Fungal Community Structure of Cotton-Field Soil Under Different Incidences of Cotton Verticillium Wilt
LIU HaiYang1, WANG Wei1, ZHANG RenFu1, RAXIDA ·ABDURAHMAN2, YAO Ju1通讯作者:
收稿日期:2018-08-7接受日期:2018-11-16网络出版日期:2019-02-13
| 基金资助: |
Received:2018-08-7Accepted:2018-11-16Online:2019-02-13
作者简介 About authors
刘海洋,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (2976KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
刘海洋, 王伟, 张仁福, 热西达·阿不都热合曼, 姚举. 黄萎病不同发生程度棉田土壤中的真菌群落特征分析[J]. 中国农业科学, 2019, 52(3): 455-465 doi:10.3864/j.issn.0578-1752.2019.03.006
LIU HaiYang, WANG Wei, ZHANG RenFu, RAXIDA ·ABDURAHMAN, YAO Ju.
0 引言
【研究意义】真菌是土壤微生物系统的重要组成部分,其种群结构的变化能够反映土壤生态的状态及变化趋势[1]。真菌在土壤生态系统中作为分解者[2],其分泌的酶可以降解易分解化合物,有利于植物吸收氮、磷等营养元素[3],在促进土壤生态系统物质循环、形成土壤团聚体和改良土壤结构等方面具有重要作用[4,5]。与细菌相比,真菌能更好地降解复杂化合物[6],对土壤环境的变化更敏感[7],其动态变化可作为土壤生态变化规律的指示因子[8,9]。棉花黄萎病菌(大丽轮枝菌Verticillium dahliae)可在土壤中长期存活,一旦入侵土壤便难以消除。作为土传病害,棉花黄萎病的发生与土壤微生物群落结构、土壤养分指标等因素密切相关。对新疆棉花黄萎病不同发生程度棉田土壤中的真菌群落特征进行分析,研究黄萎病菌入侵对棉田土壤中真菌多样性和群落组成的影响,有助于揭示病菌侵染与土壤中真菌群落的互作关系。【前人研究进展】顾美英等[10]研究表明,发病棉花根际土壤中的微生物总量、真菌数量均大于健康棉株,认为大丽轮枝菌侵染棉花能导致其根际土壤中的真菌数量大幅度增加;LUAN等[11]报道发病棉花根际土壤中镰孢菌属(Fusarium)、轮枝菌属(Verticillium)数量较多,而在健康棉花根际土中木霉属(Trichoderma)等有益菌的数量显著高于发病棉花。其他植物受病原菌侵染后亦有较为一致的研究结论,邓晓等[12]研究表明香蕉枯萎病危害程度中、重度土壤中真菌种类无明显改变,但是数量较危害程度轻和无危害土壤却明显增加。植物根腐病的发生能够引起土壤中微生物群落结构的明显改变,王玲娜等[13]研究表明,芹菜根腐病发病初期病株根区真菌的数量较健康株有所减少,而在发病中、晚期病株真菌数量均较健康株有所增加;李雪萍等[14]研究发现,青稞根腐病的发生会使根际土壤细菌和放线菌数量减少,真菌数量增多;官会林等[15]研究表明,三七根腐病的发生与土壤中厌氧细菌、真菌及放线菌数量的增加及其种群数量比例变化有密切的关联性。叶部病害的发生同样能够引起土壤中真菌的富集,吴斌等[16]研究发现,作物地上组织发病如小麦黄花叶病毒病发病严重地块的真菌数量和种类显著增加。【本研究切入点】目前,利用平板计数等传统方法分析棉花黄萎病株根际土壤中可培养微生物有少量研究。然而利用Illumina Hiseq高通量测序技术研究棉花黄萎病不同发生程度棉田土壤中真菌多样性的研究鲜见报道。【拟解决的关键问题】对新疆不同区域黄萎病发生程度不同的棉田土壤中真菌群落结构进行深入分析,探明棉田黄萎病发生程度由重至无、由无至重过程中土壤真菌群落的变化特征,以期揭示土壤中的真菌群落在棉花黄萎病发生过程中的生态作用,为利用微生态调控手段防控棉花黄萎病提供理论依据。1 材料与方法
1.1 材料
2016年在阿克苏市(40°34′ N,81°19′ E)、库尔勒市(41°45′ N,85°48′ E)和石河子市(44°18′ N,85°59′ E)分别选择具有代表性的棉花黄萎病重度发生棉田与无病(轻度)对照棉田各1块,3地共计6块棉田(表1)。重病田采集发病等级3以上棉株根围土,对照田采集健康棉株根围土,于4月与8月份每块棉田3点取样2次,每取样点间隔50 m,每点取0—10 cm土层小样5份,每份200 g,混合为1份样品,每采样时期计18份混合样品,共36份混合样品,经2 mm孔径过样筛处理,保存于-80℃冰箱备用。Table 1
表1
表1棉田土壤样品
Table 1
| 采集地点 Sampling location | 棉田类型 Cotton field type | 采集时期 Sampling time | 样品编号 Sample ID |
|---|---|---|---|
