 ,3, 李中安,11
,3, 李中安,11 2
3
The Resistance Prediction of Wheat Hybrids Based on the Sensibility of Their Parents to Stripe Rust
ZHOU TianYu1, LI JiangLing1, YANG Lan1, RUAN RenWu2, YANG YuHeng,3, LI ZhongAn,1通讯作者:
责任编辑: 岳梅
收稿日期:2019-12-17接受日期:2020-02-12网络出版日期:2020-05-16
| 基金资助: |
Received:2019-12-17Accepted:2020-02-12Online:2020-05-16
作者简介 About authors
周天宇,E-mail:1522619059@qq.com。

摘要
关键词:
Abstract
Keywords:
PDF (2671KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
周天宇, 李姜玲, 杨澜, 阮仁武, 杨宇衡, 李中安. 基于亲本对条锈病敏感性预测小麦杂交种的抗性[J]. 中国农业科学, 2020, 53(9): 1806-1819 doi:10.3864/j.issn.0578-1752.2020.09.009
ZHOU TianYu, LI JiangLing, YANG Lan, RUAN RenWu, YANG YuHeng, LI ZhongAn.
0 引言
【研究意义】小麦是全球第二大粮食作物,随着世界人口不断增加而耕地面积逐年减少,预计到2050年全球粮食需求将增长一倍,因此粮食安全非常重要[1,2,3,4]。小麦条锈病严重威胁小麦安全生产,因此筛选小麦抗条锈育种材料并预测杂交小麦的抗锈性,对我国杂交小麦抗病育种以及粮食安全具有重要意义。【前人研究进展】20世纪60年代发现小麦雄性不育系和恢复系,这给小麦的杂种优势利用带来了希望[5]。近年来,杂交小麦迅速发展,强优势组合在生产上开始大面积示范[6]。杂种优势是指杂合体在一种或多种性状上优于两个亲本的现象,在自然界广泛存在,其表现形式为高产、抗病、较高的肥料利用率、根系渗透性、种子结实率,以及对非生物胁迫的强忍耐性等[7,8]。利用杂种优势一直被认为是提高粮食产量的重要途径,在水稻、玉米、高粱等作物上已取得显著效果[9,10]。70年来中国水稻品种实现了矮杆化、杂交化、优质化的3次跨越,完成6次新品种更新换代[11]。小麦的杂交化经历了漫长的过程,但仍未取得质的突破,还处于研究的初始阶段。小麦杂交化除了提高小麦单位面积产量外,降低生产成本已成为一个重要的育种目标,以小麦基因组为基础,利用分子标记辅助育种实现了部分农艺性状的早期选择,可以更好地培育生产上需要的新品种[12]。此外,通过亲本和F1代多个性状指标(如粒长、粒重等)建立线性回归关系对F1代进行优势预测已经在杂交水稻上得以证明[13]。小麦抗病育种对于小麦稳产、增产具有重要意义,农作物抗病基因的分离鉴定直接用于品种改良和病害绿色防控[14]。关于杂交小麦抗叶锈病、条锈病、黑穗病及白粉病的规律研究已有相关报道[15]。杂交小麦F1代抗白粉病研究表明,杂种一代与亲本的抗性表现基本一致,符合遗传规律[16]。将感小麦叶锈病的父本材料分别与不同反应型的母本材料杂交发现,抗叶锈母本的杂交后代大多数都表现抗病,并且这些杂合体有很强的杂种优势[17]。通过对单杂交种群体抗赤霉病分析,发现亲本对赤霉病表现的中亲值可以作为单杂交种对赤霉病表现的指标,并且亲本综合配合力与杂交种对赤霉病抗性有很高相关性[18]。ALI等[19]通过引入外源抗小麦条纹花叶病毒基因,并与其他基因组合提高了后代的抗性。利用分子标记技术将含有小麦抗白粉病基因(Pm21、PmV)和抗小麦黄花叶病基因(Wss1)的亲本进行杂交,构建F2代群体,在F3代群体中筛选出的23个双抗基因聚合体对白粉病免疫,对黄花叶病高抗,可用于双抗聚合品种的筛选或作为抗病中间亲本[20]。崔彩红等[21]利用分子标记技术定向培育了4个含有Sr25、4个含有Sr26、1个同时含有Sr25、Sr26的小麦新品系,这些品系农艺性状良好,可作为今后小麦抗秆锈育种的亲本。以上研究表明杂交小麦在抗病育种中能够发挥巨大作用,有助于加速小麦抗病育种的进程。小麦条锈病是由小麦条锈菌(Puccinia striiformis f. sp. tritici,Pst)引起的、由气流传播的世界性重要病害,严重危害小麦产量[22]。该病原菌具有活体专性寄生、喜冷凉的特点,随降雨、露水等侵染小麦[23]。利用抗病品种是防治小麦条锈病最经济、最环保的方式,但小麦品种抗病基因单一化、病原菌生理小种变异强,再加上抗病品种选育慢,故造成小麦条锈病大流行[24],而且在杂交小麦的抗条锈病研究上未见系统报道。【本研究切入点】以小麦条锈病和小麦核不育杂交组合及双亲为研究对象,筛选含有不同的抗条锈病基因或对不同条锈病生理小种有抗性的亲本配制杂交组合,通过抗性评价明确杂交种抗性与双亲抗性间的关系。【拟解决的关键问题】通过分析亲本及F1代的反应型,鉴定抗条锈病基因及亲本、部分F1代成株期不同小种侵染量,预测杂交组合对条锈病的抗性,为杂交小麦亲本筛选和选育提供理论依据。1 材料与方法
试验于2018—2019年在西南大学完成。1.1 试验材料
供试小麦恢复系包括小偃22、陕987、高大1号、伟隆121、15CA50、丰德存5号、西农807、MR1101、MY13-3、川14品16、川13品6、川麦93、川麦98。不育系包括21份不育系,以及91份亲本杂交产生的F1代。条锈病感病对照品种为铭贤169,小偃22、高大1号、丰德存5号、西农807、伟隆121由陕西杨凌伟隆农业科技有限公司提供,MR1101、MY13-3由绵阳市农业科学院小麦研究所提供,川14品16、川13品6、川麦93、川麦98由四川省农业科学院作物研究所提供,15CA50由中国农业科学院棉花研究所提供。不育系及F1代由笔者实验室选育和配制,其中不育系为蓝标型核不育,不育基因为ms1b[25]。供试菌种:CYR23、CYR31、CYR33、CYR34均在西南大学植物保护学院组培室利用感病品种铭贤169繁育获得。
1.2 方法
1.2.1 抗病基因分子鉴定 利用小麦抗条锈基因分子标记检测亲本中抗条锈基因的分布情况,所检测的抗病基因为Yr5、Yr9、Yr10、Yr15、Yr17、Yr18、Yr26,所用分子标记引物均由上海生工生物工程技术服务有限公司合成(表1[26,27,28])。采用CTAB法提取小麦基因组DNA[29],测定浓度后稀释至100 ng·μL-1备用。单个PCR检测体系总体积为20 μL,其中包含2×Taq Master Mix 10 μL,引物终浓度为2 mmol·L-1,DNA模板100 ng,ddH2O补足20 μL。产物均用2.0%的琼脂糖凝胶进行检测,并在凝胶成像仪上观察拍照。Table 1
表1
表1小麦抗条锈病基因的引物序列
Table 1
| Yr 基因 Yr genes | 引物名称 Primer name | 引物序列 Primer sequence (5′-3′) | 退火温度 Anneal temperature (℃) | 片段大小 Fragment size (bp) |
|---|---|---|---|---|
| Yr5 | Yr5-InsertionF[26] Yr5-InsertionR | CTCACGCATTTGACCATATACAACT TATTGCATAACATGGCCTCCAGT | 52 | +1281 |
| Yr9 | AF1[27] AF4 | F: GGAGACATCATGAAACATTTG R: CTGTTGTTGGGCAGAAAG | 46 | +1500 |
| Yr10 | Yr10F1[27] Yr10R1 | TTGGAATTGGCGACAAGCGT GTGATGATTACCCACTTCCTC | 50 | +755 |
| Yr15 | Y15K1_F2[28] uhw301R | GGAGATAGAGCACATTACAGAC TTTCGCATCCCACCCTACTG | 55 | +992 |
| W_2F | TGCACGCGGATATTAGGTAGG | 55 | +2014 | |
| W_2R | TGATGAAGAGGACCAACGCA | |||
| Yr17 | VENTRIUP[27] LN2 | AGGGGCTACTGACCAAGGCT TGCAGCTACAGCAGTATGTACACAAAA | 55 | +262 |
| Yr18 | L34DINT9F[27] L34PLUSR | TTGATGAAACCAGTTTTTTTTCTA GCCATTTAACATAATCATGATGGA | 45 | +517 |
| Yr26 | WE173F[27] WE173R | GGGACAAGGGGAGTTGAAGC GAGAGTTCCAAGCAGAACAC | 50 | +451 -730 |
新窗口打开|下载CSV
1.2.2 抗条锈病鉴定 每份小麦材料种植1行,行长1 m,行距0.25 m,每隔20个品种种植1行铭贤169作为感病对照,供试品种中间垂直种植1列感病品种铭贤169作为诱发行。分别称取0.1 g不同小麦条锈菌生理小种,混合配成1 L的孢子悬浮液,在小麦拔节期对诱发行进行混合接种。小麦进入抽穗期后,按0—9级标准记载反应型(infection type,IT),累计调查3次,以最高等级作为病情级数,根据IT抗病级别分为4个类型[30]:抗病(resistance,R,IT:0—3级)、中抗(moderate resistance,MR,IT:4—6级)、中感(moderate susceptibility,MS,IT:7级)、感病(susceptibility,S,IT:8—9级)。
1.2.3 亲本-F1代反应型的二元回归分析 以亲本成株期反应型为自变量,F1代成株期反应型为因变量,用SPSS软件对反应型之间的相关性进行二元回归分析。
1.2.4 亲本成株期小种侵染量鉴定 根据半定量PCR方法[31],利用小麦条锈菌生理小种特异分子标记引物鉴定不同生理小种对小麦亲本材料的侵染量。所鉴定的小麦条锈菌生理小种为CYR23、CYR31、CYR33、CYR34,所用分子标记引物均由上海生工生物工程技术服务有限公司合成。利用小麦条锈菌延伸因子特异引物pst_EF作为参照[32](表2)。
