 ,1, 蔡更元,1,2
,1, 蔡更元,1,2Optimizing the Electroporation Condition of Porcine Fetal Fibroblasts for Large Plasmid
ZHONG CuiLi1, LI GuoLing1, WANG HaoQiang1, MO JianXin1, QUAN Rong1, ZHANG XianWei2, LI ZiCong1, WU ZhenFang1,2, GU Ting,1, CAI GengYuan,1,2通讯作者:
收稿日期:2018-09-18接受日期:2018-12-29网络出版日期:2019-02-13
| 基金资助: |
Received:2018-09-18Accepted:2018-12-29Online:2019-02-13
作者简介 About authors
钟翠丽,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (721KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
钟翠丽, 李国玲, 王豪强, 莫健新, 全绒, 张献伟, 李紫聪, 吴珍芳, 顾婷, 蔡更元. 大载体转染猪胎儿成纤维细胞的电转条件优化[J]. 中国农业科学, 2019, 52(3): 530-538 doi:10.3864/j.issn.0578-1752.2019.03.013
ZHONG CuiLi, LI GuoLing, WANG HaoQiang, MO JianXin, QUAN Rong, ZHANG XianWei, LI ZiCong, WU ZhenFang, GU Ting, CAI GengYuan.
0 引言
【研究意义】为满足日益复杂的生物研究需求,运用的载体分子量越来越大(如本试验所用到26 kb载体),其主要原因有以下两点:(1)载体中两侧同源臂较大:运用同源重组的原理,需要在载体上根据靶点上下序列设计约600—6 000 bp两侧同源臂以定点敲入外源DNA[1,2,3];(2)多基因共表达的应用:多基因共表达系统能够满足研究复杂的生理机制和实现多种生物功能的需求,在肿瘤多基因治疗和蛋白工程等方面具有重大的应用价值。目前主要提高经济性质的转基因模型载体都在10 kb以上,如腮腺生物反应器的转基因载体[4]。因此优化大载体转染条件对多基因共表达系统、基因编辑、转基因育种等具有重要的意义。【前人研究进展】转基因猪的制备效率与大载体转染供体细胞PFFs的效率有关,大载体转染至PFFs后外源基因可通过随机整合和定点整合两种基因整合方式制备转基因动物。随机整合如piggyBac系统具有基因转移能力强、整合效率高、转基因稳定表达和遗传等优点,而且整合位点相对安全[5,6,7,8]。据统计48.8%的piggyBac整合位点位于已注释的基因位点[9];而杜新华等(2013)获得的8个piggy Bac转座子的有效整合位点中有5个位于牛基因组的非调控区内[10]。因此可借助增强型绿色荧光蛋白(enhanced green fluorescent protein, EGFP)基因报告载体和piggyBac系统获得有效整合位点,分析外源基因的表达效果和对内源基因表达的影响等以评估位点的安全性[11]。而基于同源重组的定点整合技术整合效率较低,效率仅0.5%—20%,可选的安全位点较少,主要以Rosa 26为靶点插入外源基因制备转基因动物,如小鼠[12]、羊[13]、猪[11, 14],对20 kb以上的大片段定点整合仍存在极大的技术难度。目前转染方法主要有碳酸钙化学法、脂质体转染法和电穿孔法等。虽然有研究人员通过脂质体法和碳酸钙法可获得阳性PFFs细胞,但是碳酸钙转染效率低,脂质体转染法细胞毒性大[15]。而电穿孔法主要将额外加入的遗传物质从电击形成的瞬时孔隙导入细胞膜甚至细胞核内的一种转染方式,这种方式快速简易、毒性低、而且转染效率相对较高,约30%—90%[16,17,18]。故相比之下,电穿孔法更适合大载体转染PFFs。市面上电转仪种类繁多,如ECM? 830、NEPA 21和NucleofectorTM 2b等。