| 阿克苏市 Aksu City | 对照田Control field | 2016-04 | a4w2、a4w4、a4w6 |
| 2016-08 | a8w1、a8w2、a8w3 | ||
| 重病田Serious disease field | 2016-04 | an4b1、an4b3、an4b6 | |
| 2016-08 | an8b1、an8b2、an8b3 | ||
| 石河子市 Shihezi City | 对照田Control field | 2016-04 | bn4w2、bn4w4、bn4w6 |
| 2016-08 | bn8w1、bn8w2、bn8w3 | ||
| 重病田Serious disease field | 2016-04 | sn4b2、sn4b4、sn4b6 | |
| 2016-08 | sn8b1、sn8b2、sn8b3 | ||
| 库尔勒市 Korla City | 对照田Control field | 2016-04 | k4c1、k4c2、k4c3 |
| 2016-08 | k8c1、k8c2、k8c3 | ||
| 重病田Serious disease field | 2016-04 | k4v1、k4v2、k4v3 | |
| 2016-08 | k8v1、k8v2、k8v3 |
新窗口打开|下载CSV
1.2 棉田发病程度调查
每块取样棉田调查5点,每点调查100株棉花,按照国内棉花黄萎病统一分级标准进行统计,计算每块取样棉田病情指数。病情指数=[∑(各级病株数×各级代表值)/(调查总株数×4)]×100。1.3 采样棉田信息
阿克苏棉花黄萎病重病田(Aksu disease,AD)为自然棉田,以相邻的棉花黄萎病重病田经水稻轮作后的无病棉田(Aksu control,ACK)为对照,种植棉花品种均为中棉49号;石河子棉花黄萎病重病田(Shihezi disease,SD)、轻病对照田(Shihezi control,SCK)均为自然棉田,种植棉花品种均为新陆早64号;库尔勒重病田(Korla disease,KD)为连续3年在无病田中人工接种棉花黄萎病菌改造而来,以相邻无病田(Korla control,KCK)为对照,种植品种均为新陆中66号。以上棉田均为覆膜滴灌棉田,每个区域重病田与对照田的管理措施一致。棉田发病程度、土壤中微菌核量、土壤养分指标等数据已另文发表[17]。1.4 土壤总DNA的提取与文库构建、测序
利用北京百泰克生物技术公司生产的DP4001土壤试剂盒,严格按照操作步骤提取土壤总DNA,利用1%琼脂糖凝胶电泳检测提取的DNA质量,使用Nano Drop 2000 UV-Vis光谱仪测定DNA浓度,总量满足3次及以上建库要求。所用引物为真菌ITS1区域引物:5′-CTTGGTCATTTAGAGGAAGTAA-3′,5′-GCTGCG TTCTTCATCGATGC-3′;内部ITS1F:AACCTGCGG AAGGATCATT,内部ITS1R:GARCCAAGAGATCC RTTG。PCR扩增程序:95℃预变性5 min;95℃变性30 s,50℃退火30 s,72℃延伸40 s,25个循环;72℃延伸7 min。由北京百迈客生物科技有限公司构建DNA文库,采用Illumina Hiseq 2500 PE250模式进行测序。1.5 数据处理与分析
对测序获得的原始数据使用FLASH V1.2.7软件,按照最小重叠长度10 bp、重叠区最大错配比率0.2对每个样品的序列进行拼接,得到的拼接序列即原始序列。将拼接得到的序列用Trimmomatic软件设置50 bp的窗口,如果窗口内的平均质量值低于20,截去后端碱基,过滤质控后长度小于标签长度75%的标签,使用UCHIME软件并去除嵌合体,得到高质量的序列。在相似性97%的水平上使用UCLUST软件对序列进行聚类,以所有序列数的0.005%作为阈值过滤OTU。在Unite真菌ITS数据库进行比对。在置信度阈值为0.8利用RDP Classifier软件进行物种注释分类,作图时将丰度低于1%的物种合并为Others在图中显示,Unknown代表未得到分类学注释的OTU。利用ClustalW2软件进行多重比对,邻接法(neighbor- joining,NJ)建树。利用Mothurversion V.1.30软件进行Alpha多样性指数分析,包括Chao1丰富度估计量(Chao1 richness estimator)、Ace丰富度估计量(Ace richness estimator)、香农-威纳多样性指数(Shannon- wiener diversity index)与辛普森多样性指数(Simpson diversity index);基于Binary jaccard、Bray curtis多种算法呈现物种多样性矩阵,进行Beta多样性分析(n≥3),基于R语言平台绘制样本非度量多维尺度分析(NMDS)以及环境因子与样本组成相关性分析(RDA)。利用LefSe分析方法设定显著差异的LDA值为4.0,寻找组间丰度差异显著物种,采用线性判别分析(LDA)来估算每个组分(物种)丰度对差异效果的影响。