Table 2
表2
表2小麦条锈菌生理小种特异性引物序列
Table 2
| 引物名称 Primer name | 引物序列 Primer sequence (5′-3′) | 退火温度 Anneal temperature (℃) | |
|---|---|---|---|
| CYR23 | S360[33] S413 | F: AAGCGGCCTC R: GGTGGTCCAAG | 50 |
| CYR31 | CY31SP-1[34] CY31SP-2 | F: GCTACGTCAAGATGCGATACACC R: TGTCAGAAGCAAGTGGTAAACTAGG | 50 |
| CYR33 | CYR33SP-1[35] CYR33SP-2 | F: TGTCGTCTCGCCAATCTTT R: GCGGGTGTCAGTTTCTCC | 50 |
| CYR34 | V26SP-1[36] V26SP-2 | F: CTGTAAAGCGGATAAAGGAA R: CATAAGAGCCACACTTGACC | 57 |
| Pst elongation factor | Pst_EF | F: TTCGCCGTCCGTGATATGAGACAA R: ATGCGTATCATGGTGGTGGAGTGA | 55 |
新窗口打开|下载CSV
小麦条锈菌DNA提取:根据成株期鉴定结果,采集发病的小麦叶片,准确称取叶片发病部位0.02 g,利用CTAB法提取总DNA,测定浓度后稀释至100 ng·μL-1备用。同时利用CTAB法提取条锈菌生理小种CYR23、CYR31、CYR33、CYR34的DNA作为阳性对照,测定浓度后稀释至100 ng·μL-1备用。
半定量PCR:将提取的发病叶片DNA用引物pst_EF进行扩增,寻找不同材料扩增至相同亮度时的循环数,将每份材料按照确定的循环数分别用小种特异引物进行扩增,同时将纯小种模板进行所需循环数的扩增,单个PCR体系为2×Taq Master Mix 10 μL,引物终浓度2 mmol·L-1,DNA模板100 ng,ddH2O补足20 μL,重复3次。用软件Image J将条带转化为灰度值,计算每份材料中每个小种的侵染量。
2 结果
2.1 亲本抗病基因分子检测
恢复系中不含Yr5、Yr10、Yr18,小偃22、陕987、高大1号含Yr9,陕987、高大1号、伟隆121、川麦98、MY13-3含Yr17,川14品16、川13品6、川麦93、川麦98、MR1101含Yr26(图1)。不育系中不含Yr5、Yr10、Yr15、Yr26抗病基因,Yr9在不育系中检测率比较高,占91%,17L9154、17L9160、17L9210、17L6065、17L6067含Yr17,17L6085、17L7030、17L7106、17L7123、17L7152含Yr18(图2)。来自四川的恢复系及其F1代表现出优良抗性,推测其中含有对CYR34未知的抗条锈基因。MY13-3及一些不育系未检测到Yr26但抗性优良,推测其含有未知的抗性基因(表3)。图1
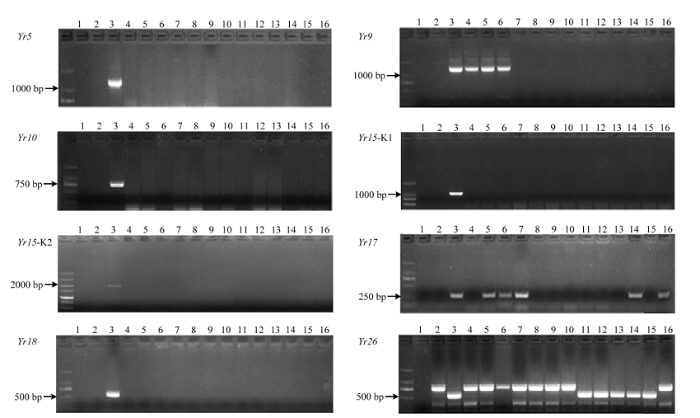 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1恢复系小麦抗条锈病基因分子鉴定
1:ddH2O;2:Avocet S;3:Yr;4:小偃22 Xiaoyan 22;5:陕987 Shaan 987;6:高大1号Gaoda No.1;7:伟隆121 Weilong 121;8:15CA50;9:丰德存5号 Fengdecun No.5;10:西农807 Xinong 807;11:川14品16 Chuan 14 pin 16;12:川13品6 Chuan 13 pin 6;13:川麦93 Chuanmai 93;14:川麦98 Chuanmai 98;15:MR1101;16:MY13-3
Fig. 1Detection of genes resistant to wheat stripe rust in restorer lines
图2
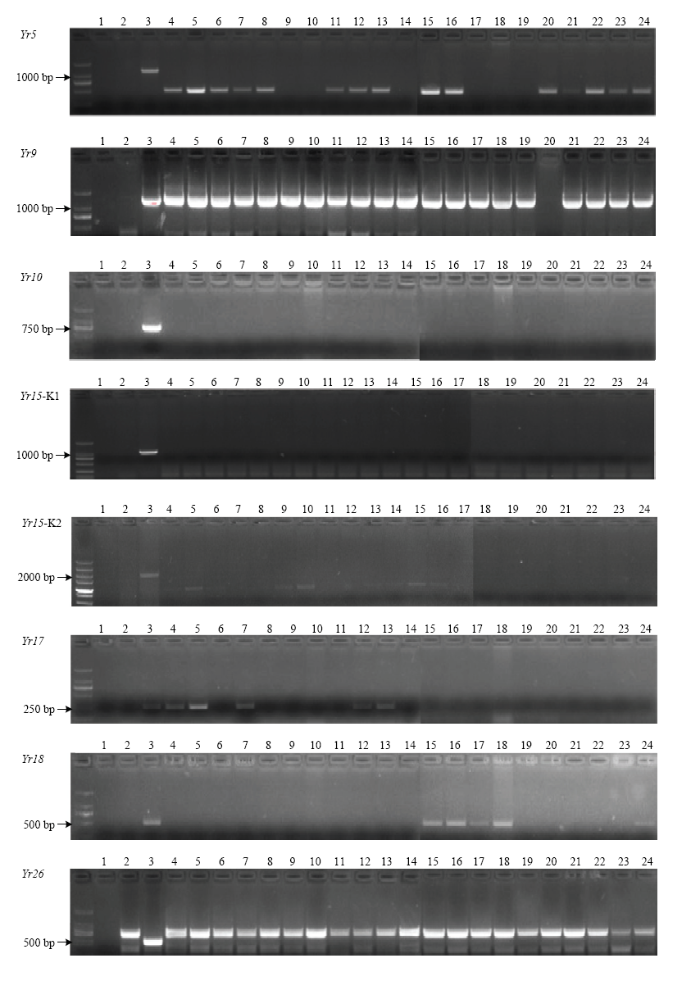 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2不育系小麦抗条锈病基因分子鉴定
1:ddH2O;2:Avocet S;3:Yr;4:17L9154;5:17L9160;6:17L9163;7:17L9210;8:17L9217;9:17L9195;10:17L6019;11:17L6062;12:17L6065;13:17L6067;14:17L6078;15:17L6085;16:17L7030;17:17L7106;18:17L7123;19:17L7140;20:17L9066;21:08L5070;22:15L7109;23:17L7128;24:17L7152
Fig. 2Detection of genes resistant to wheat stripe rust in sterile lines
Table 3
表3
表3亲本抗病表现
Table 3
| 品系 Line | 反应型 Infection type | 抗病评价 Resistance evaluation | Yr 基因 Yr genes | |
|---|---|---|---|---|
| 恢复系 Restorer lines | 小偃22 Xiaoyan 22 | 9 | S | Yr9 |
| 陕987 Shaan 987 | 5 | MR | Yr9+Yr17 | |
| 高大1号 Gaoda No.1 | 7 | MS | Yr9+Yr17 | |
| 伟隆121 Weilong 121 | 4 | MR | Yr17 | |
| 15CA50 | 7 | MS | - | |
| 丰德存5号 Fengdecun No.5 | 7 | MS | - | |
| 西农807 Xinong 807 | 7 | MS | - | |
| 川14品16 Chuan 14 pin 16 | 2 | R | Yr26 | |
| 川13品6 Chuan 13 pin 6 | 2 | R | Yr26 | |
| 川麦93 Chuanmai 93 | 0 | R | Yr26 | |
| 川麦98 Chuanmai 98 | 0 | R | Yr17+Yr26 | |
| MR1101 | 2 | R | Yr26 | |
| MY13-3 | 2 | R | Yr17 | |
| 不育系 Sterile lines | 08L5070 | 6 | MR | Yr9 |
| 15L7109 | 4 | MR | Yr9 | |
| 15L7128 | 6 | MR | Yr9 | |
| 15L7152 | 2 | R | Yr9+Yr18 | |
| 17L6019 | 6 | MR | Yr9 | |
| 17L6062 | 3 | R | Yr9 | |
| 17L6065 | 3 | R | Yr9+Yr17 | |
| 17L6067 | 3 | R | Yr9+Yr17 | |
| 17L6078 | 6 | MR | Yr9 | |
| 17L6085 | 5 | MR | Yr9+Yr17+Yr18 | |
| 17L7030 | 6 | MR | Yr9+Yr17+Yr18 | |
| 17L7106 | 3 | R | Yr9+Yr18 | |
| 17L7123 | 3 | R | Yr9+Yr18 | |
| 17L7140 | 3 | R | Yr9 | |
| 17L9066 | 6 | MR | - | |
| 17L9154 | 4 | MR | Yr9+Yr17 | |
| 17L9160 | 3 | R | Yr9+Yr17 | |
| 17L9163 | 6 | MR | Yr9 | |
| 17L9195 | 3 | R | Yr9 | |
| 17L9210 | 2 | R | Yr9+Yr17 | |
| 17L9217 | 4 | MR | Yr9 |
新窗口打开|下载CSV
2.2 亲本与F1代抗病表型
恢复系中反应型为抗病的包括川14品16、川13品6、川麦93、川麦98、MR1101、MY13-3;中抗的包括陕987、伟隆121;中感的包括高大1号、15CA50、丰德存5号、西农807;感病的为小偃22。不育系中反应型为抗病的包括15L7152、17L6062、17L6065、17L6067、17L7106、17L7123、17L7140、17L9160、17L9195、17L9210;中抗的包括08L5070、15L7109、15L7128、17L6019、17L6078、17L6085、17L7030、17L9066、17L9154、17L9163、17L9217(表3);F1代杂交种的反应型趋于亲本反应型的平均值(表4)。通过对亲本和F1代杂交种的反应型进行二元回归分析,得到R2=0.812,表明F1代杂交种反应型和亲本有很大的相关性(图3)。综合以上结果,双亲都表现抗病,则杂交种也表现抗病;双亲均表现感病或抗性较差,其杂交种的抗性就差,但双亲互补的例外;双亲之一感病,则杂交种抗病表现介于两个亲本之间,绝大多数趋于中亲值。Table 4
表4
表4F1代杂交种抗病表现
Table 4
| 编号 Number | F1代 F1 hybrids | 反应型 Infection type | 抗病评价 Resistance evaluation | Yr基因 Yr genes | 亲本反应型平均值 Infection type (average of parents) |
|---|---|---|---|---|---|
| 1 | 17L6065×小偃22 17L6065×Xiaoyan 22 | 5 | MR | Yr9+Yr17 | 6 |
| 2 | 17L6067×小偃22 17L6067×Xiaoyan 22 | 5 | MR | Yr9+Yr17 | 6 |
| 3 | 17L9160×小偃22 17L9160×Xiaoyan 22 | 5 | MR | Yr9+Yr17 | 6 |
| 4 | 17L9154×陕987 17L9154×Shaan 987 | 4 | MR | Yr9+Yr17 | 4.5 |
