 ,1,2, 肖斌,1
,1,2, 肖斌,1Cloning and Functional Analysis of CsWRKYIIcs Transcription Factors in Tea Plant
XIAO LuoDan1, TANG Lei1, WANG WeiDong1, GAO YueFang1, HUANG YiFan1, MENG Yang1, YANG YaJun,1,2, XIAO Bin,1通讯作者:
责任编辑: 杨鑫浩
收稿日期:2019-12-13网络出版日期:2020-06-16
| 基金资助: |
Received:2019-12-13Online:2020-06-16
作者简介 About authors
肖罗丹,E-mail:1023279673@qq.com。

摘要
关键词:
Abstract
Keywords:
PDF (6550KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
肖罗丹, 唐磊, 王伟东, 高岳芳, 黄伊凡, 孟阳, 杨亚军, 肖斌. 茶树CsWRKYIIcs转录因子的克隆及功能分析[J]. 中国农业科学, 2020, 53(12): 2460-2476 doi:10.3864/j.issn.0578-1752.2020.12.013
XIAO LuoDan, TANG Lei, WANG WeiDong, GAO YueFang, HUANG YiFan, MENG Yang, YANG YaJun, XIAO Bin.
0 引言
【研究意义】茶树(Camellia sinensis (L.)O. kuntze)起源于我国西南地区,是一种重要的叶用经济作物[1]。全国的茶树种植分为四大茶区,跨度大、范围广,茶树在生长过程中易遭受高温、高盐、干旱等非生物胁迫的伤害,如干旱胁迫使茶树叶片脱水变枯,导致茶氨酸、儿茶素等含量减少,严重影响其品质成分的积累,降低经济效益[2]。因此,鉴定出茶树中的抗性基因,对利用分子育种技术定向筛选和培育抗性品种有重要研究价值。【前人研究进展】WRKY是植物重要的转录因子家族之一,因具有高度保守的WRKY结构域得名。根据不同数量的WRKY结构域和不同类型的锌指结构将WRKY分成I、II、III家族,I家族有2个WRKY结构域和C2H2(C-X4-5-C-X22-23-H-X-H)型锌指结构;II家族有1个WRKY结构域及与I家族相同的锌指结构类型;III家族有1个WRKY结构域,且含与I、II家族不同的C2HC(C-X7-C-X23-H-X-C)型锌指结构。依据系统发育分析和氨基酸序列差异,II家族可进一步分为IIa—e等5个亚家族,其中IIc亚家族具有1个高度保守的WRKY结构域和C-X4-C-X22-23-H-X-H型锌配体[3,4,5]。近年来,大量研究表明,植物中不同WRKY家族成员受病菌、干旱、高盐、高温和外源激素等诱导表达,参与多种植物对生物或非生物胁迫的抗性[6,7,8,9,10],其中IIc亚家族成员也参与对多种胁迫的响应过程,如拟南芥AtWRKY8、AtWRKY48、AtWRKY50、AtWRKY51和AtWRKY57对茉莉酸和水杨酸介导的信号通路有应答作用[11],棉花GhWRKY33、GhWRKY39、GhWRKY93、GhWRKY110和GhWRKY114参与低温、盐碱、干旱、外源脱落酸(ABA)等非生物胁迫的响应[12]等。【本研究切入点】目前,茶树中的WRKY基因家族已有报道[13,14,15],研究了不同WRKY的功能[16,17,18,19,20,21,22],表明WRKY与茶树的生长发育和胁迫响应密切相关。茶树的WRKY家族基因众多,笔者课题组通过对茶树高盐、干旱和高温胁迫下转录组数据分析,发现多个IIc亚家族基因差异表达,推测其在茶树响应非生物胁迫的过程中具有重要作用。【拟解决的关键问题】基于茶树基因组数据库(http://tpia.teaplant.org/)中注释的WRKY序列设计特异性引物,从‘陕茶1号’茶树中克隆CsWRKYIIcs的cDNA序列,并进行一系列的生物信息学分析,探究不同组织和胁迫下的表达模式,验证其转录活性,为后续深入研究抗逆机理提供理论参考。1 材料与方法
试验于2018—2019年在西北农林科技大学园艺学院茶学实验室进行。1.1 植物材料、生长条件和胁迫处理
2018年6月底于人工气候室对‘陕茶1号’一年生扦插苗进行水培预培养,生长条件:昼夜温度25℃/20℃,相对湿度70%—80%,光强度300 μmol·m-2·s-1,光周期为昼夜12 h/12 h。8月底时茶苗生长状况良好且萌发出新芽,选取生长势一致的健壮茶苗随机分成4组,每组30株,分别置于4个水培箱中进行高盐、模拟干旱、高温及外源脱落酸胁迫处理。高盐处理:将茶苗的根部全部浸入含200 mmol?L-1 NaCl的溶液中;模拟干旱处理:将茶苗的根部全部浸入含20% PEG 6000的溶液中;高温处理:茶苗置于40℃的人工气候室;外源脱落酸处理:将ABA(少量95%乙醇溶解)溶于蒸馏水配制成100 μmol?L-1的溶液,均匀喷施于茶苗叶片。每个处理在0、1、2、4、8、12、24和48 h时取不同株的芽下二至三叶3—5片混匀分成3份,液氮速冻于-80℃冰箱备用。另从多株未处理的茶苗中,取初开的花朵、未木质化的嫩茎、嫩根和顶芽下第二叶各0.1 g用于组织表达分析,材料液氮速冻于-80℃冰箱保存[23,24]。1.2 茶树CsWRKYIIcs的克隆
基于茶树基因组数据库(http://tpia.teaplant.org/)中注释的WRKY序列,以拟南芥IIc亚家族(https:// www.arabidopsis.org/)的序列作为查询序列,经本地BLAST检索及冗余性检查后,获得9个CsWRKYIIcs亚家族基因。利用在线IDT网站(https://sg.idtdna. com/calc/analyzer)从基因序列起始密码子前和终止密码子后的非编码区设计特异性引物(表1),以‘陕茶1号’叶片cDNA为模板进行RT-PCR扩增,反应程序为94℃ 5 min;94℃ 30 s,58℃ 30 s,72℃ 70 s,35个循环;72℃延伸7 min。将胶回收得到的PCR产物连接至pMD-19T克隆载体,并转化大肠杆菌DH5α,挑选阳性克隆送北京奥科鼎盛生物科技有限公司测序。Table 1
表1
表1克隆、荧光定量和构建酵母载体引物
Table 1
| 引物 Primer | 上游序列(5′-3′) Upstream primer | 下游序列(5′-3′) Downstream primer | 作用 Function |
|---|---|---|---|
| CsWRKYIIc1 | ATC CTT CCA AAC GAT GAC ATT | ACCATTGTAAGTTGTACACATGG | 克隆 Cloning |
| CsWRKYIIc2 | TGAAGAAGAATCTGTGCTCCT | TCTAAAGATAACCCAACAAACCTC | |
| CsWRKYIIc3 | AGAAACTGCACTTGAAGGAGCT | CTCAACCTCAAGACCAGTCAGT | |
| CsWRKYIIc4 | TGATGTAGAAGAAATTGGAGTTGTG | ACATACCAGTAATTGGGTTTCCA | |
| CsWRKYIIc5 | TGAGATAACGCGTAGTCCCAATAG | CTGTGGTGAATTAGTTTAGTGCAT | |
| CsWRKYIIc6 | CGTGGTGGCTACAGAGACAT | GTACTTACGTGAGACTGGTTAGCC | |
| CsWRKYIIc7 | TTCATACTTTGCTCTCTCCCTT | TGAGTTGTTCTTGTACAAAGAGGT | |
| CsWRKYIIc8 | AGAGACCTTAGACAAATCTTCCTGT | ATAAAATTAGCAATTGAAAGGGGCT | |
| CsWRKYIIc9 | TGACCTTAGAGTCAAACCTATCT | TATGAATTGTCCCATACCTGACT | |
| qRT- CsWRKYIIc1 | GAGGAAATATGGACAGAAAGCTG | GCATTCCTTCGTAAGTTGTCAC | 荧光定量qRT-PCR |
| qRT- CsWRKYIIc2 | GAATCATGCCAGAAAGTGCC | GGTGTTGGGTTGTGAAAATAGG | |
| qRT- CsWRKYIIc3 | TCCACAACCTCAATTCCAACCG | AGCAAAGACGATGACAGTGA | |
| qRT- CsWRKYIIc4 | ACCACAATCTCAACCTCAACC | CGACACAATGACAGAGCAAAAG | |
| qRT- CsWRKYIIc5 | CAGCATTGTCACCATACAGTTG | AGGCAGCTTCATGATTGACTAG | |
| qRT- CsWRKYIIc6 | AAGCCACTTGACTCCTTCAG | TGGGAAGTTGAGGATTTGGC | |
| qRT- CsWRKYIIc7 | TTCGGGTTCATGGACTTACTG | AGTCATGGGCTGGTTATTCAAG | |
| qRT- CsWRKYIIc8 | AGGTCGTTTGAAGATCCATCAG | CCACATTACCCCTCAGAGTTG | |
| qRT- CsWRKYIIc6 | TCGATCTCTTCCTCGTCTACTG | CCTCCATCTTCCACCTCTTTT | |
| qRT- CsWRKYIIc7 | GCCATCTTTGATTGGAATGG | GGTGCCACAACCTTGATCTT | |
| qRT- CsWRKYIIc8 | AGGTCGTTTGAAGATCCATCAG | CCACATTACCCCTCAGAGTTG | |
| qRT- CsWRKYIIc9 | TCGATCTCTTCCTCGTCTACTG | CCTCCATCTTCCACCTCTTTT | |
| qRT- Csβ-actin | GCCATCTTTGATTGGAATGG | GGTGCCACAACCTTGATCTT | |
| pGBKT7-CsWRKYIIc1 | TCAGAGGAGGACCTGCATATGATGGACAACTACTCATCAAT | CGGCCTCCATGGCCATATGAAATGCCGTGTAAATTTG | 构建酵母载体 Constructing yeast vectors |
| pGBKT7-CsWRKYIIc2 | TCAGAGGAGGACCTGCATATGATGGAGAAGAAGAAACAGGA | CGGCCTCCATGGCCATATGCTCTTCTTTTGGCTCCTT | |
| pGBKT7-CsWRKYIIc3 | TCAGAGGAGGACCTGCATATGATGCTTGTTGTTGTGAGTGA | CGGCCTCCATGGCCATATGCTCGGGTTTTGCCTCCTT | |
| pGBKT7-CsWRKYIIc4 | TCAGAGGAGGACCTGCATATGATGGAGAGTAAAGAAGCTGT | CGGCCTCCATGGCCATATGGTCATCCTTTGTAACAAG | |