利用Excel软件对常规数据进行整理、汇总,利用SPSS19.0软件的ANOVA程序中Duncan分析法对数据进行差异显著性分析,P<0.05为差异显著。
2 结果
2.1 黄萎病不同发生程度棉田真菌多样性
在97%相似度水平下,AD和SD样品4月和8月份采集的土壤中真菌的OTU数量、Chao1丰富度指数、Ace丰富度指数均分别高于其对照ACK、SCK样品,而辛普森指数均分别低于对照。其中SD与SCK样品的OTU数量存在显著性差异。人工接菌的KD样品与对照在OTU数量、Chao1丰富度指数、Ace丰富度指数、辛普森指数方面均极为接近,两组样品之间无显著差异。分析表明,自然发病的重病田中真菌的丰度相比轻病或无病田有所上升而多样性下降,而人工接入病原菌短期内未对土壤中真菌的丰度和多样性产生较大影响(表2)。Table 2
表2
表2土壤中真菌的Alpha多样性指数
Table 2
| 采样地点Sampling location | 采样时期 Sampling time | 处理 Treatment | 分类单元 OTU | Chao1丰富度 Chao1 richness estimator | Ace丰富度 Ace richness estimator | 辛普森指数 Simpson diversity index |
|---|---|---|---|---|---|---|
| 阿克苏市 Aksu City | 2016-04 | AD | 240.0±1.0a | 241.8±1.5a | 243.6±3.5a | 0.172±0.06a |
| 2016-04 | ACK | 223.3±9.6a | 226.7±9.4a | 226.8±9.8a | 0.173±0.03a | |
| 2016-08 | AD | 232.0±14.2a | 236.1±11.0a | 236.9±12.0a | 0.167±0.03a | |
| 2016-08 | ACK | 226.3±6.6a | 232.5±5.3a | 233.6±5.3a | 0.176±0.01a | |
| 石河子市 Shihezi City | 2016-04 | SD | 248.0±1.0c | 249.5±1.40b | 251.6±1.26b | 0.132±0.02a |
| 2016-04 | SCK | 237.0±5.3b | 239.9±5.6ab | 241.7±5.5ab | 0.251±0.05b | |
| 2016-08 | SD | 239.3±4.0bc | 240.6±4.8ab | 240.1±4.7ab | 0.191±0.02b | |
| 2016-08 | SCK | 219.3±7.6a | 232.1±11.4a | 232.3±9.5a | 0.211±0.03b | |
| 库尔勒市 Korla City | 2016-04 | KD | 234.6±5.0a | 237.0±7.6a | 236.3±7.0a | 0.132±0.05a |
| 2016-04 | KCK | 234.3±5.0a | 237.8±5.1a | 238.5±6.9a | 0.127±0.03a | |
| 2016-08 | KD | 220.6±20.6a | 229.2±17.2a | 231.1±19.6a | 0.117±0.01a | |
| 2016-08 | KCK | 223.6±6.4a | 232.4±5.3a | 231.5±8.1a | 0.129±0.02a |
新窗口打开|下载CSV
NMDS分析发现,当应力值=0.1396时,阿克苏样品主要分布在第Ⅲ象限,其中AD样品的4月与8月份样品距离较近,表现出较高的相似性;ACK样品的4月与8月份样品同样表现出较高的相似性。石河子样品主要分布在第Ⅱ象限,其中SD样品的4月、8月份样品距离近,相似性较高;而SCK样品的4月与8月份样品距离较远,样品中真菌群落相似差异较大。库尔勒样品主要集中在第Ⅰ、Ⅳ象限,KD与KCK两组样品的4月份样品距离较近,相似性较高,但与8月份样品距离较远,差异较大。不同区域土壤样品之间表现出不同的时空趋向性(图1)。AD、SD中土壤样品首先表现为明显的空间趋向性,之后表现为时间趋向性,而库尔勒土壤样品则受采样时间因素影响较大,其次是空间趋向性。
图1
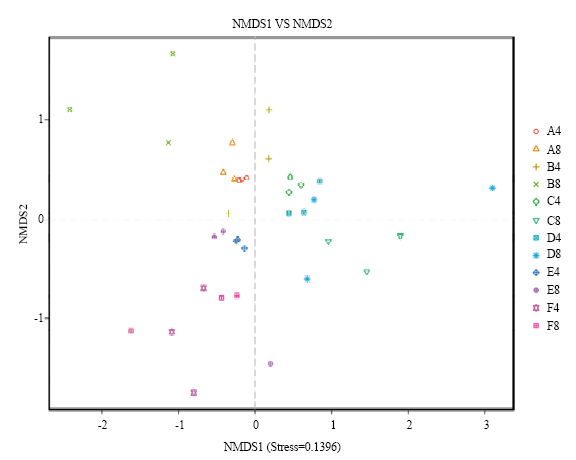 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1土壤中真菌群落基于属水平的NMDS分析