ECM? 830具有方形波电穿孔系统,通过击穿细胞膜将质粒导入细胞质内来实现高效转染,而NEPA 21和 Nucleofector? 2b均能击穿细胞膜及核膜。三者相比,ECM? 830转染成本较低;NEPA 21不需要特殊转染试剂盒,多数用于活体转染[19,20],甚少用于体外转染,ISHINO等[21]使用NEPA 21成功转染牛耳成纤维细胞,但效率很低(0.35%)。Nucleofector? 2b虽然根据细胞类型使用专用转染试剂盒,但无需自行摸索电转条件。【本研究切入点】研究表明上述3种电转仪转染PFFs的小载体(10 kb以内)转染效率可以达到50%以上[16-17, 22],但大载体转染PFFs的效率较低,且相关研究甚少,因此可通过优化电转仪(如ECM? 830、NEPA 21和Nucleofector? 2b)、电转参数、质粒用量和拓扑结构的转染条件来提高大载体转染PFFs的效率,为新型转基因环保猪的制备提供参考。【拟解决的关键问题】本文比较上述3种常用电转仪的电转参数、质粒用量和拓扑结构对26 kb质粒转染PFFs的影响,以寻找针对猪胎儿成纤维细胞的更高效、合适的大载体转染条件,为多基因共表达系统、基因编辑、转基因育种等的应用提供参考。1 材料与方法
1.1 材料
电转仪:ECM? 830(BTX,美国);Nucleofector? 2b(LONZA,德国);NEPA 21(NEPA GENE,日本)。PFFs细胞:由广东温氏食品集团股份有限公司提供。质粒pPXAT-EGFP(26 kb):由国家生猪种业工程技术研究中心提供。1.2 试验时间和地点
2016年7月广东省广州市华南农业大学国家生猪种业工程技术研究中心。1.3 方法
1.3.1 质粒准备 pPXAT-EGFP质粒依照Endo-free Plasmid Maxi Kit(Omega,美国)说明书进行抽提,取部分超螺旋质粒使用FastDigest?NotI(Thermo Scientific,美国)进行酶切线性化,纯化回收备用(图1)。图1
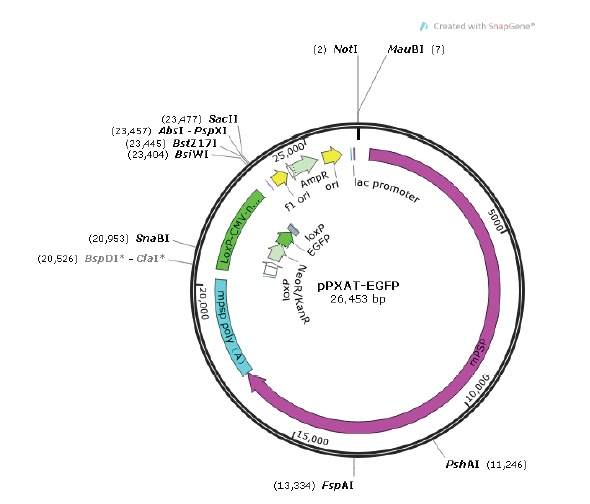 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1pPXAT-EGFP质粒图谱
Fig. 1The plasmid profile of pPXAT-EGFP
1.3.2 电转染 将PFFs细胞于39℃、12%胎牛血清(Fetal bovine serum,FBS)的DMEM完全培养基中培养至融合度至80%—90%,胰酶消化后取1×106个细胞悬液至新的离心管中,弃上清,根据不同试验分组添加电转预混液(由电转液和质粒组成的,不电转的空白组以相应体积的电转液代替电转预混液),轻柔打散成单细胞悬浮液,全部转至无菌的2 mm间距电转杯中,根据不同电转参数进行电转,然后转移至含有12% FBS的DMEM完全培养基6孔板,置于39℃,5% CO2的培养箱中培养。电转6 h后更换成含有1×青链霉素(penicillin-streptomycin solution,PS)、12% FBS的DMEM完全培养基继续培养。电转48 h后0.05%胰酶消化,90×g离心5 min,添加1 mL/孔PBS重悬后用流式细胞仪检测转染效率[转染效率=(绿色荧光细胞/细胞总数)×100%]。
1.3.3 电转参数优化 ECM? 830电转PFFs的最佳电转参数可参考ROSS等[18]研究结果(脉冲电压300 V,脉冲长度1 ms,脉冲次数3次),而NEPA 21和Nucleofector? 2b转染PFFs细胞的电转参数有待优化。
为获得NEPA 21最佳电转参数,分别采用广州市华粤行仪器有限公司(NEPA 21供货商)提供的4种电转参数转染PFFs,即分为4个处理组(NEPA-1、NEPA-2、NEPA-3和NEPA-4,表1)和一个空白组(不电转,100 μL NEPA电转液代替电转预混液),每个组3个重复,其他电转步骤及检测见1.3.2。此外,NEPA 21电转参数见表1,其电转预混液由100 μL NEPA 21电转液和8 μg质粒组成,而NEPA 21电转液为无血清Opti-MEM。