| 5 | 17L9160×陕987 17L9160×Shaan 987 | 4 | MR | Yr9+Yr17 | 4 |
| 6 | 17L7123×高大1号 17L7123×Gaoda No.1 | 5 | MR | Yr9+Yr17+Yr18 | 5 |
| 7 | 17L7140×高大1号 17L7140×Gaoda No.1 | 5 | MR | Yr9+Yr17 | 5 |
| 8 | 17L7030×高大1号 17L7030×Gaoda No.1 | 7 | MS | Yr9+Yr17+Yr18 | 6.5 |
| 9 | 17L6078×高大1号 17L6078×Gaoda No.1 | 6 | MR | Yr9+Yr17 | 6.5 |
| 10 | 17L9066×高大1号 17L9066×Gaoda No.1 | 6 | MR | Yr9+Yr17 | 6.5 |
| 11 | 17L9154×高大1号 17L9154×Gaoda No.1 | 6 | MR | Yr9+Yr17 | 5.5 |
| 12 | 17L6067×丰德存5号 17L6067×Fengdecun No.5 | 6 | MR | Yr9+Yr17 | 5 |
| 13 | 17L6019×丰德存5号 17L6019×Fengdecun No.5 | 5 | MR | Yr9 | 6.5 |
| 14 | 17L6065×丰德存5号 17L6065×Fengdecun No.5 | 5 | MR | Yr9+Yr17 | 5 |
| 15 | 17L7106×丰德存5号 17L7106×Fengdecun No.5 | 7 | MS | Yr9+Yr18 | 5 |
| 16 | 17L7030×丰德存5号 17L7030×Fengdecun No.5 | 7 | MS | Yr9+Yr17+Yr18 | 6.5 |
| 17 | 17L7123×丰德存5号 17L7123×Fengdecun No.5 | 7 | MS | Yr9+Yr18 | 7 |
| 18 | 17L9154×丰德存5号 17L9154×Fengdecun No.5 | 5 | MR | Yr9+Yr17 | 5.5 |
| 19 | 15L7109×丰德存5号 15L7109×Fengdecun No.5 | 6 | MR | Yr9 | 5.5 |
| 20 | 17L6067×西农807 17L6067×Xinong 807 | 6 | MR | Yr9+Yr17 | 5 |
| 21 | 17L6019×西农807 17L6019×Xinong 807 | 6 | MR | Yr9 | 6.5 |
| 22 | 17L7030×西农807 17L7030×Xinong 807 | 5 | MR | Yr9+Yr17+Yr18 | 6.5 |
| 23 | 17L7123×西农807 17L7123×Xinong 807 | 5 | MR | Yr9+Yr18 | 5 |
| 24 | 17L7140×西农807 17L7140×Xinong 807 | 5 | MR | Yr9 | 5 |
| 25 | 17L9154×西农807 17L9154×Xinong 807 | 6 | MR | Yr9+Yr17 | 5.5 |
| 26 | 15L7109×西农807 15L7109×Xinong 807 | 5 | MR | Yr9 | 5.5 |
| 27 | 17L6067×川14品16 17L6067×Chuan 14 pin 16 | 2 | R | Yr9+Yr17+Yr26 | 2.5 |
| 28 | 15L7109×川14品16 15L7109×Chuan 14 pin 16 | 2 | R | Yr9+Yr26 | 3 |
| 29 | 15L7128×川14品16 15L7128×Chuan 14 pin 16 | 2 | R | Yr9+Yr26 | 4 |
| 30 | 15L7152×川14品16 15L7152×Chuan 14 pin 16 | 2 | R | Yr9+Yr18+Yr26 | 2 |
| 31 | 17L6065×川13品6 17L6065×Chuan 13 pin 6 | 2 | R | Yr9+Yr17+Yr26 | 3 |
| 32 | 17L6067×川13品6 17L6067×Chuan 13 pin 6 | 2 | R | Yr9+Yr17+Yr26 | 3 |
| 33 | 17L9154×川13品6 17L9154×Chuan 13 pin 6 | 2 | R | Yr9+Yr17+Yr26 | 3.5 |
| 34 | 17L6065×MR1101 | 2 | R | Yr9+Yr17+Yr26 | 2.5 |
| 35 | 17L6067×MR1101 | 2 | R | Yr9+Yr17+Yr26 | 2.5 |
| 36 | 17L6085×MR1101 | 2 | R | Yr9+Yr17+Yr18+Yr26 | 3.5 |
| 37 | 17L7106×MR1101 | 2 | R | Yr9+Yr26+Yr18 | 2.5 |
| 38 | 17L7030×MR1101 | 2 | R | Yr9+Yr17+Yr26+Yr18 | 4 |
| 39 | 17L7123×MR1101 | 2 | R | Yr9+Yr26+Yr18 | 2.5 |
| 40 | 17L7140×MR1101 | 2 | R | Yr9+Yr26 | 2.5 |
| 41 | 17L9154×MR1101 | 0 | R | Yr9+Yr17+Yr26 | 3 |
| 42 | 15L7109×MR1101 | 0 | R | Yr9+Yr26 | 3 |
| 编号 Number | F1代 F1 hybrids | 反应型 Infection type | 抗病评价 Resistance evaluation | Yr基因 Yr genes | 亲本反应型平均值 Infection type (average of parents) |
| 43 | 15L7128×MR1101 | 1 | R | Yr9 +Yr26 | 4 |
| 44 | 15L7152×MR1101 | 2 | R | Yr9+Yr18+Yr26 | 2 |
| 45 | 08L5070×MR1101 | 2 | R | Yr9+Yr26 | 4 |
| 46 | 17L6065×MY13-3 | 2 | R | Yr9+Yr17 | 2.5 |
| 47 | 17L6067×MY13-3 | 2 | R | Yr9+Yr17 | 2.5 |
| 48 | 17L6085×MY13-3 | 2 | R | Yr9+Yr17+Yr18 | 3.5 |
| 49 | 17L7106×MY13-3 | 2 | R | Yr9+Yr17+Yr18 | 2.5 |
| 50 | 17L7030×MY13-3 | 2 | R | Yr9+Yr17+Yr18 | 4 |
| 51 | 17L7123×MY13-3 | 2 | R | Yr9+Yr17+Yr18 | 2.5 |
| 52 | 17L7140×MY13-3 | 2 | R | Yr9+Yr17 | 2.5 |
| 53 | 17L9154×MY13-3 | 2 | R | Yr9+Yr17 | 3 |
| 54 | 17L9163×MY13-3 | 2 | R | Yr9+Yr17 | 4 |
| 55 | 15L7109×MY13-3 | 2 | R | Yr9+Yr17 | 3 |
| 56 | 15L7128×MY13-3 | 2 | R | Yr9+Yr17 | 4 |
| 57 | 15L7152×MY13-3 | 2 | R | Yr9+Yr17+Yr18 | 2 |
| 58 | 08L5070×MY13-3 | 2 | R | Yr9+Yr17 | 4 |
| 59 | 17L6065×川麦93 17L6065×Chuanmai 93 | 1 | R | Yr9+Yr17+Yr26 | 1.5 |
| 60 | 17L6067×川麦93 17L6067×Chuanmai 93 | 1 | R | Yr9+Yr17+Yr26 | 1.5 |
| 61 | 17L6085×川麦93 17L6085×Chuanmai 93 | 1 | R | Yr9+Yr26 | 2.5 |
| 62 | 17L7106×川麦93 17L7106×Chuanmai 93 | 1 | R | Yr9+Yr26+Yr18 | 1.5 |
| 63 | 17L7030×川麦93 17L7030×Chuanmai 93 | 2 | R | Yr9+Yr17+Yr26+Yr18 | 3 |
| 64 | 17L7123×川麦93 17L7123×Chuanmai 93 | 1 | R | Yr9+Yr26+Yr18 | 1.5 |
| 65 | 17L7140×川麦93 17L7140×Chuanmai 93 | 1 | R | Yr9+Yr26 | 1.5 |
| 66 | 17L9154×川麦93 17L9154×Chuanmai 93 | 1 | R | Yr9+Yr17+Yr26 | 2 |
| 67 | 17L9163×川麦93 17L9163×Chuanmai 93 | 2 | R | Yr9+Yr26 | 3 |
| 68 | 15L7109×川麦93 15L7109×Chuanmai 93 | 2 | R | Yr9+Yr26 | 2 |
| 69 | 15L7128×川麦93 15L7128×Chuanmai 93 | 2 | R | Yr9+Yr26 | 3 |
| 70 | 15L7152×川麦93 15L7152×Chuanmai 93 | 2 | R | Yr9+Yr18+Yr26 | 1 |
| 71 | 08L5070×川麦93 08L5070×Chuanmai 93 | 2 | R | Yr9+Yr26 | 3 |
| 72 | 17L6065×川麦98 17L6065×Chuanmai 98 | 2 | R | Yr9+Yr17+Yr26 | 1.5 |
| 73 | 17L6067×川麦98 17L6067×Chuanmai 98 | 2 | R | Yr9+Yr17+Yr26 | 1.5 |