A4:石河子重病田(4月) Serious disease fields of Shihezi (April); B4:A4对照The control of A4;A8:石河子重病田(8月)Serious disease fields of Shihezi (August);B8:A8对照The control of A8;C4:库尔勒重病田(4月)Serious disease fields of Korla (April);D4:C4对照The control of C4;C8:库尔勒重病田(8月)Serious disease fields of Korla (August);D8:C8对照The control of C8;E4:阿克苏重病田(4月)Serious disease fields of Aksu (April);F4:E4对照The control of E4;E8:阿克苏重病田(8月)Serious disease fields of Aksu (August);F8:E8对照The control of E8
Fig. 1NMDS analysis of fungal communities in the cotton-field soil based on genus level
2.2 黄萎病不同发生程度棉田土壤真菌群落组成
在门水平上,Ascomycota(64.15%—82.10%)的丰度在不同样品中均占绝对优势,在AD、KD、SD样品中丰度均分别低于其对照;而Basidiomycota和Mortierellomycota则在AD、KD、SD样品中丰度均分别高于其对照。Ascomycota、Basidiomycota以及Mortierellomycota在库尔勒KD(74.14%、15.07%、3.87%)与其对照KCK样品(75.07%、14.59%、3.57%)之间以及石河子SD(76.42%、12.96%、3.58%)和其对照SCK样品(82.10%、10.34%、2.78%)之间丰度相差较小,而在阿克苏AD(64.15%、18.84%、5.62%)样品和其对照ACK(80.27%、9.69%、2.67%)样品之间丰度差异较大,表明病原菌的接入对土壤中门水平优势真菌群落的影响较小,而ACK样品取样田前茬种植水稻则明显地影响了土壤中门水平优势真菌的丰度(图2)。图2
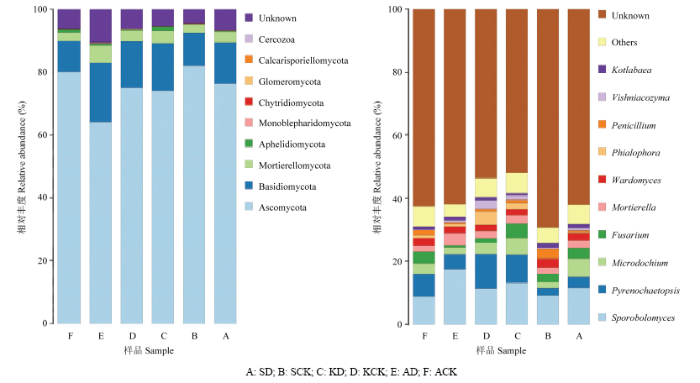 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2土壤中门(左图)和属(右图)分类水平的真菌群落结构
Fig. 2Community structure of fungi at taxonomic levels of phylum (left fig.) and genus (right fig.) in the cotton-field soil
A: SD; B: SCK; C: KD; D: KCK; E: AD; F: ACK
在属水平上,土壤中Sporobolomyces的丰度最高,其与Mortierella在不同区域重病田土壤中丰度均高于对照,其中Sporobolomyces在AD样品中的丰度(17.53%)约为ACK样品(8.79%)的2倍;而Wardomyces在不同处理的对照田中均高于重病田。此外,AD样品中Pyrenochaetopsis、Microdochium、Fusarium、Wardomyces、Penicillium均低于ACK样品,而Pyrenochaetopsis、Microdochium、Fusarium在SD样品中的丰度均高于SCK(图2)。
组间差异显著性分析发现,当设定LDA值>4时,不同组样品之间具有显著性差异的标记极少。当设定LDA值>3时,库尔勒土壤中的显著差异标记均富集于KD样品组中,包括Aphelidiomycota、Aphelidiomycetes、Fusarium、Septofusidium等。石河子土壤SD样品中富集的显著差异标记为10个,包括Dothideomycetes、Stachybotrys、Kernia和Microdochium等,而SCK样品中仅3个,包括Sordariales、Lasiosphaeriaceae和Petriella。阿克苏土壤中显著差异标记主要富集于ACK样品中,有别于石河子和库尔勒土壤,ACK组样品中的差异标记包括Hypocreales、Nectriaceae、Septofusidium和Fusarium等,表明该棉田前茬种植水稻对土壤中真菌的组成产生了较大影响(图3)。
图3
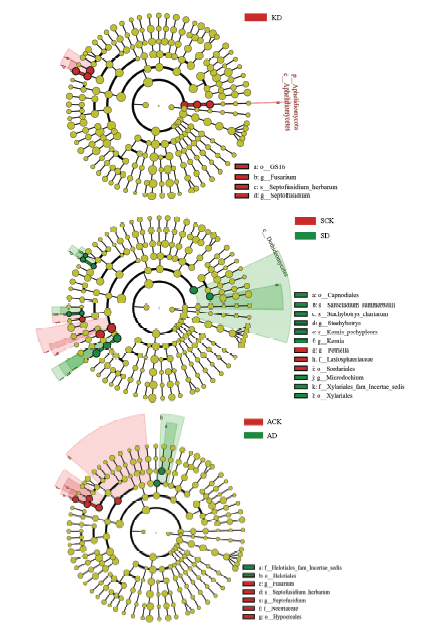 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3不同处理组间差异显著性分析
Fig. 3Significance analysis of differences among different treatment groups
2.3 真菌多样性、群落结构与土壤养分的相关性
相关性分析表明,土壤的pH以及全磷、全钾含量与真菌的辛普森指数的相关系数分别为0.7799、0.6209和0.8848,总盐与辛普森指数显著负相关,相关系数为-0.7604,但以上指标均与OTU数量、Chao1丰富度、Ace丰富度的相关性低。有机质、全氮量与真菌的OTU数量、Chao1丰富度、Ace丰富度呈显著正相关,但与辛普森指数相关性低(表3)。Table 3
表3
表3土壤养分、pH及有机质与真菌多样性指数的相关性
Table 3