Table 1
表1
表1NEPA21的穿孔脉冲参数
Table 1
| 编号 Number | 脉冲电压 Voltage (V) | 脉冲长度 Length (ms) | 脉冲间隔 Continuous (ms) | 脉冲次数 Times | 衰减幅度 Voltage attenuation range (%) | 电极 Electrode |
|---|---|---|---|---|---|---|
| NEPA-1 | 150 | 5 | 50 | 2 | 10 | + |
| NEPA-2 | 200 | 3 | 50 | 3 | 10 | + |
| NEPA-3 | 275 | 1.5 | 50 | 2 | 10 | + |
| NEPA-4 | 300 | 1 | 50 | 3 | 10 | + |
新窗口打开|下载CSV
NucleofectorTM 2b则选取文献报道中适用于PFFs的电转参数进行优化[17],即分为2个处理组(LONZA-A:A-033和LONZA-U:U-023,表2)和一个空白组(不电转,100 μL NucleofectorTM 2b电转液代替电转预混液),每个处理组重复3次,其他电转步骤及检测见1.3.2。此外,NucleofectorTM 2b电转参数为A-033和U-023,其电转预混液由100 μL NucleofectorTM 2b电转液和8 μg质粒组成,而 NucleofectorTM 2b电转液则来自电转试剂盒Basic Nucleofector Kit for Primary Mammalian Fibroblasts (Amaxa,德国)中Nucleofector? Solution 和supplement按比例4.5﹕1混合的液体。
Table 2
表2
表2优化Nucleofector? 2b电转参数的试验分组
Table 2
| 组别 Group | 电转参数 Parameters | 电转预混液 Transfection mixture |
|---|---|---|
| 处理组1 Group 1 | LONZA-A1) | 100 μL Nucleofector? 2b电转液3)+8 μg质粒 100 μL the transfection mixture of Nucleofector? 2b and 8 μg plasmid |
| 处理组2 Group 2 | LONZA-U2) | |
| 空白组Blank group | 不电转Non transfection | 100 μL Nucleofector? 2b电转液Only 100 μL the transfection mixture of Nucleofector? 2b |
新窗口打开|下载CSV
1.3.4 优化质粒用量 使用上述优化的NEPA 21和Nucleofector? 2b电转仪参数,及ECM? 830电转参数(脉冲电压300 V,脉冲长度1 ms,脉冲次数3次)[18],分别使用6、8、10和12 μg的超螺旋质粒进行转染,即根据电转仪分为3个处理组(ECM? 830、NEPA 21和Nucleofector? 2b)和对应的3个空白组(不电转,相应电转液代替电转预混液),每个处理组根据质粒用量分为4个小组(6、8、10和12 μg的超螺旋质粒),每个处理小组重复3次,电转步骤及检测见1.3.2。此外ECM? 830电转预混液由200 μL的ECM? 830电转液和质粒组成,其中电转液是25% Opti-MEM和75% cytosalts(120 mmol·L-1 KCl,0.15 mmol·L-1 CaCl 2,10 mmol·L-1 K2 HPO 4;pH 7.6,5 mmol·L-1 MgCl2)的混合液[18]。
1.3.5 质粒拓扑结构的影响 使用3种电转仪的最优电转参数和摸索的最适质粒用量,分别使用超螺旋和线性化的质粒进行转染,即根据电转仪分为3个处理组(ECM? 830、NEPA 21和Nucleofector? 2b)和对应的3个空白组(不电转,相应电转液代替电转预混液),每个处理组根据质粒分子结构分为2个小组(超螺旋质粒和线性化质粒),每个处理小组重复3次,电转步骤及检测见1.3.2。