| 74 | 17L6085×川麦98 17L6085×Chuanmai 98 | 2 | R | Yr9 +Yr17+Yr18+Yr26 | 2.5 |
| 75 | 17L7106×川麦98 17L7106×Chuanmai 98 | 2 | R | Yr9+Yr18+Yr26 | 1.5 |
| 76 | 17L7030×川麦98 17L7030×Chuanmai 98 | 2 | R | Yr9+Yr17+Yr18+Yr26 | 3 |
| 77 | 17L7123×川麦98 17L7123×Chuanmai 98 | 2 | R | Yr9+Yr18+Yr26 | 3 |
| 78 | 17L7140×川麦98 17L7140×Chuanmai 98 | 2 | R | Yr9+Yr26 | 1.5 |
| 79 | 17L9160×15CA50 | 6 | MR | Yr9+Yr17 | 5 |
| 80 | 17L6062×15CA50 | 7 | MS | Yr9 | 5 |
| 81 | 17L7106×15CA50 | 6 | MR | Yr9+Yr18 | 5 |
| 82 | 17L7140×15CA50 | 5 | MR | Yr9 | 5 |
| 83 | 17L9160×伟隆121 17L9160×Weilong 121 | 4 | MR | Yr9+Yr17 | 3.5 |
| 84 | 17L6062×伟隆121 17L6062×Weilong 121 | 4 | MR | Yr9+Yr17 | 3.5 |
| 85 | 17L6065×伟隆121 17L6065×Weilong 121 | 4 | MR | Yr9+Yr17 | 3.5 |
| 86 | 17L7106×伟隆121 17L7106×Weilong 121 | 3 | R | Yr9+Yr17+Yr18 | 3.5 |
新窗口打开|下载CSV
图3
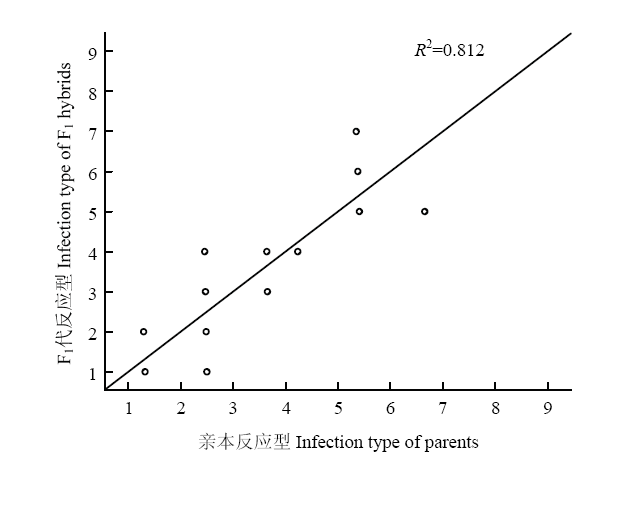 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3亲本-F1代杂交种反应型的二元回归分析
Fig. 3Binary regression analysis of infection type between parents and F1 hybrids
2.3 亲本成株期小种侵染量鉴定
通过半定量PCR方法对中抗、感病亲本及一些F1代杂交种的CYR23、CYR31、CYR33、CYR34侵染量进行鉴定,发现CYR33、CYR34在发病亲本中具有不同侵染量,CYR31在恢复系15CA50、不育系17L6078和15L7128上有少量侵染,没有检测到CYR23的侵染。随机选取表4中的F1代杂交种进行成株期菌量鉴定,发现亲本对不同小种的抗性在F1代杂交种上得到互补,例如15L7109中CYR33、CYR34侵染量较丰德存5号低,其F1代CYR34侵染量较丰德存5号明显降低。CYR34对不育系15L7109侵染量较西农807低,其F1代杂交种CYR34侵染量较西农807明显降低。恢复系川13品6、MR1101、川麦98及其F1代杂交种不发病,也没有检测到条锈菌侵染(图4)。图4
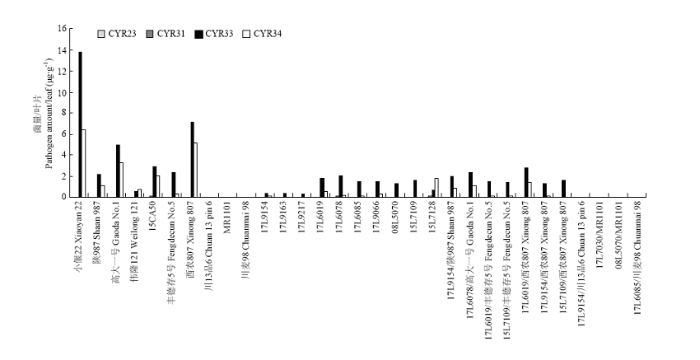 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4CYR23、CYR31、CYR33、CYR34在植株中侵染量
Fig. 4Amount of CYR23, CYR31, CYR33, CYR34 in infected plants
3 讨论
小麦抗病育种对小麦安全生产具有重要意义。MIEDANER等[37]将亲本和杂交种对小麦赤霉病的反应型进行相关性分析,发现亲本反应型的平均值与杂交种反应型有很大相关性,可以用来预测杂交种对赤霉病的抗性,这与本研究的结论一致;史丽丽等[16]在杂交小麦抗白粉病研究中利用成株期抗性的中国春与抗病材料杂交产生的F1代杂交种抗病性接近或略高于抗病亲本,其与感病品种杂交的F1代杂交种抗病性介于双亲之间,并且正反交实验结果为抗病性表现基本一致,表明杂交种抗性由双亲共同决定;LIU等[17]对1 749份小麦材料(15份感病父本,120份部分抗病母本,1 604份F1代杂交种,10份对照)进行多年多点的抗叶锈表型数据分析,发现源自抗病母本的F1代杂交种大多数抗病,总体反应型属于双峰分布,据此得出抗病的F1代杂交种有可能来自于特定亲本的假设。但根据本研究结果,认为更大的原因是部分双亲结合发生了抗性互补[38]。另外,许多材料在经过适当的组合和筛选后会在抗病方面有显著的超亲优势,双亲都显示高抗则F1代杂交种也高抗,双亲抗性有差异但抗性互补,F1代杂交种也能表现优良抗性。双亲抗性基因的显隐性与贡献都直接影响着F1代杂交种的抗性表现。通过对亲本及F1代杂交种成株期菌量的鉴定,发现F1代杂交种能够保持亲本的抗性,扩大对病原菌小种的抗谱范围。CYR33对Yr26无毒性[39,40],本研究中含有Yr26的材料均未发现CYR33侵染。CYR34是2016年正式命名的新小种,其毒谱比CYR32、CYR33更广,并且在全国的小种流行趋势中所占比例逐年增加[39]。恢复系川14品16、川麦93、川麦98、MR1101、MY13-3、伟隆121和不育系17L9154、17L9163、17L9217、17L9066、08L5070等表现优良抗性,表明其中含有对CYR34未知的抗性基因,同时这些材料可以用于我国小麦抗条锈育种。CYR23对Yr9、Yr17、Yr26无毒性,对Yr18弱毒性[27],因此不育系和F1代杂交种中均未检测到该小种,但恢复系15CA50、丰德存5号、西农807未鉴定到抗性基因和CYR23侵染,表明这些材料含有抗CYR23的基因。当前小麦品种存在抗性与农艺性状协同性差的问题,农家品种蕴含优良的抗条锈病基因资源[41],但因综合农艺性状不佳,难以作为亲本直接用于小麦品种改良,因此在育种中好用并发挥重要作用的抗原极少,而具有优异农艺性状的材料往往抗病性不佳,只能达到中感或中抗水平。本研究发现部分不育系和恢复系可以互补,其F1代杂交种明显高于抗性中亲值或与最抗亲本相当,需要进一步对这些亲本的抗性基因进行鉴定,为今后的杂交小麦亲本选择提供帮助,快速、高效地培育新的杂交种应用于生产。
培育持久抗病品种是小麦抗病育种的重要目标,培育早熟品种可以减少锈病对产量的影响,小麦的一些生理特征如气孔大小、蜡质层存在与否、角质层厚度、叶片角度和宽度等与决定持久抗性的特征非常相似[42]。育种过程中常用的选择方式有标记辅助选择、基因组选择以及表型选择等[43,44]。AGOSTINELLI等[45]通过表型选择和标记辅助选择的方法更大效率地提高了小麦杂交种对赤霉病的抗性;SALAMEH等[46]通过标记辅助选育叠加了两个抗赤霉病的QTL位点Fhb1和Qfhs.ifa-5A,抗性比较结果为Fhb1<Qfhs.ifa-5A≤Fhb1+Qfhs.ifa-5A;KIM等[47]在杂交水稻抗病育种方面,利用标记辅助选育出多个抗白叶枯病、稻瘟病、褐飞虱并且农艺性状良好的杂交组合,为水稻的抗病虫育种提供了理论依据。与单个基因控制主要抗性相比,多基因型抗性基因的叠加可以达到持久抗性的目的,研究表明,一般叠加4—5个效应较大微效基因可达到接近免疫的水平,叠加2—3个达到中抗水平[48]。主效基因和微效基因有效结合也能产生良好抗性,如兼抗3种锈病及白粉病的多抗位点Yr18/Lr34/Sr57/Pm38、Yr29/Lr46/Sr58/ Pm39和Yr46/Lr67/Sr55/Pm46与两个微效基因共同存在时会增加成株期抗性[49]。品种抗性基因单一和小种变异是导致小麦条锈病暴发的主要原因,例如由于来自于黑麦的1B/1R易位系抗病基因的大量使用曾导致某些地区小麦条锈病、白粉病大面积流行[50]。Yr9和Yr26分别在北方品系和川渝品系中有较高检出率,由于CYR34毒性较广,应减少对之前单一抗条锈基因的依赖。寻找新抗原迫在眉睫,分子标记能够精确地选择所需性状的材料,并且受环境的影响不大。没有与目的基因连锁的分子标记很难进行抗病基因累加,因此需加强抗病基因定位[51,52]。易感基因是使用基因组编辑工具进行抗性育种的重要目标,与抗性基因相比,有望为农作物提供更持久和更广谱的抗性[53,54]。
4 结论
F1代杂交种对条锈菌的反应型绝大多数趋于双亲反应型的平均值,因此可根据双亲反应型平均值预测F1代杂交种抗性。建议组合具有多个抗条锈病基因和对不同小种有抗性互补的亲本材料,以扩大F1代杂交种对条锈菌生理小种的抗性范围。应选择高产、优质、抗性良好的材料作为杂交种的亲本,才能获得适合生产上大面积推广应用的优势杂交组合。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1126/science.1185383URLPMID:20110467 [本文引用: 1]

Continuing population and consumption growth will mean that the global demand for food will increase for at least another 40 years. Growing competition for land, water, and energy, in addition to the overexploitation of fisheries, will affect our ability to produce food, as will the urgent requirement to reduce the impact of the food system on the environment. The effects of climate change are a further threat. But the world can produce more food and can ensure that it is used more efficiently and equitably. A multifaceted and linked global strategy is needed to ensure sustainable and equitable food security, different components of which are explored here.