| pGBKT7-CsWRKYIIc5 | TCAGAGGAGGACCTGCATATGATGGATGAGAACGACAGAGT | CGGCCTCCATGGCCATATGTTGATTGCGCATTCCAGG | |
| pGBKT7-CsWRKYIIc6 | TCAGAGGAGGACCTGCATATGATGGATGATGATGATAAGGA | CGGCCTCCATGGCCATATGCTGATTGCGCATTCCAGG | |
| pGBKT7-CsWRKYIIc7 | TCAGAGGAGGACCTGCATATGATGGAGAGGAAACAAGCTGT | CGGCCTCCATGGCCATATGTTTGATGAGCCAAATTTC | |
| pGBKT7-CsWRKYIIc8 | TCAGAGGAGGACCTGCATATGATGTCTGATGAACACAGAGA | CGGCCTCCATGGCCATATGTGGCTCTTGTTTAAGAAA | |
| pGBKT7-CsWRKYIIc9 | TCAGAGGAGGACCTGCATATGATGTCTGATGAACCAGGAGG | CGGCCTCCATGGCCATATGTGGCTCTTGTTTAAAAAA |
新窗口打开|下载CSV
1.3 生物信息学分析
在ORF finder网站(http://www.bioinformatics.org/ sms2/orf_find.html)分析开放阅读框。利用ExPASy网站(https://web.expasy.org/)分析理化性质。在WOLF PSORT网站(https://wolfpsort.hgc.jp/)进行亚细胞定位预测,并利用cNLS Mapper网站(http://nls-mapper. iab.keio.ac.jp/cgi-bin/NLS_Mapper_form.cgi)进行核定位信号预测。利用Meme suite 5.0.5(http://meme- suite.org/)在线预测motif基序。DNAMAN 6.0.3.99进行氨基酸序列比对,并用WebLogo(http://weblogo)生成保守域序列logo图。利用MEGA7软件中邻接法(NJ)构建系统进化树,bootstrap值设置为1 000,并利用EvolView(http://120.202.110.254:8280/ evolview/)修饰进化树。利用GSDS2.0网站构建外显子-内含子结构(http://gsds.cbi.pku.edu.cn/)。截取基因ATG上游约2 000 bp启动子区域,利用PlantCARE(http://bioinformatics.psb.ugent.be/webtools/plantcare/html/)预测顺式作用元件。1.4 实时荧光定量分析
采用CTAB法[24]提取茶树不同组织及胁迫处理的总RNA,保存于-80℃备用。用5×All-In-One RT Master Mix试剂盒(abm,加拿大)合成cDNA第一链作为模板,茶树基因CsActin作为内参[16],设计9个CsWRKYIIcs的定量引物(表1),按照Eva Green 2×qPCR Master Mix-ROX试剂盒(abm,加拿大)使用说明在QuantStudio?5荧光定量PCR仪进行qRT-PCR分析,反应条件为95℃ 3 min;95℃ 30 s,60℃ 70 s,40个循环。每个样品设3个技术重复,用2-ΔΔCT法分析数据[25],用SPSS 26统计分析,GraphPad Prism 8.0.1作图。1.5 转录活性验证
利用NovoRec? Plus PCR一步定向克隆试剂盒(Novoprotein,中国)分别将9个茶树CsWRKYIIcs的cDNA序列融合至pGBKT7表达载体上的GAL4 DNA结合域,引物见表1,构建pGBKT7-CsWRKYIIcs重组表达载体。通过PEG/LiAc法将重组表达载体转入Y2HGold酵母感受态细胞中,在SD/-Trp缺陷固体培养基上30℃倒置培养3—5 d,观察酵母菌落生长情况。挑取单菌落接种于SD/-Trp缺陷液体培养基中,在30℃下250 r/min振荡培养约24 h,取3 μL点在SD/-Ade/-His及含X-α-gal的SD/-Ade/-His缺陷固体培养基上,于30℃倒置培养3—5 d,根据菌落生长情况及是否变蓝来验证转录活性。以pGBKT7-53+pGADT7-T作为阳性对照,pGBKT7作为阴性对照[17,19]。2 结果
2.1 茶树CsWRKYIIcs的克隆及理化性质分析
以‘陕茶1号’cDNA为模板进行RT-PCR扩增,得到9条大小在500—1 000 bp不等的特异性条带(图1),将其纯化后分别连接到pMD-19T克隆载体,挑取阳性单克隆测序,获得9条CsWRKYIIcs的cDNA序列,依次命名为CsWRKYIIc1—CsWRKYIIc9,用于后续试验分析。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1CsWRKYIIcs的RT-PCR扩增
Fig. 1RT-PCR amplification of CsWRKYIIcs
M: Marker; W1: CsWRKYIIc1; W2: CsWRKYIIc2; W3: CsWRKYIIc3; W4: CsWRKYIIc4; W5: CsWRKYIIc5; W6: CsWRKYIIc6; W7: CsWRKYIIc7; W8: CsWRKYIIc8; W9: CsWRKYIIc9
理化性质分析表明,9个茶树CsWRKYIIcs的开放阅读框长度分别为561、960、936、978、897、912、720、1 008和969 bp,分别编码186、319、311、325、298、303、239、335、322个氨基酸。蛋白质相对分子质量为21.098(CsWRKYIIc1)—36.952 kD(CsWRKYIIc8)。CsWRKYIIc1、CsWRKYIIc4、CsWRKYIIc7的理论等电点大于7.0,是碱性蛋白;而CsWRKYIIc2、CsWRKYIIc3、CsWRKYIIc5、CsWRKYIIc6、CsWRKYIIc8、CsWRKYIIc9的理论等电点小于7.0,是酸性蛋白。平均疏水性指标均为负值,说明它们是亲水性蛋白。另外,核定位值区间及亚细胞定位预测显示9个基因均定位于细胞核(表2)。
Table 2
表2
表2CsWRKYIIcs的理化性质分析
Table 2
| 转录因子 Transcription factor | 基因ID Gene ID | 开放阅读框 Open Reading Frame | 氨基酸数量Amino acids number | 相对分子质量 Molecular weight | 理论等电点 Isoelectric point | 平均疏水性Grand average of hydropathy | 核定位预测值 NLSs prediction score | 亚细胞定位预测Subcellular localization prediction |
|---|---|---|---|---|---|---|---|---|
| CsWRKYIIc1 | TEA006586.1 | 561 | 186 | 21.098 | 9.28 | -0.854 | 5.6 | 细胞核 Nucleus |
| CsWRKYIIc2 | TEA028473.1 | 960 | 319 | 35.606 | 6.83 | -0.872 | 4.6 | 细胞核 Nucleus |
| CsWRKYIIc3 | TEA008513.1 | 936 | 311 | 34.324 | 6.54 | -0.692 | 7.7 | 细胞核 Nucleus |
| CsWRKYIIc4 | TEA017544.1 | 978 | 325 | 36.203 | 8.21 | -0.602 | 4.4 | 细胞核 Nucleus |
| CsWRKYIIc5 | TEA001162.1 | 897 | 298 | 32.070 | 5.79 | -0.790 | 4.3 | 细胞核 Nucleus |
| CsWRKYIIc6 | TEA022377.1 | 912 | 303 | 33.502 | 5.49 | -0.894 | 4.3 | 细胞核 Nucleus |
| CsWRKYIIc7 | TEA007197.1 | 720 | 239 | 27.123 | 8.06 | -0.789 | 4.5 | 细胞核 Nucleus |
| CsWRKYIIc8 | TEA023233.1 | 1008 | 335 | 36.952 | 6.00 | -0.840 | 7.9 | 细胞核 Nucleus |
| CsWRKYIIc9 | TEA001873.1 | 969 | 322 | 35.717 | 6.31 | -0.762 | 5.5 | 细胞核 Nucleus |
新窗口打开|下载CSV
2.2 CsWRKYIIcs的序列比对分析
9个茶树CsWRKYIIcs蛋白的氨基酸序列比对结果显示(图2-A),9个蛋白均具有一段高度保守的WRKYGQK序列,CsWRKYIIc7的C2H2型锌指基序缺少“H-X-H”在内的部分氨基酸残基,其他蛋白均具有完整的C-X4-C-X23-H-X-H型锌配体。此外,WebLogo分析WRKYGQK序列及C2H2型锌指结构出现频率高(图2-B),进一步验证了WRKY结构域和锌指结构的高度保守性。图2
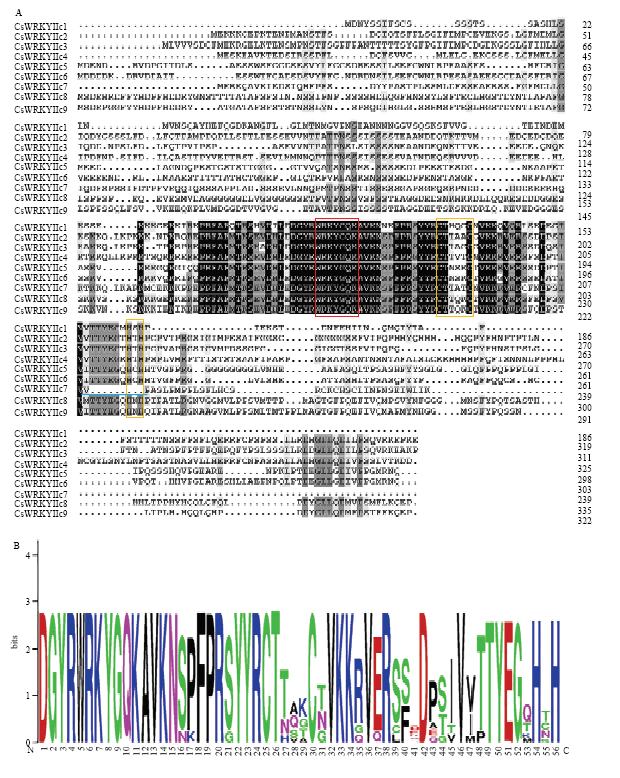 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2CsWRKYIIcs的氨基酸序列比对分析
A:红色方框表示WRKYGQK序列,橙色方框表示C2H2型锌指结构,蓝色下划线表示锌指结构缺失部分
B:纵坐标表示氨基酸的保守性,字母高度表示出现频率(以位为单位测量);横坐标表示该氨基酸在序列中的位置
Fig. 2Amino acid sequence alignment of CsWRKYIIcs
The red box represents WRKYGQK sequence and the orange box represents the C2H2 zinc finger structure and the blue underline indicates the missing part of the zinc finger structure
The vertical coordinate represents the conservatism of amino acids and the letter height represents the frequency of occurrence (measured in bits); The horizontal coordinate represents the position of the amino acids in the sequences
2.3 进化树及保守基序分析
9个茶树CsWRKYIIcs亚家族成员与拟南芥AtWRKY家族成员(https://www.arabidopsis.org/)系统进化树分析见图3,茶树CsWRKYIIcs与拟南芥IIc亚家族成员聚集,除CsWRKYIIc1外,其他茶树成员两两成对,说明9个CsWRKYIIcs均属于IIc亚家族,并且在茶树中可能发生了复制。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3CsWRKYIIcs和AtWRKYs的进化树分析