| 分析指标 Analysis index | 分类单元 OTU | Chao1丰富度 Chao1 richness estimator | Ace丰富度 Ace richness estimator | 辛普森指数 Simpson diversity index |
|---|---|---|---|---|
| 酸碱度 pH | 0.3157 | 0.2802 | 0.3294 | 0.7799* |
| 总盐 Total salt | -0.3371 | -0.2936 | -0.3471 | -0.7604* |
| 有机质 Organic matter | 0.7558* | 0.7657* | 0.7528* | 0.2136 |
| 全氮 Total nitrogen | 0.7322* | 0.7575* | 0.7498* | 0.3109 |
| 全磷 Total phosphorus | 0.3257 | 0.4119 | 0.4251 | 0.6209* |
| 全钾 Total potassium | -0.3892 | -0.2962 | -0.2389 | 0.8848** |
新窗口打开|下载CSV
冗余分析表明,土壤中的真菌种群主要与土壤养分明显相关,其中Chaetomium、Alternaria、Cryptococcus、Acremonium、Thielaviopsis等与全盐量呈正相关,主要集中在第Ⅲ象限,Lentinula、Metarhizium、Cladosporium、Wardomyces与含盐量成负相关,集中在第Ⅰ象限;而Gibberella、Fusarium、Leptosphaeria、Davidiella、Tetracladium与土壤中有机质、全氮量呈正相关,集中在第Ⅳ象限,Pseudeurotium与之呈显著负相关(图4)。
图4
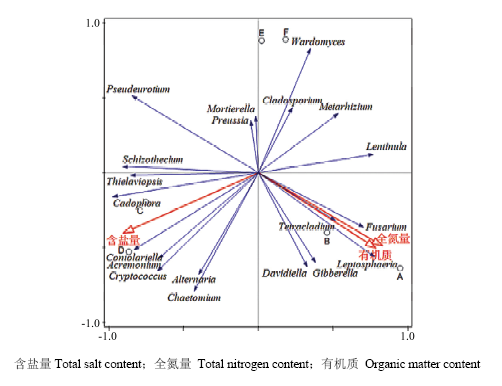 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4有机质、总盐、全氮与真菌属分类水平的冗余分析
Fig. 4RDA analysis of fungal communities with organic matter, total salt and total nitrogen at genus level
含盐量Total salt content;全氮量 Total nitrogen content;有机质 Organic matter content
3 讨论
病原菌侵染作物后,能使其根部发生组织结构变化的同时,也可改变其分泌物中某些成分及含量,根系分泌物是植物与土壤进行物质交换的载体,在调节植物与土壤微生物互作方面发挥着重要作用[18],当改变的根系分泌物扩散到土壤后,影响了作物根际的土壤环境,从而致使作物根际微生物数量及种类发生特定变化[19,20]。已有研究表明,棉花黄萎病发生能够引起其根际土壤中真菌的富集,土壤中真菌数量、致病真菌种类均明显增加[10,11],在粮食、蔬菜等作物上亦得出一致的结论[12,13,14,15,16]。本研究发现,阿克苏、石河子重病棉田发病棉花根围土中真菌的OTU数量、丰度在两个采样时期均高于无病田或轻病田中的健康棉株根围土,而真菌的多样性降低,其中阿克苏重病田与种植过水稻的对照棉田之间存在显著差异,该结果与前人研究基本一致。但是,棉田接种病原菌处理(KD)土壤中真菌的OUT数量、丰度与其对照相比虽无显著差异,却略低于对照,且AD、SD样品中真菌群落首先表现为空间趋向性,之后为时间趋向性,而库尔勒土壤样品则受采样时间因素影响更大,其次是空间因素,两方面结果均与阿克苏、石河子自然发病棉田数据不一致,反映了病原菌入侵短期内对土壤中真菌的丰度、多样性和组成影响有限,推测可能与接种年限或植物根际分泌物的影响范围有关,还需进一步验证。前人研究认为,土壤养分、pH、有机质等是影响土壤微生物群落结构的重要因素[21,22],YU等[23]研究发现有机肥处理后土壤中细菌、放线菌的群落多样性、香农指数升高,氮肥处理后真菌水平升高。本研究发现不同分类水平真菌的组成存在差异,在门分类水平上,土壤中丰度高的真菌子囊菌门、担子菌门与Mortierellomycota在库尔勒人工接菌棉田与其对照之间以及石河子自然发病棉田与其对照之间的丰度差异较小,而在阿克苏重病田与其水稻轮作处理的对照之间丰度差异较大。在属分类水平上,不同区域土壤中除掷孢酵母属、被孢霉属和Wardomyces的丰度在重病田与对照田之间变化一致外,其余菌属在不同处理组内丰度无一致变化规律。作为验证病害发生对土壤中真菌影响程度的人工接菌土壤(KD、KCK),其在土壤真菌的丰度、多样性和组成的变化方面均与自然发病棉田处理组间无明显差异,因此认为发病程度可能对棉花根围土壤微生物群落结构的影响有限,为此,研究了土壤养分等指标与真菌的多样性以及群落结构的相关性。利用相关性和RDA分析表明,全氮、有机质、含盐量、全盐、pH等指标对土壤中真菌多样性、群落组成的影响显著,这与王巍巍等研究结果一致[24,25,26]。
