P<0.0001), 对抗病材料的选择效率达到92%, GmSNAP11-5149可作为一个实用的分子标记应用于辅助抗大豆胞囊线虫品种选育和抗病种质资源鉴定。
DOI:10.3724/SP.J.1006.2015.01147URL [本文引用: 1]
大麦麦芽作为啤酒酿造的主要原料之一,其纯度决定了麦芽原料的均一性,进而影响加工工艺和啤酒品质。为高效准确地鉴定麦芽纯度,在啤酒企业进行麦芽原料采购和质量监测时提供参考依据。本研究分别利用EST-SSR和SNP标记定性检测了按比例预混的麦芽样品纯度,并利用SNP标记定量检测了4份送检的麦芽盲样纯度。结果表明,EST-SSR标记能定性检测混杂度高于10%的麦芽样品,而SNP标记能够有效鉴定混杂度低至5%的麦芽样品。SNP标记对纯度定量检测的单次抽样的测定值与真实值之间的误差在3%以内。比较发现,本研究所用的两类分子标记均可用于麦芽样品的纯度检测,但基于KASP技术的SNP标记可以满足麦芽纯度的快速定量检测需要。
DOI:10.3724/SP.J.1006.2015.01147URL [本文引用: 1]
大麦麦芽作为啤酒酿造的主要原料之一,其纯度决定了麦芽原料的均一性,进而影响加工工艺和啤酒品质。为高效准确地鉴定麦芽纯度,在啤酒企业进行麦芽原料采购和质量监测时提供参考依据。本研究分别利用EST-SSR和SNP标记定性检测了按比例预混的麦芽样品纯度,并利用SNP标记定量检测了4份送检的麦芽盲样纯度。结果表明,EST-SSR标记能定性检测混杂度高于10%的麦芽样品,而SNP标记能够有效鉴定混杂度低至5%的麦芽样品。SNP标记对纯度定量检测的单次抽样的测定值与真实值之间的误差在3%以内。比较发现,本研究所用的两类分子标记均可用于麦芽样品的纯度检测,但基于KASP技术的SNP标记可以满足麦芽纯度的快速定量检测需要。
DOI:10.1007/s00122-019-03354-9URLPMID:31049633 [本文引用: 1]
KEY MESSAGE: Introgressing one-eighth of synthetic hexaploid wheat genome through a double top-cross plus a two-phase selection is an effective strategy to develop high-yielding wheat varieties. The continued expansion of the world population and the likely onset of climate change combine to form a major crop breeding challenge. Genetic advances in most crop species to date have largely relied on recombination and reassortment within a relatively narrow gene pool. Here, we demonstrate an efficient wheat breeding strategy for improving yield potentials by introgression of multiple genomic regions of de novo synthesized wheat. The method relies on an initial double top-cross (DTC), in which one parent is synthetic hexaploid wheat (SHW), followed by a two-phase selection procedure. A genotypic analysis of three varieties (Shumai 580, Shumai 969 and Shumai 830) released from this program showed that each harbors a unique set of genomic regions inherited from the SHW parent. The first two varieties were generated from very small populations, whereas the third used a more conventional scale of selection since one of bread wheat parents was a pre-breeding material. The three varieties had remarkably enhanced yield potential compared to those developed by conventional breeding. A widely accepted consensus among crop breeders holds that introducing unadapted germplasm, such as landraces, as parents into a breeding program is a risky proposition, since the size of the breeding population required to overcome linkage drag becomes too daunting. However, the success of the proposed DTC strategy has demonstrated that novel variation harbored by SHWs can be accessed in a straightforward, effective manner. The strategy is in principle generalizable to any allopolyploid crop species where the identity of the progenitor species is known.
[本文引用: 1]
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
DOI:10.1021/acs.jafc.7b03316URLPMID:29016122 [本文引用: 1]
A pot experiment was conducted to investigate the interactive effects of cadmium (Cd) and selenium (Se) on their accumulation in three rice cultivars, which remains unclear. The results showed that Se reduced Cd-induced growth inhibition, and increased and decreased Se and Cd concentrations in brown rice, respectively. Cadmium concentrations in all tissues of the hybrid were similar to those in its male parent yet significantly lower than those in its female parent. Selenium reduced Cd accumulation in rice when Cd concentration exceeded 2.0 mg kg(-1); however Se accumulation depended on the levels of Cd exposure. Finally, Cd had minimal effect on Se translocation within the three cultivars. We concluded that Cd concentration in brown rice is a heritable trait, making crossbreeding a feasible method for cultivating high-yield, low-Cd rice cultivars. Selenium effectively decreased the toxicity and accumulation of Cd, and Cd affected Se uptake but not translocation.
DOI:10.1007/s11104-015-2418-4URL [本文引用: 1]
DOI:10.1016/j.envexpbot.2018.03.024URL [本文引用: 1]
DOI:10.1111/jipb.13007URLPMID:32877006 [本文引用: 1]
Mitogen-activated protein kinase (MAPK) cascades are highly conserved signaling modules that regulate plant immune responses. The Arabidopsis thaliana Raf-like MAPK kinase kinase ENHANCED DISEASE RESISTANCE1 (EDR1) is a key negative regulator of plant immunity that affects the protein levels of MKK4 and MKK5, two important MAPK cascade members, but the underlying mechanism is poorly understood. Here, genome-wide phosphorylation analysis demonstrated that the E3 ligase KEEP ON GOING (KEG) is phosphorylated in the edr1 mutant but not the wild type, suggesting that EDR1 negatively affects KEG phosphorylation. The identified phosphorylation sites in KEG appear to be important for its accumulation. The keg-4 mutant, a previously identified edr1 suppressor, enhances susceptibility to the powdery mildew pathogen Golovinomyces cichoracearum. In addition, MKK4 and MKK5 protein levels are reduced in the keg-4 mutant. Furthermore, we demonstrate that MKK4 and MKK5 associate with full-length KEG, but not with truncated KEG-RK or KEG-RKA, and that KEG ubiquitinates and mediates the degradation of MKK4 and MKK5. Taken together, these results indicate that MKK4 and MKK5 protein levels are regulated by KEG via ubiquitination, uncovering a mechanism by which plants fine-tune immune responses by regulating the homeostasis of key MAPK cascade members via ubiquitination and degradation.
URLPMID:32877006 [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}