Fig. 3Phylogenetic analysis of CsWRKYIIcs and AtWRKYs
9个茶树CsWRKYIIcs与水稻(7条)、玉米(3条)、葡萄(8条)和拟南芥(12条)等不同物种的WRKYIIcs亚家族成员构建系统进化树分析显示(图4-A),39个成员被分为4个组,每组都包含茶树的CsWRKYIIcs亚家族成员。其中CsWRKYIIc8和CsWRKYIIc9与VvWRKY33和VvWRKY37关系更近;CsWRKYIIc2与VvWRKY16亲缘关系最近,并与CsWRKYIIc3及成对的CsWRKYIIc4和CsWRKYIIc7聚类;CsWRKYIIc5和CsWRKYIIc6与VvWRKY1和AtWRKY57关系更近;CsWRKYIIc1与VvWRKY3关系最近。另外,不同物种中的WRKYIIc亚家族成员有相似的保守基序(图4-B)。motif1出现在39个WRKYIIcs亚家族成员中;除了茶树CsWRKYIIc7外,其他38个成员均拥有motif2;4个组中都出现motif3和motif5,第1组、2组、3组中均含motif4。此外,同组成员拥有更多相同基序,如motif6和motif9只出现在第2组。而有的基序只出现在某一组中,如motif7只存在第1组,motif10出现在第3组,motif8只出现在第4组,说明WRKYIIcs在进化过程中不同组成员的保守基序有一定的差异。
图4
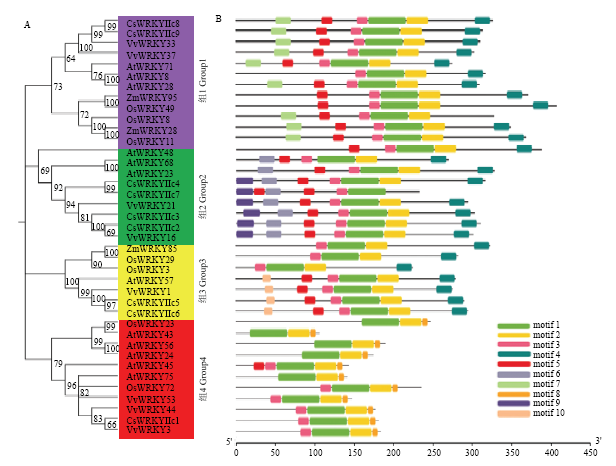 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4不同物种WRKYIIcs的进化树和保守基序分析
At:拟南芥 Arabidopsis thaliana;Cs:茶树 Camellia sinensis;Zm:玉米 Zea mays;Os:水稻 Oryza sativa;Vv:葡萄 Vitis vinifera 4个物种的WRKYIIcs蛋白ID号ID numbers of WRKYIIcs proteins from four species : AtWRKY8 (AT5G46350), AtWRKY24 (AT5G41570), AtWRKY23 (AT2G47260), AtWRKY28 (AT4G18170), AtWRKY43 (AT2G46130), AtWRKY45 (AT3G01970), AtWRKY48 (AT5G49520), AtWRKY56 (AT1G64000), AtWRKY57 (AT1G69310), AtWRKY68 (AT3G62340), AtWRKY71 (AT1G29860), AtWRKY75 (AT5G13080), ZmWRKY28 (Zm00001d011413), ZmWRKY85 (Zm00001d018656), ZmWRKY95 (Zm00001d043950), VvWRKY1 (CBI27268.3), VvWRKY3 (CBI27681.3), VvWRKY16 (CBI22862.3), VvWRKY21 (CBI36970.3), VvWRKY33 (CBI30827.3), VvWRKY37 (CBI22108.3), VvWRKY44 (CBI21329.3), VvWRKY53 (CBI15677.3), OsWRKY3 (Os03g0758000), OsWRKY8 (Os05g0583000), OsWRKY11 (Os01g0626400), OsWRKY23 (Os01g0734000), OsWRKY29 (Os07g0111400), OsWRKY49 (Os05g0565900), OsWRKY72 (Os11g0490900)
Fig. 4Analysis of phylogenetic tree and motifs of WRKYIIcs from different species
2.4 CsWRKYIIcs的结构分析
9个茶树CsWRKYIIcs亚家族成员分为4个组(图5-A)。外显子-内含子结构分析显示9个基因的编码区包含1—2个内含子和2—3个外显子(图5-B),表明它们在进化过程中可能行使相似的功能。图5
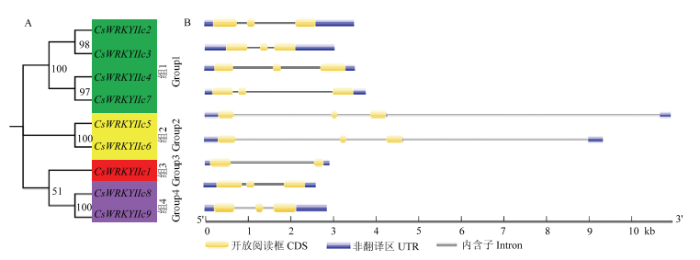 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5CsWRKYIIcs的结构分析
Fig. 5Structure analysis of CsWRKYIIcs
2.5 顺式作用元件预测
9个茶树CsWRKYIIcs的启动子区域预测到多个与非生物胁迫相关的顺式作用元件(表3),它们重复出现在一个或多个启动子区域,可能影响CsWRKYIIcs对不同非生物胁迫的应答。值得探讨的是,每个启动子序列中高频出现响应脱落酸的作用元件ABRE及受干旱诱导的作用元件MYB,在多个启动子区域频繁出现与干旱胁迫相关的作用元件MBS和MYC,由此推测9个CsWRKYIIcs可能与干旱胁迫密切相关。Table. 3
表3
表3顺式作用元件预测
Table. 3
| 顺式作用元件 Cis-element | 元件数量 Cis-element number | 功能 Function | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| W1 | W2 | W3 | W4 | W5 | W6 | W7 | W8 | W9 | ||
| ABRE | 4 | 3 | 3 | 7 | 4 | 10 | 2 | 3 | 2 | 参与脱落酸反应性的顺式作用元件 Cis-acting elements involved in abscisic acid reactivity |
| ARE | 5 | 2 | 3 | 1 | 1 | 2 | 3 | 2 | 8 | 厌氧诱导所必需的顺式调节元件 Cis-regulating elements necessary for anaerobic Induction |
| Box 4 | 4 | 5 | 2 | 1 | 4 | 7 | 5 | 4 | 光响应DNA模块部分 Optical response DNA module part | |
| CGTCA-motif | 1 | 1 | 1 | 3 | 4 | 4 | 5 | 参与MEJA反应的顺式调节元件 Cis-regulating elements involved in MEJA reaction | ||
| ERE | 4 | 1 | 3 | 1 | 4 | 2 | 参与乙烯反应的顺式调节元件 Cis-regulating elements involved in ethylene reaction | |||
| G-box | 3 | 2 | 1 | 4 | 2 | 4 | 1 | 3 | 光应答元件Cis-regulating elements involved in light reaction | |
| MBS | 2 | 1 | 1 | 2 | 1 | 1 | 1 | 干旱诱导MYB结合位点Drought-induced MYB binding site | ||
| MYB | 2 | 6 | 3 | 1 | 2 | 1 | 3 | 2 | 4 | 参与干旱、高盐和低温诱导Drought/high salt/low temperature-induced |
| MYC | 2 | 7 | 3 | 2 | 5 | 2 | 4 | 2 | 干旱应答元件 Cis-regulating elements involved in drought reaction | |
| STRE | 3 | 4 | 4 | 5 | 1 | 2 | 参与热响应 Heat stress responsiveness | |||
| TGACG-motif | 1 | 1 | 1 | 3 | 1 | 4 | 4 | 5 | 参与MEJA反应的顺式调节元件 Cis-regulating elements involved in MEJA reaction | |
| W box | 4 | 2 | 3 | 防卫和胁迫应答元件 Cis-acting elements involved in defense and stress response | ||||||
新窗口打开|下载CSV
2.6 CsWRKYIIcs的表达分析
qRT-PCR结果显示(图6),CsWRKYIIc1、CsWRKYIIc8和CsWRKYIIc9在花中优势表达,根中次之,在茎和叶中的表达量最低;而CsWRKYIIc2和CsWRKYIIc3、CsWRKYIIc4和CsWRKYIIc7在根中表达量显著,花中次之,在茎和叶中的表达量相对较低;CsWRKYIIc5在根中的表达量最高,茎中次之,在叶和花中相对较低;CsWRKYIIc6在根中的表达显著高于茎、叶和花。这9个CsWRKYIIcs在根和花中的表达量明显高于茎和叶,表明其组织表达存在差异。图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6CsWRKYIIcs在不同组织中的表达分析
R:根 Root;S:茎 Stem;L:叶 Leaf;F:花 Flower不同小写字母表示差异显著(P<0.05)
Fig. 6Expression analysis of CsWRKYIIcs in different tissues
Different lowercase letters indicate significant difference (P<0.05)
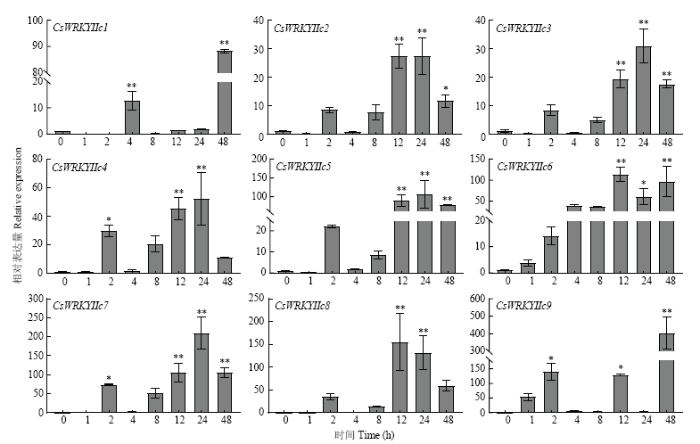
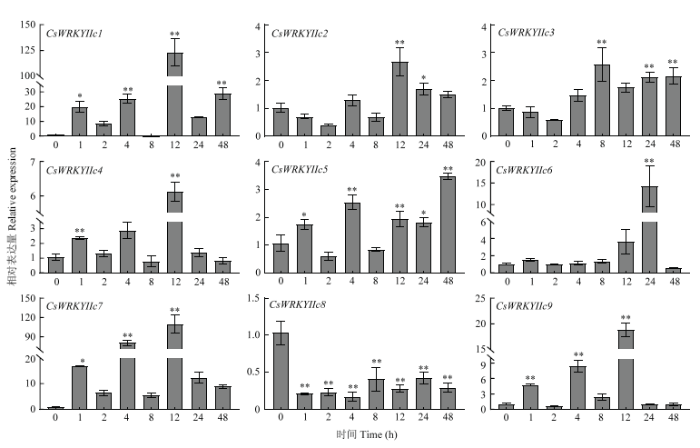
在干旱、ABA、高盐、高温等不同胁迫处理下,CsWRKYIIcs主要呈不同的表达模式。干旱胁迫下,不同CsWRKYIIcs被阶段性诱导(图7),CsWRKYIIc5和CsWRKYIIc6的表达量先升高后降低,在8 h时达到峰值,达0 h的8倍和10倍;其他基因在1—4 h受干旱影响较小,仅有CsWRKYIIc1、CsWRKYIIc3和CsWRKYIIc7在1 h明显上调;而8—48 h时间段总体表达水平高于前期,其中CsWRKYIIc2和CsWRKYIIc3均显著上调,CsWRKYIIc1在48 h的表达量为对照的36倍。在ABA诱导下,CsWRKYIIcs均强烈地上调表达(图8),除CsWRKYIIc1和CsWRKYIIc7稍显迟缓外,其他基因均能在1 h时快速作出反应,在随后时间段也有较高表达水平,其中CsWRKYIIc1在48 h表达量最高,达处理前的60倍以上,CsWRKYIIc7在4和24 h均达对照的70倍。在盐处理下,不同CsWRKYIIcs的表达水平不等(图9),CsWRKYIIc1仅在4和48 h时被显著诱导;CsWRKYIIc2和CsWRKYIIc3、CsWRKYIIc5和CsWRKYIIc6、CsWRKYIIc8于1—8 h变化较小,在12 h后表达量显著增加;而CsWRKYIIc4和CsWRKYIIc7在24 h表达量达最高,其中CsWRKYIIc7表达量为处理前200倍。CsWRKYIIc9分别在2、12和48 h上调显著,表达量最高可达处理前的400倍。高温胁迫下CsWRKYIIcs的表达也存在差异(图10),CsWRKYIIc2和CsWRKYIIc3在处理前期无显著变化,分别在12和8 h时上调表达,后期表达量小幅度下降;CsWRKYIIc5在各阶段上调表达,除2和8 h外;CsWRKYIIc6仅在24 h时上调表达;CsWRKYIIc1、CsWRKYIIc4和CsWRKYIIc7、CsWRKYIIc9均在12 h时表达量达到最高,特别是CsWRKYIIc1和CsWRKYIIc7可达对照的100倍;而CsWRKYIIc8在高温下的表达受到强烈的抑制。这些结果表明,CsWRKYIIcs能快速应答ABA信号,不同程度地响应干旱、高盐、高温胁迫,高盐胁迫下相对表达水平高于干旱、高温胁迫,表明其可能对盐胁迫有更高的敏感性。其中CsWRKYIIc1和CsWRKYIIc7表达量更显著,可能参与了茶树对这些胁迫的抗逆反应。