1.3.6 比较电转仪的转染效率 根据上述探索的ECM? 830,NEPA 21和Nucleofector? 2b的最佳电转条件(电转参数、质粒用量和结构)转染PFFs以探索电转仪对转染效率的影响,即分为3个处理组和对应的3个空白组(不电转,相应电转液代替电转预混液),每个处理组重复3次,电转步骤及检测见1.3.2。
1.3.7 统计学处理 采用 SPSS 18软件进行单因素分析(Duncan)。 数据以 Mean ± SD表示,显著性水平P<0. 05则表示差异有统计学显著性意义。
2 结果
2.1 不同电转参数的比较
分别在不同的电转参数下以8 μg质粒转染PFFs,研究结果表明NEPA 21的最佳电转参数为NEPA-4(脉冲电压300 V,脉冲长度1 ms,脉冲间隔50 ms,脉冲次数3次),转染效率为13.24%±1.63%(P>0.05,图2-A)。在Nucleofector? 2b的最佳电转参数为U-023,其转染效率高达46.36%±3.95%(P<0.05,图2-B)。图2
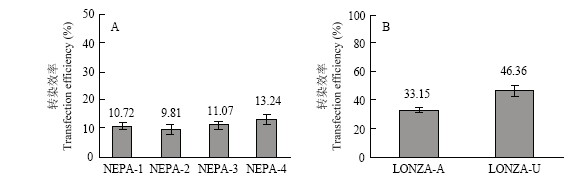 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2不同电转参数条件下的转染效率
A:4种NEPA21电转参数的转染效率比较,NEPA-1~NEPA-4的电转参数见
Fig. 2The transfection efficiency of different parameters
A: Comparison of transfection efficiency between 4 kinds of NEPA21 electroporation parameters which are shown in
2.2 不同质粒用量的比较
在最佳电转参数下使用3种电转仪分别转染6、8、10和12 μg的26 kb超螺旋质粒。由图3可知当质粒用量为12 μg时,ECM? 830和Nucleofector ? 2b转染效率最高,分别为8.44%±0.90%(P<0.05)和14.63%±3.21%(P>0.05)。而NEPA 21使用10 μg质粒转染PFFs时效率达到最高(6.09%±0.72%),但与12 μg质粒的转染效率差异不显著(5.12%±1.96%,P>0.05)。图3
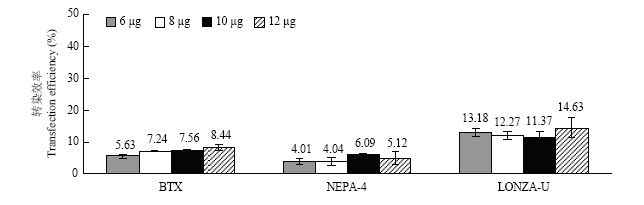 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3不同质粒用量的转染效率
BTX:ECM? 830的最佳电转参数(脉冲电压300 V,脉冲长度1 ms,脉冲次数3次);NEPA-4:NEPA 21的最佳电转参数(脉冲电压200 V,脉冲长度3 ms,脉冲间隔50 ms,脉冲次数3次); LONZA-U:Nucleofector? 2b的最佳电转参数(U-023)
Fig. 3The transfection efficiency of plasmid with different dosages
BTX: The better parameter of ECM? 830(voltage 200 V, continuous 3 ms and 3 times);NEPA-4: The better parameter of NEPA 21(voltage 200 V, continuous 3 ms, interval 50 ms and 3 times); LONZA-U: The better parameter of Nucleofector? 2b(U-023)
2.3 不同质粒拓扑结构的比较
在上述摸索的最佳参数下ECM? 830,NEPA 21和Nucleofector? 2b分别将12 μg超螺旋和线性化质粒转染PFFs细胞。结果显示3种电转仪分别使用超螺旋和线性化质粒的转染效率为:16.18%±1.45% 和7.83%±1.27%;5.64%±1.12%和2.20%±0.23%;33.15%±1.30%和11.59%±1.20%(图4)。由此可知线性化质粒的转染效率为显著低于其超螺旋结构(P<0.05),且仅为超螺旋质粒转染效率的34.96%—48.39%。图4