棉花黄萎病防治困难,明确土壤中大丽轮枝菌的动态以及大丽轮枝菌入侵棉田与土壤中真菌群落的动态关系,对于利用微生态调控措施防控棉花黄萎病具有重要意义。前期研究发现,经水稻轮作棉田(ACK)不仅提高了土壤肥力,还消灭了土壤中的大丽轮枝菌,杜绝了病害发生;而棉田地力水平最高、连作达30年的重病棉田(SD)土壤中,大丽轮枝菌菌源数量却高达193个/g土[17]。本研究表明,在土壤中含有病原菌的前提下,不改变种植作物以及栽培模式,仅改善土壤肥力可能加重病害的发生。而通过不同作物间作、轮作和种植抗病品种等,利用不同作物根系分泌的化感物质持续对土壤中微生物群落结构进行调节,增加土壤中有益菌群数量,降低病原菌数量,在“农药、化肥”双减战略下可能是防控土传病害的有效手段。
4 结论
自然发病重棉田土壤中真菌的OTU数量、丰度均高于轻病或无病田,但多样性降低;而棉田内人工接种大丽轮枝菌短期内并未对土壤中真菌的丰度、多样性和群落结构造成明显影响。相关性分析表明,土壤中真菌的多样性、丰度和组成主要受有机质、全氮、含盐量等指标决定,且采样时期同样对土壤中真菌群落结构有明显影响。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1111/nph.13116URLPMID:25348596 [本文引用: 1]

Summary The ongoing expansion of shrub cover in response to climate change represents a unique opportunity to explore the link between soil microbial communities and vegetation changes. This link is particularly important in peatlands where shrub expansion is expected to feed back negatively on the carbon sink capacity of these ecosystems. Microbial community structure and function were measured seasonally in four peatlands located along an altitude gradient representing a natural gradient of climate and associated vascular plant abundance. We show that increased soil temperature and reduced water content are associated with greater vascular plant biomass, in particular that of ericoids, and that this, in turn, is correlated with greater microbial biomass. More specifically, microbial community structure is characterized by an increasing dominance of fungi over bacteria with improved soil oxygenation. We also found that the carbon and nitrogen stoichiometry of microbial biomass differs in relation to soil microbial community structure and that this is ultimately associated with a different investment in extracellular enzymatic activity. Our findings highlight the fact that the determination of the structural identity of microbial communities can help to explain the biogeochemical dynamics of organic matter and provide a better understanding of ecosystem response to environmental changes.
[本文引用: 1]
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.apsoil.2012.03.008URL [本文引用: 1]