图7
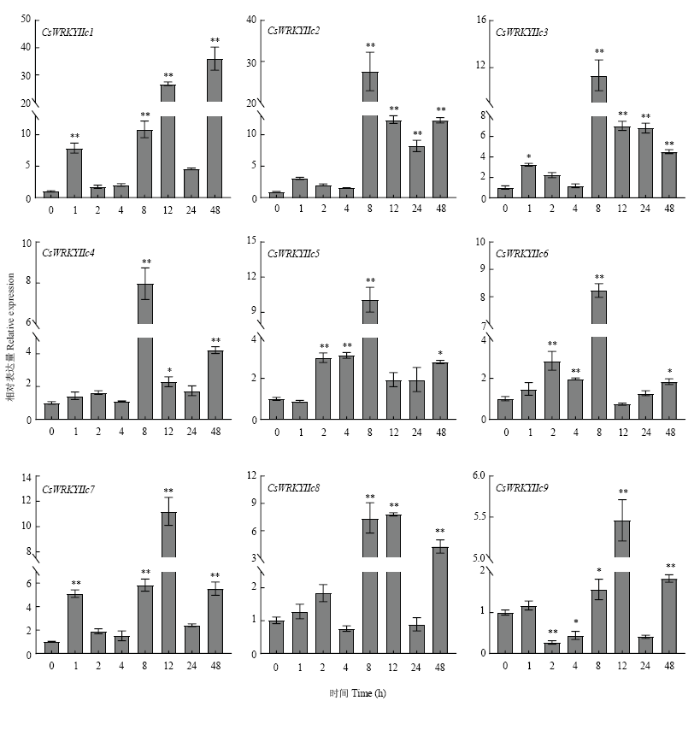 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7CsWRKYIIcs在干旱胁迫下的表达分析
*表示差异显著(P<0.05),**表示差异极显著(P<0.01)。下同
Fig. 7Expression analysis of CsWRKYIIcs under drought stress
*shows significant difference (P<0.05), **indicates extremely significant difference (P<0.01). The same as below
图8
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8CsWRKYIIcs在ABA诱导下的表达分析
Fig. 8Expression analysis of CsWRKYIIcs under ABA-induced
图9
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图9CsWRKYIIcs在盐胁迫下的表达分析
Fig. 9Expression analysis of CsWRKYIIcs under NaCl stress
图10
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图10CsWRKYIIcs在高温胁迫下的表达分析
Fig. 10Expression analysis of CsWRKYIIcs under heat stress
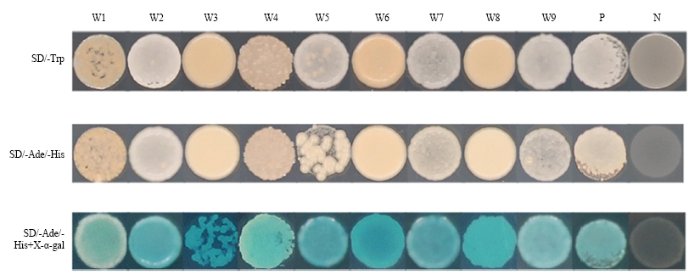
2.7 转录活性试验结果
利用酵母试验研究CsWRKYIIcs蛋白的转录活性(图11),所有重组表达载体、阳性对照及阴性对照均能在SD/-Trp培养基上生长;所有重组表达载体及阳性对照在SD/-Ade/-His培养基上正常生长,而阴性对照生长受抑制;在加入X-α-gal的培养基上,阴性对照长势微弱且无变蓝,但所有重组表达载体及阳性对照菌斑均呈现不同程度的蓝色,表明9个茶树CsWRKYIIcs重组蛋白都具有转录激活活性。图11
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图11CsWRKYIIcs的转录活性分析
W1: pGBKT7-CsWRKYIIc1; W2: pGBKT7-CsWRKYIIc2; W3: pGBKT7-CsWRKYIIc3; W4: pGBKT7-CsWRKYIIc4; W5: pGBKT7-CsWRKYIIc5; W6: pGBKT7-CsWRKYIIc6; W7: pGBKT7-CsWRKYIIc7; W8: pGBKT7-CsWRKYIIc8; W9: pGBKT7-CsWRKYIIc9; P: 阳性对照 Positive; N: 阴性对照 Negative
Fig. 11Transcription activity analysis of CsWRKYIIcs
3 讨论
WRKY转录因子在植物生长发育、防御调控和胁迫应答中发挥重要作用。有研究表明植物经常发生锌指结构丢失事件,如菠萝的AcWRKY23和‘铁观音’的CsWRKY39均出现锌指结构缺失[20,26],本研究也发现CsWRKYIIc7缺少锌指残基,推测缺失可能导致其功能差异,还需要进一步研究。基因结构分析显示9个CsWRKYIIcs含有1—2个内含子和2—3个外显子,苹果MdWRKYIIcs亚家族成员也有类似现象[27],说明它们结构组成之间密切联系,可能有相似作用。大量研究表明植物中不同WRKY的组织表达具有特异性,例如,玉米ZmWRKY58在根中表达较强[28],陆地棉GhWRKY42主要在茎中表达[29],菊花CmWRKY40在叶片中表达量最高[30]。本研究中‘陕茶1号’CsWRKYIIcs在不同组织中的表达也有一定差异,根和花中的表达量显著高于茎和叶,推测其可能在茶树的根和花中发挥重要的生物学作用。前人研究证实不同茶树品种间的WRKY存在较大差异[14],如‘龙井43号’的CsWRKY3在根中表达最强[18],CsWRKY7在老叶和根中都优势表达[22],‘铁观音’的CsWRKY48在根和茎中的表达强于叶和花,而CsWRKY6和CsWRKY31分别在老叶和花中优势表达[21],意味着不同的茶树品种和生长环境可能导致WRKY在不同组织中的表达差异。
近年来,众多研究表明植物的WRKY参与响应多种非生物胁迫[31,32,33],本研究结果也说明CsWRKYIIcs与茶树的逆境胁迫响应密切相关。在干旱胁迫下,CsWRKYIIcs被阶段性诱导表达,与菊花[30]等植物有类似的表达模式,说明它们参与干旱胁迫的响应。前人证实激活AtWRKY57表达可上调ABA3和NCED3,提高ABA水平增强拟南芥的抗旱性[34]。CsWRKYIIc5和CsWRKYIIc6受干旱胁迫于8 h时达对照的8倍和10倍,系统进化树表明其与AtWRKY57亲缘关系相近,推测其可能具有相似的生物学功能。前人研究表明WRKY是ABA信号途径的关键基因,如CsWRKY2通过参与到ABA下游信号途径中响应干旱胁迫[16];过表达GmWRKY16诱导体内ABA合成并调控胁迫相关基因表达从而提高植株的抗性[35]等。本研究结果显示ABA诱导下所有CsWRKYIIcs均上调表达,且反应迅速,这可能与其表达快速而瞬时的特点相关[36]。其中CsWRKYIIc1和CsWRKYIIc7表达量高于其他CsWRKYIIcs,预示着它们可能在ABA信号调控途径中起关键作用。另一方面,从启动子区域预测到多个响应ABA和干旱胁迫的顺式作用元件,推测CsWRKYIIcs可能通过ABA信号途径参与茶树的抗旱调控,将是下一步研究重点。
在盐胁迫下,CsWRKYIIcs被不同程度地诱导,与前人分析结果一致[15],表明其参与了茶树对盐胁迫的响应。有研究证实过量表达ZmWRKY17调节ABA和胁迫相关基因的表达,降低植株对盐胁迫的耐受性[37]。而ZHU等[38]证实葡萄VvWRKY30受盐胁迫强烈诱导,并通过活性氧清除和渗透压积累增强转基因拟南芥的耐盐性。本研究中CsWRKYIIcs被盐胁迫诱导较高水平表达,尤其是CsWRKYIIc7和CsWRKYIIc9可分别达对照的200和400倍,因此推测其可能在茶树抗盐反应中扮演重要角色。另外,WU等[13]分析表明不同茶树品种的WRKY受温度诱导存在差异,但又表现出群体一致性。本试验中CsWRKYIIcs的表达没有出现规律性的上调或下调,其中CsWRKYIIc1、CsWRKYIIc2、CsWRKYIIc4和CsWRKYIIc7、CsWRKYIIc9表达量在12 h达到峰值,尤其是CsWRKYIIc1和CsWRKYIIc7可达100倍,可能与ZmWRKY106和OsWRKY11具有相似的功能,即正向调控植物对高温胁迫的耐受性[9,39]。而CsWRKYIIc8被强烈地抑制,与茶树CsWRKY7和菊花多个CmWRKYs的表达趋势一致[22,30],可能在高温胁迫中起负调控作用。此外,本研究也发现组内基因之间有相似的表达趋势,如干旱胁迫下的CsWRKYIIc2和CsWRKYIIc3、CsWRKYIIc5和CsWRKYIIc6,盐处理下的CsWRKYIIc2和CsWRKYIIc3、CsWRKYIIc4和CsWRKYIIc7,高温胁迫下的CsWRKYIIc4和CsWRKYIIc7等,推测它们在逆境中可能有相似的响应机制,但还需进一步的试验证明。
另一个方面,WRKY是植物重要的转录因子家族,它的一个基本特征是具有转录激活活性,可以通过激活下游基因的表达参与调控作用[40,41]。因此,本文通过酵母试验验证茶树CsWRKYIIcs的转录活性。结果显示9个pGBKT7-CsWRKYIIcs重组表达载体在含X-α-gal的缺陷培养基上呈蓝色,表明它们都具有转录激活活性。前人报道茶树中不同CsWRKYs定位于细胞核内[16,19,22],本研究根据CsWRKYIIcs的核定位预测值和亚细胞定位预测结果,推测它们是定位于细胞核的调控蛋白。另外,研究表明WRKY的N端是表现活性的关键结构[38,42],因此,可进一步验证CsWRKYIIcs序列的活性部位,为后续试验提供一定理论依据。
4 结论
从‘陕茶1号’中克隆获得9个CsWRKYIIcs转录因子,除CsWRKYIIc7锌指基序缺失外,其他8个CsWRKYIIcs均含有1个高度保守的WRKY结构域和C2H2型锌指结构,属于IIc亚家族。9个蛋白均具有转录激活活性,可能作为转录激活因子参与茶树的逆境调控。这些基因受ABA、干旱、高盐和高温胁迫不同程度的诱导表达,而且从启动子序列预测到多个与非生物胁迫相关的作用元件,表明CsWRKYIIcs参与了茶树对多种非生物胁迫的响应;高盐胁迫下的总体表达水平高于干旱、高温胁迫,表明其可能对盐胁迫具有更高的敏感性。在ABA、干旱、高盐和高温处理下,CsWRKYIIc1和CsWRKYIIc7表达量变化显著,可作为茶树逆境候选基因进行深入研究。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
DOI:10.1016/s1360-1385(00)01600-9URLPMID:10785665 [本文引用: 1]

The WRKY proteins are a superfamily of transcription factors with up to 100 representatives in Arabidopsis. Family members appear to be involved in the regulation of various physio-logical programs that are unique to plants, including pathogen defense, senescence and trichome development. In spite of the strong conservation of their DNA-binding domain, the overall structures of WRKY proteins are highly divergent and can be categorized into distinct groups, which might reflect their different functions.