DOI:10.1038/nature10452URLPMID:21993620 [本文引用: 1]
Increasing population and consumption are placing unprecedented demands on agriculture and natural resources. Today, approximately a billion people are chronically malnourished while our agricultural systems are concurrently degrading land, water, biodiversity and climate on a global scale. To meet the world's future food security and sustainability needs, food production must grow substantially while, at the same time, agriculture's environmental footprint must shrink dramatically. Here we analyse solutions to this dilemma, showing that tremendous progress could be made by halting agricultural expansion, closing 'yield gaps' on underperforming lands, increasing cropping efficiency, shifting diets and reducing waste. Together, these strategies could double food production while greatly reducing the environmental impacts of agriculture.
DOI:10.1038/ncomms2296URLPMID:23250423 [本文引用: 1]
In the coming decades, continued population growth, rising meat and dairy consumption and expanding biofuel use will dramatically increase the pressure on global agriculture. Even as we face these future burdens, there have been scattered reports of yield stagnation in the world's major cereal crops, including maize, rice and wheat. Here we study data from ~2.5 million census observations across the globe extending over the period 1961-2008. We examined the trends in crop yields for four key global crops: maize, rice, wheat and soybeans. Although yields continue to increase in many areas, we find that across 24-39% of maize-, rice-, wheat- and soybean-growing areas, yields either never improve, stagnate or collapse. This result underscores the challenge of meeting increasing global agricultural demands. New investments in underperforming regions, as well as strategies to continue increasing yields in the high-performing areas, are required.
DOI:10.1371/journal.pone.0066428URLPMID:23840465 [本文引用: 1]
Several studies have shown that global crop production needs to double by 2050 to meet the projected demands from rising population, diet shifts, and increasing biofuels consumption. Boosting crop yields to meet these rising demands, rather than clearing more land for agriculture has been highlighted as a preferred solution to meet this goal. However, we first need to understand how crop yields are changing globally, and whether we are on track to double production by 2050. Using ~2.5 million agricultural statistics, collected for ~13,500 political units across the world, we track four key global crops-maize, rice, wheat, and soybean-that currently produce nearly two-thirds of global agricultural calories. We find that yields in these top four crops are increasing at 1.6%, 1.0%, 0.9%, and 1.3% per year, non-compounding rates, respectively, which is less than the 2.4% per year rate required to double global production by 2050. At these rates global production in these crops would increase by ~67%, ~42%, ~38%, and ~55%, respectively, which is far below what is needed to meet projected demands in 2050. We present detailed maps to identify where rates must be increased to boost crop production and meet rising demands.
DOI:10.1093/jxb/ert333URLPMID:24179097 [本文引用: 1]
Global food security demands the development and delivery of new technologies to increase and secure cereal production on finite arable land without increasing water and fertilizer use. There are several options for boosting wheat yields, but most offer only small yield increases. Wheat is an inbred plant, and hybrids hold the potential to deliver a major lift in yield and will open a wide range of new breeding opportunities. A series of technological advances are needed as a base for hybrid wheat programmes. These start with major changes in floral development and architecture to separate the sexes and force outcrossing. Male sterility provides the best method to block self-fertilization, and modifying the flower structure will enhance pollen access. The recent explosion in genomic resources and technologies provides new opportunities to overcome these limitations. This review outlines the problems with existing hybrid wheat breeding systems and explores molecular-based technologies that could improve the hybrid production system to reduce hybrid seed production costs, a prerequisite for a commercial hybrid wheat system.
[本文引用: 1]
[本文引用: 1]
DOI:10.2135/cropsci2004.1070URL [本文引用: 1]
DOI:10.1007/s11032-011-9555-0URL [本文引用: 1]
Efforts in hybrid breeding have made this technology one of the main factors contributing to the substantial global rise in agricultural output over the last few decades. For hybrid breeding, an efficient pollination control system is necessary to avoid the unwanted self-pollination or sib-pollination of the female parental line. This review will provide a historical overview of pollination control systems and their use in hybrid crop breeding. We outline the prerequisites for commercial hybrid breeding and summarize the most important non-biological and biological technologies. Our main focus is on hybrid systems that are based on genetically engineered plants. We describe their suitability for pollination control, propagation of the male-sterile crossing partner, fertility restoration and mixed planting. Additionally, we report on the latest findings in the development of inducible sterility systems and various technologies that enable pollination control via metabolic engineering. We discuss the pros and cons of the different pollination control strategies.
DOI:10.1038/s41467-017-00945-2URLPMID:29021581 [本文引用: 1]
The current rate of yield gain in crops is insufficient to meet the predicted demands. Capturing the yield boost from heterosis is one of the few technologies that offers rapid gain. Hybrids are widely used for cereals, maize and rice, but it has been a challenge to develop a viable hybrid system for bread wheat due to the wheat genome complexity, which is both large and hexaploid. Wheat is our most widely grown crop providing 20% of the calories for humans. Here, we describe the identification of Ms1, a gene proposed for use in large-scale, low-cost production of male-sterile (ms) female lines necessary for hybrid wheat seed production. We show that Ms1 completely restores fertility to ms1d, and encodes a glycosylphosphatidylinositol-anchored lipid transfer protein, necessary for pollen exine development. This represents a key step towards developing a robust hybridization platform in wheat.Heterosis can rapidly boost yield in crop species but development of hybrid-breeding systems for bread wheat remains a challenge. Here, Tucker et al. describe the molecular identification of the wheat Ms1 gene and discuss its potential for large-scale hybrid seed production in wheat.
DOI:10.1007/s00122-012-1967-7URLPMID:22918662 [本文引用: 1]
Hybrid breeding in autogamous cereals has a long history of attempts with moderate success. There is a vast amount of literature investigating the potential problems and solutions, but until now, market share of hybrids is still a niche compared to line varieties. Our aim was to summarize the status quo of hybrid breeding efforts for the autogamous cereals wheat, rice, barley, and triticale. Furthermore, the research needs for a successful hybrid breeding in autogamous cereals are intensively discussed. To our opinion, the basic requirements for a successful hybrid breeding in autogamous cereals are fulfilled. Nevertheless, optimization of the existing hybridization systems is urgently required and should be coupled with the development of clear male and female pool concepts. We present a quantitative genetic framework as a first step to compare selection gain of hybrid versus line breeding. The lack of precise empirical estimates of relevant quantitative genetic parameters, however, is currently the major bottleneck for a robust evaluation of the potential of hybrid breeding in autogamous cereals.
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s00122-019-03286-4URLPMID:30673804 [本文引用: 1]
Recent technological advances in wheat genomics provide new opportunities to uncover genetic variation in traits of breeding interest and enable genome-based breeding to deliver wheat cultivars for the projected food requirements for 2050. There has been tremendous progress in development of whole-genome sequencing resources in wheat and its progenitor species during the last 5?years. High-throughput genotyping is now possible?in wheat not only for routine gene introgression but also for high-density genome-wide genotyping. This is a major transition phase to enable genome-based breeding to achieve progressive genetic gains to parallel to projected wheat production demands. These advances have intrigued wheat researchers to practice less pursued analytical approaches which were not practiced due to the short history of genome sequence availability. Such approaches have been successful in gene discovery and breeding applications in other crops and animals for which genome sequences have been available for much longer. These?strategies include, (i) environmental?genome-wide association studies in wheat genetic resources stored in genbanks to identify genes for local adaptation?by using agroclimatic traits as phenotypes, (ii) haplotype-based analyses to improve the statistical power and resolution of genomic selection and gene mapping experiments, (iii) new breeding strategies for genome-based prediction of heterosis patterns in wheat, and (iv) ultimate use of genomics information to develop more efficient and robust genome-wide genotyping platforms to precisely predict higher yield potential and stability with greater precision. Genome-based breeding has potential to achieve the ultimate objective of ensuring sustainable wheat production through developing high yielding, climate-resilient wheat cultivars with high nutritional quality.
DOI:10.1111/pbi.13170URLPMID:31124256 [本文引用: 1]
Hybrid breeding is the main strategy for improving productivity in many crops, especially in rice and maize. Genomic hybrid breeding is a technology that uses whole-genome markers to predict future hybrids. Predicted superior hybrids are then field evaluated and released as new hybrid cultivars after their superior performances are confirmed. This will increase the opportunity of selecting true superior hybrids with minimum costs. Here, we used genomic best linear unbiased prediction to perform hybrid performance prediction using an existing rice population of 1495 hybrids. Replicated 10-fold cross-validations showed that the prediction abilities on ten agronomic traits ranged from 0.35 to 0.92. Using the 1495 rice hybrids as a training sample, we predicted six agronomic traits of 100 hybrids derived from half diallel crosses involving 21 parents that are different from the parents of the hybrids in the training sample. The prediction abilities were relatively high, varying from 0.54 (yield) to 0.92 (grain length). We concluded that the current population of 1495 hybrids can be used to predict hybrids from seemingly unrelated parents. Eventually, we used this training population to predict all potential hybrids of cytoplasm male sterile lines from 3000 rice varieties from the 3K Rice Genome Project. Using a breeding index combining 10 traits, we identified the top and bottom 200 predicted hybrids. SNP genotypes of the training population and parameters estimated from this training population are available for general uses and further validation in genomic hybrid prediction of all potential hybrids generated from all varieties of rice.