Soil microorganisms play a crucial role in mineralization and breakdown of complex organic compounds in soil. Microbial populations and functional diversity are greatly influenced by quantity and quality of crop residue and other incorporate organic amendments. This study investigated the effect of cover crops (rye or a mixture of rye-vetch) and compost on soil microflora and microfauna under an organic tomato production system. Each cover crop treatment was used in conjunction with or without compost in a split-plot experimental design. Data on soil respiration, microbial biomass, metabolic quotient, and nematode populations were measured at the end of the growing season. Metabolic characteristics of the soil microbial community were determined using 31 C substrates on Biolog-EcoPlate64. Community level physiological profile (CLPP) was assessed by calculating average well color development (AWCD), richness (S), Shannon–Wiener diversity index (H), and evenness (E). Effect of compost was more pronounced on soil respiration than cover crop treatment. Highest microbial biomass was found in the soils amended with rye and compost (195–210μggdrysoil611). Regression analysis between microbial biomass and soil organic matter (SOM) showed strong correlation (R2 value of 0.68–0.56) in two out of the three growing seasons. Calcium, magnesium, and potassium concentrations in soil also positively correlated with microbial biomass. There were significant differences among soils in numbers of plant parasitic, bacterial, and fungal feeding nematodes during the initial years of the study but the differences were not evident later. Shannon–Wiener diversity index was significantly affected by cover crop treatment with rye treatments generally exhibiting higher degree of soil microbial functional diversity. Biolog-EcoPlate64 assay was sensitive to changes in the short-term. Principal component analysis of the Biolog data allowed differentiation of treatments but distribution patterns varied from year to year. We conclude that both rye and rye-vetch mixture can affect the functional diversity of soil microbial community but differences between them are marginal when compared to compost and no-compost treatments. Microbial communities were more responsive to compost applications than cover crop effects.