[本文引用: 1]
DOI:10.1186/s13062-015-0076-3URL [本文引用: 1]
DOI:10.3864/j.issn.0578-1752.2013.19.009URL [本文引用: 1]
【Objective】 The objective of this study is to isolate rice WRKY80, to analyze the characteristics of its coding sequence and to investigate its expression patterns in different organs and under pathogen inoculation and hormone application, thus providing a basis for its function identification. 【Method】Specific primer sequences were designed according to the annotated gene Loc_Os03g63810 released in rice genome database. RT-PCR was used to amplify WRKY80 cDNA sequence from RNA pools generated from methyl jasmonate (MeJA)-treated rice leaves. Bioinformatical tools were employed to analyze its deduced protein sequence and cis-elements in its promoter. Northern blot or real-time fluorescence quantification PCR was used to investigate its expression patterns. 【Result】 The obtained cDNA sequence of WRKY80 was 1 392 bp in length, containing an entire open reading frame of 1 164 bp, encoding a polypeptide of 387 amino acid residues consisting of one classic conserved WRKY domain with a zinc finger motif of C2H2, belonging to the WRKY subgroup Ⅱ. WRKY80 possessed an acidic C terminus with consecutive 6 glutamines and 8 threonines, an acidic region possibly responsible for transcription activating activity, and was predicted to be localized in nucleus. WRKY80 shared high identity at the amino acid level with those from monocotyledons Zea mays and Sorghum bicolor. WRKY80 was constitutively expressed in all tested organs. The transcript abundance was relatively higher in leaves, roots and panicles, next was in flowers, and less was in stems and grains. WRKY80 expression was higher in mature leaves and roots than in young leaves and roots, respectively, indicating a developmental stage-related feature. It was rapidly induced by inoculation with Magnaporthe oryzae and Rhizoctonia solani, and also by application of exogenous MeJA and ethephon, whereas salicylic acid exerted no effects on its expression. Consistent with the expression profiles was the prediction result of cis-elements in its promoter. 【Conclusion】WRKY80 possesses structure characteristics as a putative transcription factor. These findings suggest that it may be involved in defense response to the fungal pathogens and also in development regulation by jasmonic acid/ethylene-dependent signaling pathway.
DOI:10.3864/j.issn.0578-1752.2013.19.009URL [本文引用: 1]
【Objective】 The objective of this study is to isolate rice WRKY80, to analyze the characteristics of its coding sequence and to investigate its expression patterns in different organs and under pathogen inoculation and hormone application, thus providing a basis for its function identification. 【Method】Specific primer sequences were designed according to the annotated gene Loc_Os03g63810 released in rice genome database. RT-PCR was used to amplify WRKY80 cDNA sequence from RNA pools generated from methyl jasmonate (MeJA)-treated rice leaves. Bioinformatical tools were employed to analyze its deduced protein sequence and cis-elements in its promoter. Northern blot or real-time fluorescence quantification PCR was used to investigate its expression patterns. 【Result】 The obtained cDNA sequence of WRKY80 was 1 392 bp in length, containing an entire open reading frame of 1 164 bp, encoding a polypeptide of 387 amino acid residues consisting of one classic conserved WRKY domain with a zinc finger motif of C2H2, belonging to the WRKY subgroup Ⅱ. WRKY80 possessed an acidic C terminus with consecutive 6 glutamines and 8 threonines, an acidic region possibly responsible for transcription activating activity, and was predicted to be localized in nucleus. WRKY80 shared high identity at the amino acid level with those from monocotyledons Zea mays and Sorghum bicolor. WRKY80 was constitutively expressed in all tested organs. The transcript abundance was relatively higher in leaves, roots and panicles, next was in flowers, and less was in stems and grains. WRKY80 expression was higher in mature leaves and roots than in young leaves and roots, respectively, indicating a developmental stage-related feature. It was rapidly induced by inoculation with Magnaporthe oryzae and Rhizoctonia solani, and also by application of exogenous MeJA and ethephon, whereas salicylic acid exerted no effects on its expression. Consistent with the expression profiles was the prediction result of cis-elements in its promoter. 【Conclusion】WRKY80 possesses structure characteristics as a putative transcription factor. These findings suggest that it may be involved in defense response to the fungal pathogens and also in development regulation by jasmonic acid/ethylene-dependent signaling pathway.
DOI:10.1104/pp.16.00747URLPMID:27268959 [本文引用: 1]
Although necrotrophic pathogens cause many devastating plant diseases, our understanding of the plant defense response to them is limited. Here, we found that loss of function of WRKY57 enhanced the resistance of Arabidopsis (Arabidopsis thaliana) against Botrytis cinerea infection. Further investigation suggested that the negative regulation of WRKY57 against B cinerea depends on the jasmonic acid (JA) signaling pathway. Chromatin immunoprecipitation experiments revealed that WRKY57 directly binds to the promoters of JASMONATE ZIM-DOMAIN1 (JAZ1) and JAZ5, encoding two important repressors of the JA signaling pathway, and activates their transcription. In vivo and in vitro experiments demonstrated that WRKY57 interacts with nuclear-encoded SIGMA FACTOR BINDING PROTEIN1 (SIB1) and SIB2. Further experiments display that the same domain, the VQ motif, of SIB1 and SIB2 interact with WRKY33 and WRKY57. Moreover, transient transcriptional activity assays confirmed that WRKY57 and WRKY33 competitively regulate JAZ1 and JAZ5, SIB1 and SIB2 further enhance these competitions of WRKY57 to WRKY33. Therefore, coordinated regulation of Arabidopsis against B cinerea by transcription activators and repressors would benefit plants by allowing fine regulation of defense.
DOI:10.1016/j.jplph.2018.04.007URLPMID:29689430 [本文引用: 1]
Plant WRKY transcription factors play a vital role in abiotic stress tolerance and regulation of plant defense responses. This study examined AtWRKY11 and AtWRKY17 expression under ABA, salt, and osmotic stress at different developmental stages in Arabidopsis. We used reverse transcriptase PCR, quantitative real-time PCR, and promoter:GUS lines to analyze expression. Both genes were upregulated in response to abiotic stress. Next, we applied the same stressors to seedlings of T-DNA insertion wrky11 and 17 knock-out mutants (single and double). Under stress, the mutants exhibited slower germination and compromised root growth compared with the wild type. In most cases, double-mutant seedlings were more affected than single mutants. These results suggest that wrky11 and wrky17 are not strictly limited to plant defense responses but are also involved in conferring stress tolerance.
DOI:10.3390/ijms19010030URL [本文引用: 2]
DOI:10.3864/j.issn.0578-1752.2019.12.001URL [本文引用: 1]
【Objective】 There are nearly 100 WRKY transcription factor members in rice genome, many of them are involved in plant growth and development, biotic and abiotic stress responses. Molecular biology & bioinformatics lab identified that the expression of OsWRKY68 protein was induced after inoculation with Xanthomonas oryzae pv. oryzae (Xoo) in rice. The aim of this study is attempt to further explore the function of OsWRKY68. 【Method】Rice TP309 samples of different tissues at different developmental stages, including germination, seedling, tillering, booting and flowering stages of root, stem, leaf, sheath, cushion, panicle, anther, husk, seed, abiotic stress (4℃, 44℃, 48℃, submerge, NaCl, PEG, constant light, constant dark) and hormone treatments (abscisic acid, methyl jasmonate, salicylic acid, ethephon) were collected. Total protein were extracted and analyzed by Western blot (WB) systematically using OsWRKY68-specific antibody. The expression patterns of OsWRKY68 protein isolated from different tissues at different developmental stages, and tissues obtained from abiotic stresses and hormone treatments were investigated. RNA interfering vector was constructed and transformed to wildtype TP309 rice variety via Agrobacterium tumefaciens strategy. Identification of transgenic plants were carried out by PCR and WB. The phenotype of OsWRKY68 RNAi transgenic plants were monitored and plant height, tiller number, spike length, spikelet number and seed-setting rate were measured.【Result】By comparing the abundance of OsWRKY68 protein in different tissues, it was found that OsWRKY68 protein was expressed in a constitutive way during the normal growth and development of rice, the abundance of OsWRKY68 protein expressed among different tissues were not varied too much. However, different levels of OsWRKY68 were observed. The expression level of OsWRKY68 in anthers at flowering stage was higher than that in mature panicles, panicle axis and husk. It was not expressed in sheaths at tillering and booting stages, but it was expressed in sheaths at flowering stage. In panicles, the abundance of OsWRKY68 was decreased gradually along with the growth of the young panicle. By investigating the expression patterns of OsWRKY68 protein under abiotic stress and hormone treatments, it was found that the abundance of OsWRKY68 protein decreased steadily under salt stress. The expression of OsWRKY68 protein increased steadily at constant light treatment, a specific band (designated as OsWRKY68 +) with higher molecular weight appeared at three days and enhanced in the following timepoints. After methyl jasmonate (MeJA) and ethephon (ET) treatments, OsWRKY68 + band appeared also and its intensity increased as the treatments continues. Four homozygous OsWRKY68 RNAi transgenic lines (Y316, Y317, Y326 and Y337) were checked by PCR and WB analyses and verified at T3 generation. The abundance of OsWRKY68 protein in RNAi transgenic plants was lower than that in wildtype TP309. Phenotypic investigation revealed significant reduction in plant height, tiller number and seed setting rate in transgenic plants.【Conclusion】Rice OsWRKY68 protein plays an important role in the process of normal growth and development of rice. Knocking down the abundance of OsWRKY68 protein via RNAi affected the normal growth of rice. In addition, the data of expression patterns suggested that the function of OsWRKY68 protein may be involved with salt stress, light, MeJA and ethene-mediated signal transduction pathways.