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s00122-019-03397-yURLPMID:31321476 [本文引用: 1]
The review outlines past failures, present status and future prospects of hybrid wheat, and includes information on CMS/CHA/transgenic approaches for male sterility, heterotic groups and cost-effective hybrid seed production. Hybrid varieties give increased yield and improved grain quality in both cross- and self-pollinated crops. However, hybrid varieties in self-pollinated crops (particularly cereals) have not been very successful, except for hybrid rice in China. In case of hybrid wheat, despite the earlier failures, renewed efforts in recent years have been made and hybrid varieties with desirable attributes have been produced and marketed in some European countries. This review builds upon previous reviews, with a new outlook and improved knowledge base, not covered in earlier reviews. New technologies have been described, which include the Hordeum chilense-based CMS-fertility restorer system, chromosomal XYZ-4E-ms system and the following transgenic technologies: (1) conditional male sterility involving use of tapetum-specific expression of a gene that converts a pro-toxin into a phytotoxin causing male sterility; (2) barnase-barstar SeedLink system of Bayer CropScience; (3) split-barnase system that obviates the need of a barstar-based male restorer line; and (4) seed production technology of DuPont-Pioneer that makes use of transgenes in production of male-sterile lines, but gives hybrid seed with no transgenes. This review also includes a brief account of studies for discovery of heterotic QTL, genomic prediction of hybrid vigour and the development of heterotic groups/patterns and their importance in hybrid wheat production. The problem of high cost of hybrid seed due to required high seed rate in wheat relative to hybrid rice has also been addressed. The review concludes with a brief account of the current efforts and future possibilities in making hybrid wheat a commercial success.
[本文引用: 2]
[本文引用: 2]
DOI:10.1111/pbi.13303URLPMID:31782598 [本文引用: 2]
Resistance breeding is crucial for a sustainable control of leaf rust (Puccinia triticina) in wheat (Triticum aestivum L.) while directly targeting functional variants is the Holy Grail for efficient marker-assisted selection and map-based cloning. We assessed the limits and prospects of exome association analysis for severity of leaf rust in a large hybrid wheat population of 1574 single-crosses plus their 133 parents. After imputation and quality control, exome sequencing revealed 202?875 single-nucleotide polymorphisms (SNPs) covering 19.7% of the high-confidence annotated gene space. We performed intensive data mining and found significant associations for 2171 SNPs corresponding to 50 different loci. Some of these associations mapped in the proximity of the already known resistance genes Lr21, Lr34-B, Lr1 and Lr10, while other associated genomic regions, such as those on chromosomes 1A and 3D, harboured several annotated genes putatively involved in resistance. Validation with an independent population helped to narrow down the list of putative resistance genes that should be targeted by fine-mapping. We expect that the proposed strategy of intensive data mining coupled with validation will significantly influence research in plant genetics and breeding.
DOI:10.1007/s10681-015-1498-9URL [本文引用: 1]
DOI:10.1038/hdy.2016.36URLPMID:27245423 [本文引用: 1]
Pyramiding of alien-derived Wheat streak mosaic virus (WSMV) resistance and resistance enhancing genes in wheat is a cost-effective and environmentally safe strategy for disease control. PCR-based markers and cytogenetic analysis with genomic in situ hybridisation were applied to identify alien chromatin in four genetically diverse populations of wheat (Triticum aestivum) lines incorporating chromosome segments from Thinopyrum intermedium and Secale cereale (rye). Out of 20 experimental lines, 10 carried Th. intermedium chromatin as T4DL*4Ai#2S translocations, while, unexpectedly, 7 lines were positive for alien chromatin (Th. intermedium or rye) on chromosome 1B. The newly described rye 1RS chromatin, transmitted from early in the pedigree, was associated with enhanced WSMV resistance. Under field conditions, the 1RS chromatin alone showed some resistance, while together with the Th. intermedium 4Ai#2S offered superior resistance to that demonstrated by the known resistant cultivar Mace. Most alien wheat lines carry whole chromosome arms, and it is notable that these lines showed intra-arm recombination within the 1BS arm. The translocation breakpoints between 1BS and alien chromatin fell in three categories: (i) at or near to the centromere, (ii) intercalary between markers UL-Thin5 and Xgwm1130 and (iii) towards the telomere between Xgwm0911 and Xbarc194. Labelled genomic Th. intermedium DNA hybridised to the rye 1RS chromatin under high stringency conditions, indicating the presence of shared tandem repeats among the cereals. The novel small alien fragments may explain the difficulty in developing well-adapted lines carrying Wsm1 despite improved tolerance to the virus. The results will facilitate directed chromosome engineering producing agronomically desirable WSMV-resistant germplasm.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1111/mpp.12116URLPMID:24373199 [本文引用: 1]
Stripe (yellow) rust, caused by Puccinia striiformis f. sp. tritici (Pst), is a serious disease of wheat occurring in most wheat areas with cool and moist weather conditions during the growing season. The basidiomycete fungus is an obligate biotrophic parasite that is difficult to culture on artificial media. Pst is a macrocyclic, heteroecious fungus that requires both primary (wheat or grasses) and alternate (Berberis or Mahonia spp.) host plants to complete its life cycle. Urediniospores have the capacity for wind dispersal over long distances, which may, under high inoculum pressure, extend to thousands of kilometres from the initial infection sites. Stripe rust, which is considered to be the current major rust disease affecting winter cereal production across the world, has been studied intensively for over a century. This review summarizes the current knowledge of the Pst-wheat pathosystem, with emphasis on the life cycle, uredinial infection process, population biology of the pathogen, genes for stripe rust resistance in wheat and molecular perspectives of wheat-Pst interactions. Taxonomy The stripe rust pathogen, Puccinia striiformis Westend. (Ps), is classified in kingdom Fungi, phylum Basidiomycota, class Urediniomycetes, order Uredinales, family Pucciniaceae, genus Puccinia. Ps is separated below the species level by host specialization on various grass genera, comprising up to nine formae speciales, of which P. striiformis f. sp. tritici Erikss. (Pst) causes stripe (or yellow) rust on wheat. Host range Uredinial/telial hosts: Pst mainly infects common wheat (Triticum aestivum L.), durum wheat (T. turgidum var. durum L.), cultivated emmer wheat (T. dicoccum Schrank), wild emmer wheat (T. dicoccoides Korn) and triticale (Triticosecale). Pst can infect certain cultivated barleys (Hordeum vulgare L.) and rye (Secale cereale L.), but generally does not cause severe epidemics. In addition, Pst may infect naturalized and improved pasture grass species, such as Elymus canadensis L. , Leymus secalinus Hochst, Agropyron spp. Garetn, Hordeum spp. L., Phalaris spp. L and Bromus unioloides Kunth. Pycnial/aecial (alternative) hosts: Barberry (Berberis chinensis, B. koreana, B. holstii, B. vulgaris, B. shensiana, B. potaninii, B. dolichobotrys, B. heteropoda, etc.) and Oregon grape (Mahonia aquifolium). Disease symptoms Stripe rust appears as a mass of yellow to orange urediniospores erupting from pustules arranged in long, narrow stripes on leaves (usually between veins), leaf sheaths, glumes and awns on susceptible plants. Resistant wheat cultivars are characterized by various infection types from no visual symptoms to small hypersensitive flecks to uredinia surrounded by chlorosis or necrosis with restricted urediniospore production. On seedlings, uredinia produced by the infection of a single urediniospore are not confined by leaf veins, but progressively emerge from the infection site in all directions, potentially covering the entire leaf surface. Individual uredinial pustules are oblong, 0.4-0.7 mm in length and 0.1 mm in width. Urediniospores are broadly ellipsoidal to broadly obovoid, (16-)18-30(-32) x (15-)17-27(-28) mu m, with a mean of 24.5 x 21.6 mu m, yellow to orange in colour, echinulate, and with 6-18 scattered germ pores. Urediniospores can germinate rapidly when free moisture (rain or dew) occurs on leaf surfaces and when the temperatures range is between 7 and 12 degrees C. At higher temperatures or during the later growing stages of the host, black telia are often produced, which are pulvinate to oblong, 0.2-0.
7 mm in length and 0.1 mm in width. The teliospores are predominantly two-celled, dark brown with thick walls, mostly oblong-clavate, (24-)31-56(-65) x (11-)14-25(-29) mu m in length and width, and rounded or flattened at the apex.
DOI:10.3864/j.issn.0578-1752.2015.17.011URL [本文引用: 1]
Wheat stripe rust, caused by Puccinia striiformis West. f. sp. tritici Eriks. and Henn. (Pst), is an air-borne cryogenic fungal disease, which causes serious yield and economic losses and severely threatens the security of wheat production worldwide due to its wide transmission and high frequent epidemics. As an obligate parasitic pathogen, the rapid virulence variation of Pst leads to resistance breakdown of the existing resistant wheat cultivars, which then leads to the constant epidemic of the disease. Thus, the study on virulence variation mechanism of Pst has been paid much attention since 1950s. Until now, great efforts have been made and significant progress has been achieved. The progress in research of the disease made in recent years and the relationship between resistance breakdown of wheat cultivar and virulence variation, the variation ways of Pst pathogenicity, genetic structure analyses of Pst population and the molecular mechanism underlying the rapid virulence variation of Pst were summarized. Based on this review, the issues and challenges were put forward to accurately predict main predominant races, rationally use and distribute the resistance genes, and to create the new kind of disease-resistance materials through novel strategies, which will optimize and improve the comprehensive management system for wheat stripe rust, and help to realize the sustainable control of wheat stripe rust.
DOI:10.3864/j.issn.0578-1752.2015.17.011URL [本文引用: 1]
Wheat stripe rust, caused by Puccinia striiformis West. f. sp. tritici Eriks. and Henn. (Pst), is an air-borne cryogenic fungal disease, which causes serious yield and economic losses and severely threatens the security of wheat production worldwide due to its wide transmission and high frequent epidemics. As an obligate parasitic pathogen, the rapid virulence variation of Pst leads to resistance breakdown of the existing resistant wheat cultivars, which then leads to the constant epidemic of the disease. Thus, the study on virulence variation mechanism of Pst has been paid much attention since 1950s. Until now, great efforts have been made and significant progress has been achieved. The progress in research of the disease made in recent years and the relationship between resistance breakdown of wheat cultivar and virulence variation, the variation ways of Pst pathogenicity, genetic structure analyses of Pst population and the molecular mechanism underlying the rapid virulence variation of Pst were summarized. Based on this review, the issues and challenges were put forward to accurately predict main predominant races, rationally use and distribute the resistance genes, and to create the new kind of disease-resistance materials through novel strategies, which will optimize and improve the comprehensive management system for wheat stripe rust, and help to realize the sustainable control of wheat stripe rust.