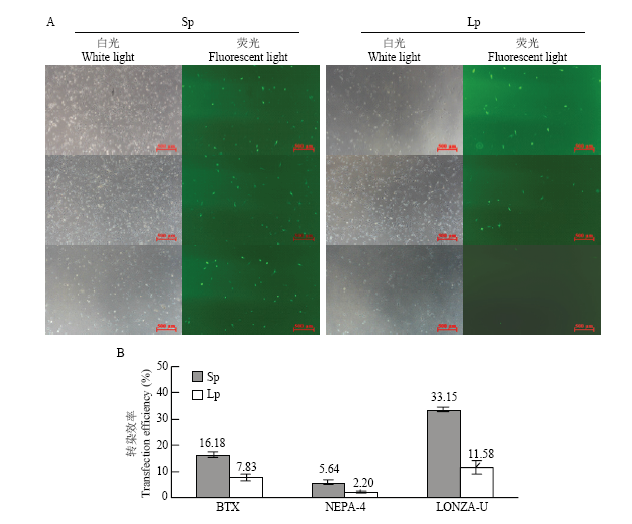 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4不同质粒拓扑结构的转染效率
A:荧光显微镜观察转染不同拓扑结构质粒的细胞;B:比较超螺旋和线性化质粒的转染效率。BTX:ECM? 830的最佳电转参数(脉冲电压300 V,脉冲长度1 ms,脉冲次数3次);NEPA-4:NEPA 21的最佳电转参数(脉冲电压200 V,脉冲长度3 ms,脉冲间隔50 ms,脉冲次数3次); LONZA-U:Nucleofector? 2b的最佳电转参数(U-023);Sp:超螺旋质粒;Lp:线性化质粒
Fig. 4The transfection efficiency of plasmid with different topological structures
A: Cells transfected with the different topological structures of plasmid were observed by fluorescence microscopy; B: Comparison of transfection efficiency between supercoiled plasmid and linearized plasmid. BTX: The better parameter of ECM? 830(voltage 200 V, continuous 3 ms and 3 times); NEPA-4: The better parameter of NEPA 21(voltage 200 V, continuous 3 ms, interval 50 ms and 3 times); LONZA-U: The better parameter of Nucleofector? 2b(U-023); Sp: Supercoiled plasmid; Lp: Linearized plasmid.*: P<0.05; **: P<0.01
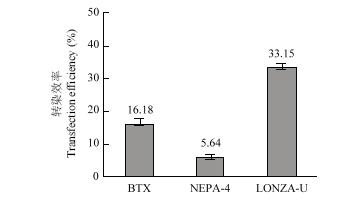
2.4 不同电转仪的比较
在最佳电转参数下比较ECM? 830、NEPA 21和Nucleofector? 2b转染12 μg超螺旋质粒的效率。结果显示3种电转仪的转染效率分别为:16.18%±1.45%、5.64%±1.12%和33.15%±1.30%,即3种电转仪中Nucleofector? 2b转染PFFs的转染效率最高(图5)。图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5不同电转仪的转染效率比较
BTX:ECM? 830的最佳电转参数(脉冲电压300 V,脉冲长度1 ms,脉冲次数3次);NEPA-4:NEPA 21的最佳电转参数(脉冲电压200 V,脉冲长度3 ms,脉冲间隔50 ms,脉冲次数3次); LONZA-U:Nucleofector? 2b的最佳电转参数(U-023)
Fig. 5The transfection efficiency of different electroporation apparatus
BTX: The better parameter of ECM? 830 (voltage 200 V, continuous 3 ms and 3 times); NEPA-4: The better parameter of NEPA 21(voltage 200 V, continuous 3 ms, interval 50 ms and 3 times); LONZA-U: The better parameter of Nucleofector? 2b(U-023)
3 讨论