DOI:10.3864/j.issn.0578-1752.2019.12.001URL [本文引用: 1]
【Objective】 There are nearly 100 WRKY transcription factor members in rice genome, many of them are involved in plant growth and development, biotic and abiotic stress responses. Molecular biology & bioinformatics lab identified that the expression of OsWRKY68 protein was induced after inoculation with Xanthomonas oryzae pv. oryzae (Xoo) in rice. The aim of this study is attempt to further explore the function of OsWRKY68. 【Method】Rice TP309 samples of different tissues at different developmental stages, including germination, seedling, tillering, booting and flowering stages of root, stem, leaf, sheath, cushion, panicle, anther, husk, seed, abiotic stress (4℃, 44℃, 48℃, submerge, NaCl, PEG, constant light, constant dark) and hormone treatments (abscisic acid, methyl jasmonate, salicylic acid, ethephon) were collected. Total protein were extracted and analyzed by Western blot (WB) systematically using OsWRKY68-specific antibody. The expression patterns of OsWRKY68 protein isolated from different tissues at different developmental stages, and tissues obtained from abiotic stresses and hormone treatments were investigated. RNA interfering vector was constructed and transformed to wildtype TP309 rice variety via Agrobacterium tumefaciens strategy. Identification of transgenic plants were carried out by PCR and WB. The phenotype of OsWRKY68 RNAi transgenic plants were monitored and plant height, tiller number, spike length, spikelet number and seed-setting rate were measured.【Result】By comparing the abundance of OsWRKY68 protein in different tissues, it was found that OsWRKY68 protein was expressed in a constitutive way during the normal growth and development of rice, the abundance of OsWRKY68 protein expressed among different tissues were not varied too much. However, different levels of OsWRKY68 were observed. The expression level of OsWRKY68 in anthers at flowering stage was higher than that in mature panicles, panicle axis and husk. It was not expressed in sheaths at tillering and booting stages, but it was expressed in sheaths at flowering stage. In panicles, the abundance of OsWRKY68 was decreased gradually along with the growth of the young panicle. By investigating the expression patterns of OsWRKY68 protein under abiotic stress and hormone treatments, it was found that the abundance of OsWRKY68 protein decreased steadily under salt stress. The expression of OsWRKY68 protein increased steadily at constant light treatment, a specific band (designated as OsWRKY68 +) with higher molecular weight appeared at three days and enhanced in the following timepoints. After methyl jasmonate (MeJA) and ethephon (ET) treatments, OsWRKY68 + band appeared also and its intensity increased as the treatments continues. Four homozygous OsWRKY68 RNAi transgenic lines (Y316, Y317, Y326 and Y337) were checked by PCR and WB analyses and verified at T3 generation. The abundance of OsWRKY68 protein in RNAi transgenic plants was lower than that in wildtype TP309. Phenotypic investigation revealed significant reduction in plant height, tiller number and seed setting rate in transgenic plants.【Conclusion】Rice OsWRKY68 protein plays an important role in the process of normal growth and development of rice. Knocking down the abundance of OsWRKY68 protein via RNAi affected the normal growth of rice. In addition, the data of expression patterns suggested that the function of OsWRKY68 protein may be involved with salt stress, light, MeJA and ethene-mediated signal transduction pathways.
DOI:10.1186/1471-2229-13-188URLPMID:24267479 [本文引用: 1]
BACKGROUND: The WRKY transcription factor is an important member of the stress-related transcription factors, which mediate diverse abiotic stresses in many plants. However, up until now, the number of WRKY members, and the regulatory mechanisms involved in abiotic stress responses in Pak-choi (Brassica campestris ssp. chinensis), remained unknown. RESULTS: We isolated and identified 56 full-length WRKY cDNAs from a Pak-choi stress-induced cDNA library. The 56 putative BcWRKY proteins were divided into three groups based on structural and phylogenetic analyses. A subcellular localization prediction indicated that the putative BcWRKY proteins were enriched in the nuclear region. Experiments involving BcWRKY25 and BcWRKY40 confirmed the prediction. A total of 22 BcWRKYs were differentially expressed in response to at least one stress condition (abscisic acid, cold, salinity, heat, or osmosis) tested on Pak-choi leaves, and a co-expression analysis indicated stress-inducible BcWRKYs co-regulated multiple abiotic stresses. BcWRKY33, BcWRKY40, BcWRKY53, and BcWRKY70 acted as key regulators and played dominant roles within co-regulatory networks of stress-inducible BcWRKYs. CONCLUSIONS: We first isolated and characterized the 56 stress-inducible WRKY transcription factor family members. A total of 22 stress-inducible BcWRKYs found in leaves can co-regulate multiple environmental stresses by integrating the potential mutual interactions of WRKYs in Pak-choi. This information will be valuable when exploring the molecular mechanisms of WRKYs in response to abiotic stresses in plants.
DOI:10.1007/s00438-014-0872-yURLPMID:24942461 [本文引用: 1]
WRKY proteins are major transcription factors involved in regulating plant growth and development. Although many studies have focused on the functional identification of WRKY genes, our knowledge concerning many areas of WRKY gene biology is limited. For example, in cotton, the phylogenetic characteristics, global expression patterns, molecular mechanisms regulating expression, and target genes/pathways of WRKY genes are poorly characterized. Therefore, in this study, we present a genome-wide analysis of the WRKY gene family in cotton (Gossypium raimondii and Gossypium hirsutum). We identified 116 WRKY genes in G. raimondii from the completed genome sequence, and we cloned 102 WRKY genes in G. hirsutum. Chromosomal location analysis indicated that WRKY genes in G. raimondii evolved mainly from segmental duplication followed by tandem amplifications. Phylogenetic analysis of alga, bryophyte, lycophyta, monocot and eudicot WRKY domains revealed family member expansion with increasing complexity of the plant body. Microarray, expression profiling and qRT-PCR data revealed that WRKY genes in G. hirsutum may regulate the development of fibers, anthers, tissues (roots, stems, leaves and embryos), and are involved in the response to stresses. Expression analysis showed that most group II and III GhWRKY genes are highly expressed under diverse stresses. Group I members, representing the ancestral form, seem to be insensitive to abiotic stress, with low expression divergence. Our results indicate that cotton WRKY genes might have evolved by adaptive duplication, leading to sensitivity to diverse stresses. This study provides fundamental information to inform further analysis and understanding of WRKY gene functions in cotton species.
DOI:10.1007/s00438-015-1107-6URLPMID:26308611 [本文引用: 2]
Tea plant [Camellia sinensis (L.) O. Kuntze] is a leaf-type healthy non-alcoholic beverage crop, which has been widely introduced worldwide. Tea is rich in various secondary metabolites, which are important for human health. However, varied climate and complex geography have posed challenges for tea plant survival. The WRKY gene family in plants is a large transcription factor family that is involved in biological processes related to stress defenses, development, and metabolite synthesis. Therefore, identification and analysis of WRKY family transcription factors in tea plant have a profound significance. In the present study, 50 putative C. sinensis WRKY proteins (CsWRKYs) with complete WRKY domain were identified and divided into three Groups (Group I-III) on the basis of phylogenetic analysis results. The distribution of WRKY family transcription factors among plantae, fungi, and protozoa showed that the number of WRKY genes increased in higher plant, whereas the number of these genes did not correspond to the evolutionary relationships of different species. Structural feature and annotation analysis results showed that CsWRKY proteins contained WRKYGQK/WRKYGKK domains and C2H2/C2HC-type zinc-finger structure: D-X18-R-X1-Y-X2-C-X4-7-C-X23-H motif; CsWRKY proteins may be associated with the biological processes of abiotic and biotic stresses, tissue development, and hormone and secondary metabolite biosynthesis. Temperature stresses suggested that the candidate CsWRKY genes were involved in responses to extreme temperatures. The current study established an extensive overview of the WRKY family transcription factors in tea plant. This study also provided a global survey of CsWRKY transcription factors and a foundation of future functional identification and molecular breeding.
[本文引用: 2]
[本文引用: 2]
DOI:10.1007/s13258-018-0734-9URLPMID:30238224 [本文引用: 2]
The WRKY transcription factors (TFs) family is one of the largest TF families in plants and plays a central role in diverse regulation and multiple stress responses. However, the systematical analysis of the WRKY gene family in tea plant (Camellia sinensis) based on genomic data has been lacking. The primary objective of this study was to set a systematic analysis of the WRKY gene family based on genomic data in tea plant and analyze their expression profiles under various abiotic stresses. We searched the tea plant genome using the consensus model of the WRKY domain (PF03106) and then used these search results to identify all the WRKY family members by SMART and the CDD program. Analyze their phylogeny, classification, structure, conserved motifs, Cis-elements, interactors and expression profiles. 56 putative WRKY genes were identified from the tea plant genome and divided into three main groups (I-III) and five subgroups (IIa-IIe) according to the WRKY domains and the zinc-finger structure. The gene structure and conserved motifs of the CsWRKY genes were also characterized and were consistent with the classification results. Annotation analysis showed that 34 CsWRKY genes may be involved in stress responses. Promoter analysis implied that CsWRKY genes, except for CsWRKY55, possessed at least one abiotic stress response cis-element. Expression profiles of CsWRKY genes in different tissues were analyzed with RNA-seq data. The results showed that 56 CsWRKY genes had differential expression in their transcript abundance. The expression profiles also showed that many identified CsWRKY genes were possibly involved in the response to cold, drought, salt, or ABA treatment. Tea plant genome contains at least 56 WRKY genes. These results provide useful information for further exploring the function and regulatory mechanism of CsWRKY genes in the growth, development, and adaption to abiotic stresses in tea plant.
DOI:10.1007/s10535-016-0618-2URL [本文引用: 4]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
DOI:10.1021/acs.jafc.8b02175URLPMID:30350966 [本文引用: 3]
Tea O-methylated catechins, especially (-)-epigallocatechin 3- O-(3- O-methyl)gallate (EGCG3''Me), have been attracting much attention as a result of their positive health effects. The transcription regulators of O-methylated catechin biosynthesis remain elusive. In this study, the expression pattern of genes related to O-methylated catechin biosynthesis, including CsLAR, CsANS, CsDFR, CsANR, and CCoAOMT, in three tea cultivars with different contents of EGCG3''Me was investigated. Two WRKY transcription factors (TFs), designated as CsWRKY31 and CsWRKY48, belonging to groups IIb and IIc of the WRKY family, respectively, were further identified. CsWRKY31 and CsWRKY48 were nuclear-localized proteins and possessed transcriptional repression ability. Furthermore, expression of CsWRKY31 and CsWRKY48 showed negative correlation with CsLAR, CsDFR, and CCoAOMT during EGCG3''Me accumulation in tea leaves. More importantly, W-box (C/T)TGAC(T/C) elements were located in the promoter of CsLAR, CsDFR, and CCoAOMT, and further assays revealed that CsWRKY31 and CsWRKY48 were capable of repressing the transcription of CsLAR, CsDFR, and CCoAOMT via the attachment of their promoters to the W-box elements. Collectively, our findings identify two novel negative regulators of O-methylated catechin biosynthesis in tea plants, which might provide a potential strategy to breed high-quality tea cultivar.
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
DOI:10.3390/ijms20112815URL [本文引用: 4]
DOI:10.1016/j.plaphy.2018.09.021URLPMID:30245342 [本文引用: 1]
The sucrose nonfermenting 1 (SNF1)-related protein kinase 2 (SnRK2) genes play central roles in plant stress signal transduction. In this study, 8 SnRK2 genes were identified from the tea plant genome database and named CsSnRK2.1-8. Phylogenetic analysis showed that the CsSnRK2 genes were classifiable into three groups, similar to those of Arabidopsis thaliana, Oryza sativa and maize. The coding sequences (CDSs) of all CsSnRK2s were separated by eight introns, and their exon-intron organizations exhibited high similarity to those of other plants. The fluorescence of GFP fused with CsSnRK2.3 was detected in only the cytoplasm, while the rest of the proteins showed GFP signal in both the nucleus and the cytoplasm. The results of the expression patterns of the CsSnRK2 genes showed that CsSnRK2s were differentially induced by salt, polyethylene glycol (PEG) and abscisic acid (ABA) stress. Interestingly, The expression of CsSnRK2.3 was inhibited by ABA, suggesting the complicated roles of CsSnRK2s in the ABA signal transduction pathway. Some CsSnRK2 gene pairs showed significant expression change correlations under stresses, indicating that CsSnRK2s might exhibit synergistic effects of signal regulation in response to various stresses. In summary, this comprehensive analysis will facilitate further studies of the SnRK2 family of Camellia sinensis and provide useful information for the functional validation of CsSnRK2s.