URL [本文引用: 1]
In order to investigate the yellow rust resistant genes (Yr genes) of 75 wheat cultivars approved to be released by the National Crop Variety Approval Committee (NCVAC) of China, seedlings of 75 Chinese commercial wheat cultivars were inoculated with 21 Pst isolates from different countries or regions to postulate yellow rust resistance genes (Yr) under greenhouse conditions. The results showed that a total of 16 seedling yellow rust resistance genes or gene combinations (Yr1, Yr2, Yr3, Yr5, Yr6, Yr7, Yr8, Yr9, Yr17, Yr27, YrA, Yr25, YrSu, YrSp, YrSkand YrSk) were postulated in 49 wheat cultivars. Of the Yr genes identified among the 75 wheat lines, Yr3 was the most frequent (26.7%), followed by Yr2 and Yr9 (20.0 and 16.0%, respectively). Yr2 and Yr3 were postulated at a ratio of 14.7% in combinations. Gene Yr1 and YrSp were each present in 13.6% and the other postulated Yr genes were below 10.0%. Zhenmai 8, Ningmai 15 and Shengxuan 6 were susceptible to all Pst strains, as a result of no known Yr genes could be postulated.
URL [本文引用: 1]
In order to investigate the yellow rust resistant genes (Yr genes) of 75 wheat cultivars approved to be released by the National Crop Variety Approval Committee (NCVAC) of China, seedlings of 75 Chinese commercial wheat cultivars were inoculated with 21 Pst isolates from different countries or regions to postulate yellow rust resistance genes (Yr) under greenhouse conditions. The results showed that a total of 16 seedling yellow rust resistance genes or gene combinations (Yr1, Yr2, Yr3, Yr5, Yr6, Yr7, Yr8, Yr9, Yr17, Yr27, YrA, Yr25, YrSu, YrSp, YrSkand YrSk) were postulated in 49 wheat cultivars. Of the Yr genes identified among the 75 wheat lines, Yr3 was the most frequent (26.7%), followed by Yr2 and Yr9 (20.0 and 16.0%, respectively). Yr2 and Yr3 were postulated at a ratio of 14.7% in combinations. Gene Yr1 and YrSp were each present in 13.6% and the other postulated Yr genes were below 10.0%. Zhenmai 8, Ningmai 15 and Shengxuan 6 were susceptible to all Pst strains, as a result of no known Yr genes could be postulated.
[P]. (
[本文引用: 1]
[P]. (
[本文引用: 1]
DOI:10.1038/s41477-018-0236-4URLPMID:30150615 [本文引用: 2]
Crop diseases reduce wheat yields by ~25% globally and thus pose a major threat to global food security1. Genetic resistance can reduce crop losses in the field and can be selected through the use of molecular markers. However, genetic resistance often breaks down following changes in pathogen virulence, as experienced with the wheat yellow (stripe) rust fungus Puccinia striiformis f. sp. tritici (Pst)2. This highlights the need to (1) identify genes that, alone or in combination, provide broad-spectrum resistance, and (2) increase our understanding of the underlying molecular modes of action. Here we report the isolation and characterization of three major yellow rust resistance genes (Yr7, Yr5 and YrSP) from hexaploid wheat (Triticum aestivum), each having a distinct recognition specificity. We show that Yr5, which remains effective to a broad range of Pst isolates worldwide, is closely related yet distinct from Yr7, whereas YrSP is a truncated version of Yr5 with 99.8% sequence identity. All three Yr genes belong to a complex resistance gene cluster on chromosome 2B encoding nucleotide-binding and leucine-rich repeat proteins (NLRs) with a non-canonical N-terminal zinc-finger BED domain3 that is distinct from those found in non-NLR wheat proteins. We developed diagnostic markers to accelerate haplotype analysis and for marker-assisted selection to expedite the stacking of the non-allelic Yr genes. Our results provide evidence that the BED-NLR gene architecture can provide effective field-based resistance to important fungal diseases such as wheat yellow rust.
DOI:10.1007/s10681-013-1030-zURL [本文引用: 7]
Stripe rust, caused by Puccinia striiformis f. sp. tritici (Pst), is one of the most important diseases on wheat in China. To assess resistance in wheat cultivars and breeding lines in China, 330 leading cultivars and 164 advanced breeding lines were evaluated with stripe rust. In the greenhouse tests, seedlings of the entries were inoculated separately with several Pst pathotypes. In the field tests, the entries were evaluated for stripe rust resistance in Yangling, Shaanxi Province artificially inoculated and in Tianshui, Gansu Province under natural infection of Pst. The oversummering/wintering and spring epidemic zones of resistance genes were postulated using molecular markers for Yr5, Yr9, Yr10, Yr15, Yr17, Yr18, and Yr26, in combination with resistance spectra. Out of the 494 wheat entries, 16 (3.24 %) entries had all-stage resistance (ASR) in all race tests, 99 (20.04 %) had adult-plant resistance (APR), 28 (5.67 %) were considered to have slow-rusting (SR), and 351 (71.05 %) were susceptible to one or more races in both seedling and adult-plant stages. Advanced breeding lines had a higher percentage (37.2 %) of resistant entries (The sum of ASR, APR and SR) than leading cultivars (24.85 %). Among the epidemic regions, southern Gansu had a higher percentage of resistant entries than any other regions. Based on stripe rust reactions and molecular markers, two cultivars were found to possibly have Yr5 while no entries have Yr10 or Yr15. Resistance genes Yr9, Yr17, Yr18, and Yr26 were found in 134 (29.4 %), 45 (9.1 %), 10 (2 %), and 15 (3 %) entries, respectively.
DOI:10.1038/s41467-018-06138-9URLPMID:30282993 [本文引用: 2]
Yellow rust, caused by Puccinia striiformis f. sp. tritici (Pst), is a devastating fungal disease threatening much of global wheat production. Race-specific resistance (R)-genes are used to control rust diseases, but the rapid emergence of virulent Pst races has prompted the search for a more durable resistance. Here, we report the cloning of Yr15, a broad-spectrum R-gene derived from wild emmer wheat, which encodes a putative kinase-pseudokinase protein, designated as wheat tandem kinase 1, comprising a unique R-gene structure in wheat. The existence of a similar gene architecture in 92 putative proteins across the plant kingdom, including the barley RPG1 and a candidate for Ug8, suggests that they are members of a distinct family of plant proteins, termed here tandem kinase-pseudokinases (TKPs). The presence of kinase-pseudokinase structure in both plant TKPs and the animal Janus kinases sheds light on the molecular evolution of immune responses across these two kingdoms.
[本文引用: 1]
DOI:10.3864/j.issn.0578-1752.2017.03.001URL [本文引用: 1]
【目的】重庆是中国小麦条锈病流行体系中重要冬繁区,准确评价该地区小麦品种(系)对当前小麦条锈病流行小种的抗性和了解抗条锈基因在该区的分布状况,为小麦安全生产、品种合理布局及小麦抗条锈育种工作提供依据。【方法】从该区征集了18份当地主栽品种和89份高代品系材料,应用中国小麦条锈菌流行生理小种条中32(CYR32)、条中33(CYR33)、V26/G22-9和V26/CM42,在杨凌进行苗期分小种(CYR32、CYR33、V26/G22-9和V26/CM42)温室抗病性鉴定、并于2015年和2016年连续两年分别进行杨凌成株期条锈菌混合小种(CYR32、CYR33)人工接种病圃和天水自然诱发条锈菌病圃鉴定,根据苗期和田间成株期的抗病性鉴定结果对其进行抗病类型分类和评价;结合抗谱分析、参照单基因系材料的感病结果,及以Yr5、Yr9、Yr10 、Yr15、Yr17、Yr18和Yr26等7个已知抗条锈基因的标记分别进行的分子检测分析,推测小麦材料可能携带抗病基因。【结果】在107份参鉴材料中,苗期对CYR32与CYR33均表现免疫或者近免疫的品种(系)有57份,占53.27%;对CYR32、CYR33和V26/CM42均表现免疫或者近免疫的品种(系)只有11份,占10.28%;对CYR32、CYR33和V26/G22-9均表现免疫或者近免疫的品种(系)只有9份,占8.41%。综合评价,全生育期抗性的材料仅有8份,占7.48%;成株期抗病材料仅有9份,占8.41%;感病材料90份,占84.11%。分子检测表明,供试材料中21份可能含有Yr9,39份可能含有Yr26,17份可能含有Yr17,3份可能含有Yr18。其他材料中未检测到上述Yr基因(分子标记)的存在,其中没有发现可能含Yr5、Yr10和Yr15的材料。8份具有全生育期抗性的材料,未检测到上述Yr基因(分子标记)的存在,可能含有未检测到的其他抗病基因。【结论】重庆地区小麦品种(系)对小麦条锈菌当前流行小种的抗性整体水平较低,尤其是含Yr26的材料在育种中被广泛而单一地利用。建议利用多基因聚合育种等手段提高当地小麦品种的抗条锈性。
DOI:10.3864/j.issn.0578-1752.2017.03.001URL [本文引用: 1]
【目的】重庆是中国小麦条锈病流行体系中重要冬繁区,准确评价该地区小麦品种(系)对当前小麦条锈病流行小种的抗性和了解抗条锈基因在该区的分布状况,为小麦安全生产、品种合理布局及小麦抗条锈育种工作提供依据。【方法】从该区征集了18份当地主栽品种和89份高代品系材料,应用中国小麦条锈菌流行生理小种条中32(CYR32)、条中33(CYR33)、V26/G22-9和V26/CM42,在杨凌进行苗期分小种(CYR32、CYR33、V26/G22-9和V26/CM42)温室抗病性鉴定、并于2015年和2016年连续两年分别进行杨凌成株期条锈菌混合小种(CYR32、CYR33)人工接种病圃和天水自然诱发条锈菌病圃鉴定,根据苗期和田间成株期的抗病性鉴定结果对其进行抗病类型分类和评价;结合抗谱分析、参照单基因系材料的感病结果,及以Yr5、Yr9、Yr10 、Yr15、Yr17、Yr18和Yr26等7个已知抗条锈基因的标记分别进行的分子检测分析,推测小麦材料可能携带抗病基因。【结果】在107份参鉴材料中,苗期对CYR32与CYR33均表现免疫或者近免疫的品种(系)有57份,占53.27%;对CYR32、CYR33和V26/CM42均表现免疫或者近免疫的品种(系)只有11份,占10.28%;对CYR32、CYR33和V26/G22-9均表现免疫或者近免疫的品种(系)只有9份,占8.41%。综合评价,全生育期抗性的材料仅有8份,占7.48%;成株期抗病材料仅有9份,占8.41%;感病材料90份,占84.11%。分子检测表明,供试材料中21份可能含有Yr9,39份可能含有Yr26,17份可能含有Yr17,3份可能含有Yr18。其他材料中未检测到上述Yr基因(分子标记)的存在,其中没有发现可能含Yr5、Yr10和Yr15的材料。8份具有全生育期抗性的材料,未检测到上述Yr基因(分子标记)的存在,可能含有未检测到的其他抗病基因。【结论】重庆地区小麦品种(系)对小麦条锈菌当前流行小种的抗性整体水平较低,尤其是含Yr26的材料在育种中被广泛而单一地利用。建议利用多基因聚合育种等手段提高当地小麦品种的抗条锈性。
DOI:10.1007/s00294-003-0397-0URLPMID:12740713 [本文引用: 1]
A straightforward and easy-to-apply semi-quantitative RT-PCR method was developed to study multigenic expression in the phytopathogenic fungus Botrytis cinerea. This procedure is based on the one-step reverse transcription-amplification of a specific transcript within total RNA and product amount determination by densitometric analysis of ethidium bromide fluorescence upon gel electrophoresis. The semi-quantitative analysis is achieved, at a fixed PCR cycle-number, within a range of total RNA concentrations that stays in the exponential phase of the PCR. Co-amplification of the transcript of interest with internal controls allowed comparison between different RNA samples. Using this method, we could demonstrate a differential regulation of chitin synthase genes during fungal growth and an effect of the culture carbon source on the expression of two pectin methylesterase genes in B. cinerea. Finally, the method was shown to be applicable to plant-infected tissue, making it a useful tool to detect pathogenicity genes in B. cinerea.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1094/PDIS-94-2-0221URLPMID:30754258 [本文引用: 1]