DOI:10.1016/j.apsoil.2005.05.007URL [本文引用: 1]
This study considers the relationship between microbial diversity and soil organic decomposition function of two soils whose microbial diversity was first established using molecular biology (DGGE) and in situ catabolic potential (ISCP). The soil used was a tropical ferruginous Oxisol, with samples taken from a 21-year fallow and a plot that had been cultivated for 4 years after lying fallow for 17 years. The samples from the 0 10 cm soil layer were incubated with or without the addition of Faidherbia albida litter under laboratory conditions (28 C, 100% WHC) for 240 h. During incubation, the microorganism activity was measured (CO 2, mineral N, phosphatase, glucosidase and urease). In the unamended soil, the activity of the microorganisms was greater in the fallow soil which had a greater microbial diversity than that in the cultivated soil. However, other soil properties (carbon and organic nitrogen content, total microbial biomass) may also explain this result. For the amended soil, only the first 8 h of incubation showed a difference between the fallow and cultivated soil. During this period, the CO 2 respiration in the fallow soil was higher than that recorded in the cultivated soil. This difference should be compared with the catabolic microbial diversity, which was higher in the fallow soil than in the cultivated soil. After this initial phase, the microbial community in the cultivated soil seemed to acquire similar functions to those in the fallow soil. The phosphatase and glucosidase activities of the amended soils were higher in the fallow soil. This difference was maintained for the whole incubation period. The redundancy of microbial functions is discussed.
DOI:10.1007/s00572-007-0141-6URLPMID:17638027Magsci [本文引用: 1]
We investigated the species identity of mat-forming ectomycorrhizal (EM) fungi associated with old- and second-growth Douglas-fir stands. Using molecular analyses of rhizomorphs and EM root tips, we characterized 28 unique internal transcribed spacer sequences and considered them proxies for mat-forming EM species. In both stand age classes, one Athelioid species in the genus Piloderma dominated our sample of the mat-forming fungal community. In second-growth stands, the second most frequently encountered mat-forming EM species belonged to the genus Hysterangium . In old-growth stands, several Ramaria species were associated with a frequently encountered mat morphology but no species dominated the community. After using rarefaction analysis to standardize sampling effort, the total species richness did not differ statistically between old- and second-growth habitats. Both an abundance of infrequently encountered species and incomplete sampling of the mat-forming EM community may have limited our ability to detect potential differences in species richness. Several frequently encountered Piloderma species appear to have broad (holarctic) distributions and diverse host associations and their potential importance in forest ecosystems warrants further study.
DOI:10.1007/s00572-012-0468-5URLPMID:23179900Magsci [本文引用: 1]
The impact of rest grazing on arbuscular mycorrhizal fungi (AMF) and the interactions of AMF with vegetation and soil parameters under rest grazing condition were investigated between spring and late summer in a desert steppe ecosystem with different grazing managements (rest grazing with different lengths of resting period, banned or continuous grazing) in Inner Mongolia, China. AMF diversity and colonization, vegetation biomass, soil properties and soil phosphatase activity were examined. In rest grazing areas of 60 days, AMF spore number and diversity index at a 0-10 cm soil depth as well as vesicular and hyphal colonization rates were higher compared with other grazing treatments. In addition, soil organic matter and total N contents were highest and soil alkaline phosphatase was most active under 60-day rest grazing. In August and September, these areas also had the highest amount of aboveground vegetation. The results indicated that resting grazing for an appropriate period of time in spring has a positive effect on AMF sporulation, colonization and diversity, and that under rest grazing conditions, AMF parameters are positively correlated with some soil characteristics.
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.apsoil.2014.09.013URL [本文引用: 1]
A long-term field experiment was used to evaluate the effects of different fertilizers (OIO, organic inorganic compound fertilizer specially formulated for mulberry; N, N fertilizer only; NPK, N, P and K fertilizer; NF, the control without fertilization) on microbial abundance, community structure and microbial functional diversity. The OIO treatment exhibited the highest bacterial and actinomycete levels, community diversity, Shannon indices and average well color development (AWCD). However, the N treatment resulted in the highest fungi levels. Bacillus sp. were the dominant bacteria in the OIO treatment. The carbohydrate and carboxylic acid utilization by microbes was highest in the OIO treatment, whereas the amino acid utilization was highest in the N treatment. Most microbial parameters were primarily correlated with the soil organic matter content. The principal component analysis of the DGGE profiles suggested that the culture-independent bacterial community was not really affected by the fertilization treatments but was significantly affected by the sampling time. In contrast, the carbon substrate utilization was significantly affected by the fertilization treatments but not by the sampling time. Our findings suggested that the soil microbial activities were significantly changed by long-term fertilization and that more attention should be devoted to seasonal shifts in the microbial community.
[本文引用: 1]
[本文引用: 1]
 土PLFA指纹特征
土PLFA指纹特征 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}