[本文引用: 2]
[本文引用: 2]
DOI:10.1006/meth.2001.1262URLPMID:11846609 [本文引用: 1]
The two most commonly used methods to analyze data from real-time, quantitative PCR experiments are absolute quantification and relative quantification. Absolute quantification determines the input copy number, usually by relating the PCR signal to a standard curve. Relative quantification relates the PCR signal of the target transcript in a treatment group to that of another sample such as an untreated control. The 2(-Delta Delta C(T)) method is a convenient way to analyze the relative changes in gene expression from real-time quantitative PCR experiments. The purpose of this report is to present the derivation, assumptions, and applications of the 2(-Delta Delta C(T)) method. In addition, we present the derivation and applications of two variations of the 2(-Delta Delta C(T)) method that may be useful in the analysis of real-time, quantitative PCR data.
DOI:10.1186/s12864-018-4880-xURLPMID:29940851 [本文引用: 1]
BACKGROUND: WRKY proteins comprise a large family of transcription factors that play important roles in many aspects of physiological processes and adaption to environment. However, little information was available about the WRKY genes in pineapple (Ananas comosus), an important tropical fruits. The recent release of the whole-genome sequence of pineapple allowed us to perform a genome-wide investigation into the organization and expression profiling of pineapple WRKY genes. RESULTS: In the present study, 54 pineapple WRKY (AcWRKY) genes were identified and renamed on the basis of their respective chromosome distribution. According to their structural and phylogenetic features, the 54 AcWRKYs were further classified into three main groups with several subgroups. The segmental duplication events played a major role in the expansion of pineapple WRKY gene family. Synteny analysis and phylogenetic comparison of group III WRKY genes provided deep insight into the evolutionary characteristics of pineapple WRKY genes. Expression profiles derived from transcriptome data and real-time quantitative PCR analysis exhibited distinct expression patterns of AcWRKY genes in various tissues and in response to different abiotic stress and hormonal treatments. CONCLUSIONS: Fifty four WRKY genes were identified in pineapple and the structure of their encoded proteins, their evolutionary characteristics and expression patterns were examined in this study. This systematic analysis provided a foundation for further functional characterization of WRKY genes with an aim of pineapple crop improvement.
DOI:10.3864/j.issn.0578-1752.2015.16.012URL [本文引用: 1]
【Objective】In this study, 132 putative WRKY proteins in the apple (Malus domestica Borkh.) genome were identified, so as to provide a basis for studying the theoretical roles of WRKY genes in the regulation of plant stress responses, growth and development, and to provide valuable information for WRKY genes cloning in apple.【Method】WRKY DNA-binding domain (PF03106) downloaded from Pfam protein families database was employed to identify WRKY genes from apple genome using HMMER 3.0. The obtained amino acid sequences were analyzed with the bioinformatics softwares, including DNAMAN 5.0, Weblogo 3, MEGA 5.1, MapInspect and MEME. RT-PCR was used to detect WRKY genes expression in different tissues of apple. 【Result】 Totally 132 apple WRKY genes were found in apple genome. The result of group identification and phylogenetic analysis revealed that apple WRKY genes were classified into Group I, Group II and Group III. Twenty-four MdWRKY proteins with two WRKY domains (group I-N and group I-C) containing CX4CX22-23HXH zinc-finger motif belonged to Group I. Seventy-nine Group II MdWRKY proteins had a single WRKY domain including CX4–5CX23HXH zinc-finger motif and could be further divided into five subgroups (Group II-a: 8 members, Group II-b: 12 members, Group II-c: 31 members, Group II-d: 14 members, and Group II-e: 14 members, respectively), whereas 29 Group III MdWRKY proteins contained a single WRKY domain with CX7CX23–24HXC zinc-finger motif. The results of domain analysis indicated that the WRKY regions contained a highly conserved heptapeptide stretch WRKYGQK at its N-terminus followed by a zinc-finger motif. Chromosome mapping analysis showed that apple WRKY genes were distributed with different densities on 17 chromosomes. The largest number of apple WRKY genes were found on chromosomes 1 and 9 (thirteen genes), followed by chromosome 12 (twelve genes). Only 4 genes located on chromosomes 2, 5 and 14. The results of gene structure analysis revealed that most of the WRKY gene contained 2-5 exons and WRKY gene structure were highly conserved in apple. Conserved motif analysis showed that the conserved motifs 1-6, which specify the WRKY domain, were observed in all apple WRKY proteins, motif 8 and motifs 7 and 9 as the unknown domain were observed in Group II-a and II-b and Group III, respectively. Two WRKY domains were assigned to Group I. RT-PCR results indicated that 12 MdWRKY genes were expressed in roots, stems, leaves, flowers and fruits at various expression levels.【Conclusion】These results suggested that MdWRKY gene family was highly and structurally conserved, and may be involved into the regulation of growth and development processes in apple.
DOI:10.3864/j.issn.0578-1752.2015.16.012URL [本文引用: 1]
【Objective】In this study, 132 putative WRKY proteins in the apple (Malus domestica Borkh.) genome were identified, so as to provide a basis for studying the theoretical roles of WRKY genes in the regulation of plant stress responses, growth and development, and to provide valuable information for WRKY genes cloning in apple.【Method】WRKY DNA-binding domain (PF03106) downloaded from Pfam protein families database was employed to identify WRKY genes from apple genome using HMMER 3.0. The obtained amino acid sequences were analyzed with the bioinformatics softwares, including DNAMAN 5.0, Weblogo 3, MEGA 5.1, MapInspect and MEME. RT-PCR was used to detect WRKY genes expression in different tissues of apple. 【Result】 Totally 132 apple WRKY genes were found in apple genome. The result of group identification and phylogenetic analysis revealed that apple WRKY genes were classified into Group I, Group II and Group III. Twenty-four MdWRKY proteins with two WRKY domains (group I-N and group I-C) containing CX4CX22-23HXH zinc-finger motif belonged to Group I. Seventy-nine Group II MdWRKY proteins had a single WRKY domain including CX4–5CX23HXH zinc-finger motif and could be further divided into five subgroups (Group II-a: 8 members, Group II-b: 12 members, Group II-c: 31 members, Group II-d: 14 members, and Group II-e: 14 members, respectively), whereas 29 Group III MdWRKY proteins contained a single WRKY domain with CX7CX23–24HXC zinc-finger motif. The results of domain analysis indicated that the WRKY regions contained a highly conserved heptapeptide stretch WRKYGQK at its N-terminus followed by a zinc-finger motif. Chromosome mapping analysis showed that apple WRKY genes were distributed with different densities on 17 chromosomes. The largest number of apple WRKY genes were found on chromosomes 1 and 9 (thirteen genes), followed by chromosome 12 (twelve genes). Only 4 genes located on chromosomes 2, 5 and 14. The results of gene structure analysis revealed that most of the WRKY gene contained 2-5 exons and WRKY gene structure were highly conserved in apple. Conserved motif analysis showed that the conserved motifs 1-6, which specify the WRKY domain, were observed in all apple WRKY proteins, motif 8 and motifs 7 and 9 as the unknown domain were observed in Group II-a and II-b and Group III, respectively. Two WRKY domains were assigned to Group I. RT-PCR results indicated that 12 MdWRKY genes were expressed in roots, stems, leaves, flowers and fruits at various expression levels.【Conclusion】These results suggested that MdWRKY gene family was highly and structurally conserved, and may be involved into the regulation of growth and development processes in apple.
[本文引用: 1]
DOI:10.1186/s12863-018-0653-4URLPMID:30060731 [本文引用: 1]
BACKGROUND: WRKY transcription factors (TFs) participate in various physiological processes of plants. Although WRKY genes have been well studied in model plants, knowledge of the functional roles of these genes is still extremely limited in cotton. RESULTS: In this study, a group IId WRKY gene from cotton, GhWRKY42, was isolated and characterized. Our data showed that GhWRKY42 localized to the nucleus. A transactivation assay in yeast demonstrated that GhWRKY42 was not a transcriptional activator. A beta-glucuronidase (GUS) activity assay revealed that the promoter of GhWRKY42 showed fragment deletion activity in Nicotiana tabacum and was mainly expressed in the roots, stems and leaves of ProGhWRKY42::GUS transgenic Arabidopsis plants. Quantitative real-time PCR (qRT-PCR) analysis indicated that GhWRKY42 was up-regulated during leaf senescence and was induced after exposure to abiotic stresses. Constitutive expression of GhWRKY42 in Arabidopsis led to a premature aging phenotype, which was correlated with an increased number of senescent leaves, reduced chlorophyll content and elevated expression of senescence-associated genes (SAGs). In addition, virus-induced gene silencing (VIGS) was used to silence the endogenous GhWRKY42 gene in cotton, and this silencing reduced plant height. CONCLUSIONS: Our findings indicate that GhWRKY42 is involved in abiotic stress responses, premature leaf senescence and stem development. This work establishes a solid foundation for further functional analysis of the GhWRKY42 gene in cotton.
DOI:10.1016/j.plaphy.2015.07.002URLPMID:26184088 [本文引用: 3]
Members of the large WRKY transcription factor family are responsible for the regulation of plant growth, development and the stress response. Here, five WRKY members were isolated from chrysanthemum. They each contained a single WRKY domain and a C2H2 zinc finger motif, so were classified into group II. Transient expression experiments demonstrated that all five were expressed in the nucleus, although CmWRKY42 was also expressed in the cytoplasm. When expressed heterologously in yeast, the products of CmWRKY22 and CmWRKY48 exhibited transactivation activity, while those of CmWRKY21, CmWRKY40 and CmWRKY42 did not. The transcription of the five CmWRKY genes was profiled when the plants were challenged with a variety of abiotic and biotic stress agents, as well as being treated with various phytohormones. CmWRKY21 proved to be markedly induced by salinity stress, and suppressed by high temperature exposure; CmWRKY22 was induced by high temperature exposure; CmWRKY40 was highly induced by salinity stress, and treatment with either abscisic acid (ABA) or methyl jasmonate (MeJA); CmWRKY42 was up-regulated by salinity stress, low temperature, ABA and MeJA treatment and aphid infestation; CmWRKY48 was induced by drought stress, ABA and MeJA treatment and aphid infestation. The function of CmWRKY48 was further investigated by over-expressing it transgenically. The constitutive expression of this transcription factor inhibited the aphids' population growth capacity, suggesting that it may represent an important component of the plant's defense machinery against aphids.