Wheat stripe rust, caused by Puccinia striiformis f. sp. tritici, is a devastating disease in China. Races CYR32 and CYR33 have been predominant in the recent P. striiformis f. sp. tritici population. To develop molecular markers for these races, initially 86 isolates, most of which were collected in 2007 throughout China, were tested on the set of wheat genotypes for differentiating Chinese P. striiformis f. sp. tritici races, and their genomic DNA were amplified with 94 random amplified polymorphic DNA (RAPD) primers. Twelve isolates were identified as CYR33, 14 as CYR32, and 60 as 13 other races. A 320-bp band was identified to be associated with CYR32 with primer S1271 (5'-CTTCTCGGTC-3'), and a 550-bp band was identified to be specific to CYR33 with primer S1304 (5'-AGGAGCGACA-3'). The two bands were cloned and sequenced. Based on the sequences, sequence characterized amplified region (SCAR) markers CYR32sp1/sp2 and CYR33sp1/sp2 were developed to differentiate CYR32 and CYR33, respectively, from other races. The SCAR markers were validated with DNA samples from wheat leaves inoculated with selected isolates from the 86 isolates and urediniospore DNA samples from an additional 63 isolates collected from 2006 to 2009. The detection of CYR32 and CYR33 with the SCAR markers was completely consistent with the results of the race identification with the set of differential wheat genotypes. Thus, the markers are highly reliable for identification of the two races.
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s00122-016-2826-8URLPMID:27866226 [本文引用: 1]
Mid-parent values of Fusarium head blight (FHB) resistance tested across several locations are a good predictor of hybrid performance caused by a preponderance of additive gene action in wheat. Hybrid breeding is intensively discussed as one solution to boost yield and yield stability including an enhanced biotic stress resistance. Our objectives were to investigate (1) the heterosis for Fusarium head blight (FHB) resistance, (2) the importance of general (GCA) vs. specific combining ability (SCA) for FHB resistance, and (3) the possibility to predict the FHB resistance of the hybrids by the parental means. We re-analyzed phenotypic data of a large population comprising 1604 hybrids and their 120 female and 15 male parental lines evaluated in inoculation trials across seven environments. Mid-parent heterosis of FHB severity averaged -9%, with a range from -36 to +35%. Mean better parent heterosis was 2% and 78 of the hybrids significantly (P?&lt;?0.05) outperformed the best commercial check variety included in our study. FHB resistance was not correlated with grain yield in healthy status for lines (r?=?0.01) and hybrids (r?=?0.09, P?&lt;?0.01). While a preponderance of GCA variance (P?&lt;?0.01) was found, SCA variance was not significantly different from zero. Accuracy to predict hybrid performance of FHB severity based on mid-parent values and on GCA effects was high (r?=?0.70 and 0.86, respectively; P?&lt;?0.01). Similarly, line per se performance and GCA effects were significantly correlated (r?=?0.77; P?&lt;?0.01). The substantial level of mid-parent heterosis in the desired direction of decreased susceptibility and the negligible better parent heterosis suggest that hybrids are an attractive alternative variety type to improve FHB resistance.
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1371/journal.pone.0158635URLPMID:27383841 [本文引用: 1]
Bread-making quality traits are central targets for wheat breeding. The objectives of our study were to (1) examine the presence of major effect QTLs for quality traits in a Central European elite wheat population, (2) explore the optimal strategy for predicting the hybrid performance for wheat quality traits, and (3) investigate the effects of marker density and the composition and size of the training population on the accuracy of prediction of hybrid performance. In total 135 inbred lines of Central European bread wheat (Triticum aestivum L.) and 1,604 hybrids derived from them were evaluated for seven quality traits in up to six environments. The 135 parental lines were genotyped using a 90k single-nucleotide polymorphism array. Genome-wide association mapping initially suggested presence of several quantitative trait loci (QTLs), but cross-validation rather indicated the absence of major effect QTLs for all quality traits except of 1000-kernel weight. Genomic selection substantially outperformed marker-assisted selection in predicting hybrid performance. A resampling study revealed that increasing the effective population size in the estimation set of hybrids is relevant to boost the accuracy of prediction for an unrelated test population.
[本文引用: 1]
DOI:10.1534/g3.116.032532URLPMID:27440921 [本文引用: 1]
Genomic selection (GS) is a breeding tool that estimates breeding values (GEBVs) of individuals based solely on marker data by using a model built using phenotypic and marker data from a training population (TP). The effectiveness of GS increases as the correlation of GEBVs and phenotypes (accuracy) increases. Using phenotypic and genotypic data from a TP of 470 soft winter wheat lines, we assessed the accuracy of GS for grain yield, Fusarium Head Blight (FHB) resistance, softness equivalence (SE), and flour yield (FY). Four TP data sampling schemes were tested: (1) use all TP data, (2) use subsets of TP lines with low genotype-by-environment interaction, (3) use subsets of markers significantly associated with quantitative trait loci (QTL), and (4) a combination of 2 and 3. We also correlated the phenotypes of relatives of the TP to their GEBVs calculated from TP data. The GS accuracy within the TP using all TP data ranged from 0.35 (FHB) to 0.62 (FY). On average, the accuracy of GS from using subsets of data increased by 54% relative to using all TP data. Using subsets of markers selected for significant association with the target trait had the greatest impact on GS accuracy. Between-environment prediction accuracy was also increased by using data subsets. The accuracy of GS when predicting the phenotypes of TP relatives ranged from 0.00 to 0.85. These results suggest that GS could be useful for these traits and GS accuracy can be greatly improved by using subsets of TP data.
DOI:10.1007/s11032-010-9498-xURL [本文引用: 1]
DOI:10.9787/PBB.2019.7.3.272URL [本文引用: 1]
[本文引用: 1]
DOI:10.1146/annurev-phyto-080615-095835URLPMID:27296137 [本文引用: 1]
Wheat is grown worldwide in diverse geographical regions, environments, and production systems. Although many diseases and pests are known to reduce grain yield potential and quality, the three rusts and powdery mildew fungi have historically caused major crop losses and continue to remain economically important despite the widespread use of host resistance and fungicides. The evolution and fast spread of virulent and more aggressive race lineages of rust fungi have only worsened the situation. Fusarium head blight, leaf spotting diseases, and, more recently, wheat blast (in South America and Bangladesh) have become diseases of major importance in recent years largely because of intensive production systems, the expansion of conservation agriculture, undesirable crop rotations, or increased dependency on fungicides. High genetic diversity for race-specific and quantitative resistance is known for most diseases; their selection through phenotyping reinforced with molecular strategies offers great promise in achieving more durable resistance and enhancing global wheat productivity.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/nbt.2969URLPMID:25038773 [本文引用: 1]
Sequence-specific nucleases have been applied to engineer targeted modifications in polyploid genomes, but simultaneous modification of multiple homoeoalleles has not been reported. Here we use transcription activator-like effector nuclease (TALEN) and clustered, regularly interspaced, short palindromic repeats (CRISPR)-Cas9 (refs. 4,5) technologies in hexaploid bread wheat to introduce targeted mutations in the three homoeoalleles that encode MILDEW-RESISTANCE LOCUS (MLO) proteins. Genetic redundancy has prevented evaluation of whether mutation of all three MLO alleles in bread wheat might confer resistance to powdery mildew, a trait not found in natural populations. We show that TALEN-induced mutation of all three TaMLO homoeologs in the same plant confers heritable broad-spectrum resistance to powdery mildew. We further use CRISPR-Cas9 technology to generate transgenic wheat plants that carry mutations in the TaMLO-A1 allele. We also demonstrate the feasibility of engineering targeted DNA insertion in bread wheat through nonhomologous end joining of the double-strand breaks caused by TALENs. Our findings provide a methodological framework to improve polyploid crops.
DOI:10.1016/j.tplants.2015.01.010URLPMID:25726138 [本文引用: 1]
Although genome-editing technologies facilitate efficient plant breeding without introducing a transgene, it is creating indistinct boundaries in the regulation of genetically modified organisms (GMOs). Rapid advances in plant breeding by genome-editing require the establishment of a new global policy for the new biotechnology, while filling the gap between process-based and product-based GMO regulations. In this Opinion article we review recent developments in producing major crops using genome-editing, and we propose a regulatory model that takes into account the various methodologies to achieve genetic modifications as well as the resulting types of mutation. Moreover, we discuss the future integration of genome-editing crops into society, specifically a possible response to the 'Right to Know' movement which demands labeling of food that contains genetically engineered ingredients.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}