DOI:10.3390/f10040335URL [本文引用: 1]
DOI:10.1007/s00299-018-2302-9URLPMID:29796948 [本文引用: 1]
KEY MESSAGE: Overexpression of VaWRKY14 increases drought tolerance in Arabidopsis by modulating the expression of stress-related genes, including COR15A, COR15B, COR413, KIN2, and RD29A. The WRKY family is one of a largest transcription factors in plants, and it is a key component of multiple stress responses. In this study, the drought- and cold-induced WRKY family gene VaWRKY14 was isolated and characterized. Phylogenetic analysis indicated that VaWRKY14 belongs to the WRKY IIa subfamily, of which several members participate in biotic and abiotic stress responses in plants. Fluorescence observation from Arabidopsis mesophyll protoplasts transformed with the VaWRKY14::eGFP fusion vector suggested that VaWRKY14 was localized in the nucleus. The VaWRKY14 in yeast cells did not display any transcriptional activity. The expression of VaWRKY14 could be induced by exogenous phytohormones, including salicylic acid (SA) and abscisic acid (ABA). Overexpression of VaWRKY14 enhanced the drought tolerance of transgenic Arabidopsis. Compared with wild-type Arabidopsis, the VaWRKY14-OE lines exhibited higher water content and antioxidant enzyme activities in leaves after drought treatment. RNA sequencing analysis revealed that several stress-related genes, including COR15A, COR15B, COR413, KIN2, and RD29A, were upregulated in transgenic plants relative to their expression in wild-type Arabidopsis under normal conditions. Several genes (3 upregulated and 49 down-regulated) modulated by VaWRKY14 were also affected by drought stress in wild-type plants. These data suggest that VaWRKY14 responds to drought and cold stresses and that drought tolerance may be enhanced by regulating the expression of stress-related genes in Arabidopsis.
[本文引用: 1]
[本文引用: 1]
DOI:10.1093/mp/sss080URL [本文引用: 1]
Drought is one of the most serious environmental factors that limit the productivity of agricultural crops worldwide. However, the mechanism underlying drought tolerance in plants is unclear. WRKY transcription factors are known to function in adaptation to abiotic stresses. By screening a pool of WRKY-associated T-DNA insertion mutants, we isolated a gain-of-function mutant, acquired drought tolerance (adt), showing improved drought tolerance. Under drought stress conditions, adt accumulated higher levels of ABA than wild-type plants. Stomatal aperture analysis indicated that adt was more sensitive to ABA than wild-type plants. Molecular genetic analysis revealed that a T-DNA insertion in adt led to activated expression of a WRKY gene that encodes the WRKR57 protein. Constitutive expression of WRKY57 also conferred similar drought tolerance. Consistently with the high ABA content and enhanced drought tolerance, three stress-responsive genes (RD29A, NCED3, and ABA3) were up-regulated in adt. ChIP assays demonstrated that WRKY57 can directly bind the W-box of RD29A and NCED3 promoter sequences. In addition, during ABA treatment, seed germination and early seedling growth of adt were inhibited, whereas, under high osmotic conditions, adt showed a higher seed germination frequency. In summary, our results suggested that the activated expression of WRKY57 improved drought tolerance of Arabidopsis by elevation of ABA levels. Establishment of the functions of WRKY57 will enable improvement of plant drought tolerance through gene manipulation approaches.
DOI:10.3389/fpls.2018.01979URLPMID:30740122 [本文引用: 1]
The WRKY transcription factors (TFs) are one of the largest families of TFs in plants and play multiple roles in plant development and stress response. In the present study, GmWRKY16 encoding a WRKY transcription factor in soybean was functionally characterized in Arabidopsis. GmWRKY16 is a nuclear protein that contains a highly conserved WRKY domain and a C2H2 zinc-finger structure, and has the characteristics of transcriptional activation ability, presenting a constitutive expression pattern with relative expression levels of over fourfold in the old leaves, flowers, seeds and roots of soybean. The results of quantitative real time polymerase chain reaction (qRT-PCR) showed that GmWRKY16 could be induced by salt, alkali, ABA, drought and PEG-6000. As compared with the control, overexpression of GmWRKY16 in Arabidopsis increased the seed germination rate and root growth of seedlings in transgenic lines under higher concentrations of mannitol, NaCl and ABA. In the meantime, GmWRKY16 transgenic lines showed over 75% survival rate after rehydration and enhanced Arabidopsis tolerance to salt and drought with higher proline and lower MDA accumulation, less water loss of the detached leaves, and accumulated more endogenous ABA than the control under stress conditions. Further studies showed that AtWRKY8, KIN1, and RD29A were induced in GmWRKY16 transgenic plants under NaCl treatment. The expressions of the ABA biosynthesis gene (NCED3), signaling genes (ABI1, ABI2, ABI4, and ABI5), responsive genes (RD29A, COR15A, COR15B, and RD22) and stress-related marker genes (KIN1, LEA14, LEA76, and CER3) were regulated in transgenic lines under drought stress. In summary, these results suggest that GmWRKY16 as a WRKY TF may promote tolerance to drought and salt stresses through an ABA-mediated pathway.
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s00425-017-2766-9URLPMID:28861611 [本文引用: 1]
MAIN CONCLUSION: We cloned and characterized the ZmWRKY17 gene from maize. Overexpression of ZmWRKY17 in Arabidopsis led to increased sensitivity to salt stress and decreased ABA sensitivity through regulating the expression of some ABA- and stress-responsive genes. The WRKY transcription factors have been reported to function as positive or negative regulators in many different biological processes including plant development, defense regulation and stress response. This study isolated a maize WRKY gene, ZmWRKY17, and characterized its role in tolerance to salt stress by generating transgenic Arabidopsis plants. Expression of the ZmWRKY17 was up-regulated by drought, salt and abscisic acid (ABA) treatments. ZmWRKY17 was localized in the nucleus with no transcriptional activation in yeast. Yeast one-hybrid assay showed that ZmWRKY17 can specifically bind to W-box, and it can activate W-box-dependent transcription in planta. Heterologous overexpression of ZmWRKY17 in Arabidopsis remarkably reduced plant tolerance to salt stress, as determined through physiological analyses of the cotyledons greening rate, root growth, relative electrical leakage and malondialdehyde content. Additionally, ZmWRKY17 transgenic plants showed decreased sensitivity to ABA during seed germination and early seedling growth. Transgenic plants accumulated higher content of ABA than wild-type (WT) plants under NaCl condition. Transcriptome and quantitative real-time PCR analyses revealed that some stress-related genes in transgenic seedlings showed lower expression level than that in the WT when treated with NaCl. Taken together, these results suggest that ZmWRKY17 may act as a negative regulator involved in the salt stress responses through ABA signalling.
DOI:10.1016/j.plantsci.2018.03.018URLPMID:30823991 [本文引用: 2]
High salinity severely inhibits the growth and productivity of grape plants. However, knowledge of salt-stress regulation remains limited in WRKY members of grapes. Here, we isolated a novel VvWRKY30 gene from Vitis vinifera L. and studied its role in salt-stress resistance. The VvWRKY30 protein fused with green fluorescent protein localized to the nucleus and the transcriptional activation activity of VvWRKY30 was confirmed in yeast. Moreover, VvWRKY30 showed key transcriptional activity domain at the N-terminal and specifically binds to the W-BOX. VvWRKY30 showed the highest expression in the shoot tip and functional leaves of grape plants. VvWRKY30 expression was induced by salt as well as stress signaling molecules H2S and H2O2. Overexpression of VvWRKY30 in Arabidopsis increased its resistance to salt stress at different stages of growth. Under salinity stress, VvWRKY30 overexpressing lines had higher antioxidant activities and lower reactive oxygen species contents. Soluble sugar and proline concentrations also increased in VvWRKY30 overexpressing lines in the presence of NaCl. In addition, the transcription of genes related to antioxidant biosynthesis, glyco-metabolism and proline biosynthesis increased in the VvWRKY30 overexpressing lines. Taken together, this study confirmed the important role of VvWRKY30 in increasing salt stress resistance by regulating reactive oxygen species-scavenging and the accumulation of osmoticum.
DOI:10.1007/s00299-008-0614-xURL [本文引用: 1]
An OsWRKY11 gene, which encodes a transcription factor with the WRKY domain, was identified as one of the genes that was induced by both heat shock and drought stresses in seedlings of rice (Oryza sativa L.). To determine if overexpression of OsWRKY11 confers heat and drought tolerance, OsWRKY11 cDNA was fused to the promoter of HSP101 of rice and introduced into a rice cultivar Sasanishiki. Overexpression of OsWRKY11 was induced by heat treatment. After heat pretreatment, the transgenic lines showed significant heat and drought tolerance, as indicated by the slower leaf-wilting and less-impaired survival rate of green parts of plants. They also showed significant desiccation tolerance, as indicated by the slower water loss in detached leaves. Our results indicate that the OsWRKY11 gene plays a role in heat and drought stress response and tolerance, and might be useful for improvement of stress tolerance.
[D].
[本文引用: 1]
[D].
[本文引用: 1]
DOI:10.1007/s00299-015-1793-xURLPMID:25893877 [本文引用: 1]
KEY MESSAGE: CmWRKY17 was induced by salinity in chrysanthemum, and it might negatively regulate salt stress in transgenic plants as a transcriptional repressor. WRKY transcription factors play roles as positive or negative regulators in response to various stresses in plants. In this study, CmWRKY17 was isolated from chrysanthemum (Chrysanthemum morifolium). The gene encodes a 227-amino acid protein and belongs to the group II WRKY family, but has an atypical WRKY domain with the sequence WKKYGEK. Our data indicated that CmWRKY17 was localized to the nucleus in onion epidermal cells. CmWRKY17 showed no transcriptional activation in yeast; furthermore, luminescence assay clearly suggested that CmWRKY17 functions as a transcriptional repressor. DNA-binding assay showed that CmWRKY17 can bind to W-box. The expression of CmWRKY17 was induced by salinity in chrysanthemum, and a higher expression level was observed in the stem and leaf compared with that in the root, disk florets, and ray florets. Overexpression of CmWRKY17 in chrysanthemum and Arabidopsis increased the sensitivity to salinity stress. The activities of superoxide dismutase and peroxidase and proline content in the leaf were significantly lower in transgenic chrysanthemum than those in the wild type under salinity stress, whereas electrical conductivity was increased in transgenic plants. Expression of the stress-related genes AtRD29, AtDREB2B, AtSOS1, AtSOS2, AtSOS3, and AtNHX1 was reduced in the CmWRKY17 transgenic Arabidopsis compared with that in the wild-type Col-0. Collectively, these data suggest that CmWRKY17 may increase the salinity sensitivity in plants as a transcriptional repressor.
DOI:10.1016/j.plantsci.2018.04.029URLPMID:29807593 [本文引用: 1]
Recent studies with Arabidopsis and soybean have shown that a class of valine-glutamine (VQ) motif-containing proteins interacts with some WRKY transcription factors. However, little is known about the evolution, structures, and functions of those proteins in apple. Here, we examined their features and identified 49 apple VQ genes. Our evolutional analysis revealed that the proteins could be clustered into nine groups together with their homologues in 33 species. Historically, the main characteristics of proteins in Groups I, V, VI, VII, IX, and X were thought to have been generated before the monocot-dicot split, whereas those in Groups II, III+IV, and VIII were generated after that split. In the structural analysis, apple MdVQ proteins appeared to bind only with Group I and IIc MdWRKY proteins. Meanwhile, MdVQ1, MdVQ10, MdVQ15, and MdVQ36 interacted with multiple MdVQ proteins to form heterodimers but MdVQ15 formed a homodimer. The functional analysis indicated that overexpression of some apple MdVQs in Arabidopsis and tobacco plants effected their vegetative and reproductive growth. These results provide important information about the characteristics of apple MdVQ genes and can serve as a solid foundation for further studies about the role of WRKY-VQ interactions in regulating apple developmental and defense mechanisms.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}