市面上电转仪种类繁多,除了使用广泛的ECM? 830以外,还出现了很多转染效率高的新型电转仪,如NEPA 21和NucleofectorTM 2b,本研究主要是与ECM? 830相比,探索新型电转仪转染PFFs的效果并优化大载体转染条件。ECM? 830转染条件借鉴ROSS等[18]研究,文献结果显示在优化的转染参数下将2.5 μg/200 μL的5.8 kb超螺旋质粒电转1×106个PFFs,其最高转染效率在70%左右。据报道使用NEPA 21将7 μg/100 μL的9 kb环状质粒转染至牛耳成纤维细胞,其转染效率仅为0.35%[21]。而NAKAYAMA等[17]使用Nucleofector? 2b的U-023程序转染3.5 kb超螺旋质粒,PFFs的转染效率为79%。相比之下,本试验结果显示26 kb质粒转染至PFFs的转染效率最高仅为46.36%±3.95%(Nucleofector? 2b),NEPA 21的效率不足10%,而ECM? 830在ROSS等[18]优化的电转参数下转染26 kb超螺旋质粒的效率低于16%,而明显低于上述小载体的转染效率,表明质粒大小与转染效率成反比的关系。本试验还探索不同拓扑结构的质粒对转染效率的影响,结果显示超螺旋质粒转染PFFs的效率高于线性化质粒效率的两倍(图4),这可能由于线性质粒的空间位阻较大[23]。而且有报道显示超螺旋质粒和线性化质粒的细胞死亡率和转染效率分别为:15%和35%、76%和11%[24]。推测线性化质粒对细胞的毒性较大,很可能影响后期猪体细胞克隆效率,因此超螺旋质粒更利于转染。
此外,电转仪也是显著影响转染效率的一个重要因素。3种电转仪相比,Nucleofector? 2b的转染效率显著高于ECM? 830和NEPA 21(图5),而且应用于小载体转染时效率亦可高达79%,这可能由于Nucleofector? 2b的电流同时击穿细胞膜和细胞核膜更利于外源遗传物质转染。因此转染26 kb大载体的最佳电转仪是Nucleofector? 2b,而且被广泛大量运用于转染成纤维细胞以制备转基因动物。结合钟翠丽等[22]研究结果认为电转成本较低且转染效率高达90%的ECM? 830、ECM? 2001等电转仪更适合小载体转染PFFs。此外ECM? 830还具有对细胞损伤小的优势,如图4-A中虽然ECM? 830转染12 μg线性化质粒的效率低于NEPA 21和Nucleofector? 2b,但是存活率较高且荧光细胞总数相对较多。最后,NEPA 21在本试验中应用于转染PFFs,26 kb超螺旋质粒的转染效率较低(4%—13%),而7.6 kb超螺旋质粒的效率可达80%以上[22]。相比之下,NEPA 21不适合大载体转染PFFs。
本试验主要通过优化电转参数、质粒用量和拓扑结构等条件以提高大载体转染效率,对多基因共表达系统、基因编辑和转基因育种等的应用具有重要的意义。大载体的低转染效率是上述技术的难点之一,因此除了通过探索电转条件,还可以通过降低载体大小来提高转染效率。结合本试验结果及前人研究提出以下策略:(1)构建单启动子表达多基因或无启动子基因打靶载体,其中单启动子载体需要选择一个强启动子以启动多个基因表达,而无启动子基因载体则是借助内源启动子实现多基因表达[25,26];(2)运用IRES和2A序列等元件构建多顺反子[27,28];(3)缩短同源臂长度(短至300 bp)[29],能大幅度降低载体负载量,有利于提高转染效率,或运用同源臂长度仅5—25 bp的微同源重组技术[30]。
4 结论
针对目前大载体转染PFFs的效率较低且优化转染条件的研究甚少的情况,本研究通过在不同的电转仪(ECM? 830/NEPA 21/Nucleofector? 2b)、电转参数、质粒用量和拓扑结构的条件下转染PFFs以优化大载体的转染条件,为多基因共表达系统、基因编辑、转基因育种等提供参考数据。本研究结果显示3种电转仪中,Nucleofector? 2b是大载体转染PFFs的理想电转仪,且在U-023的程序下转染12 μg的26 kb超螺旋质粒时可达到最高转染效率。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1038/srep14253URLPMID:26381350 [本文引用: 1]

Transgenic pigs play an important role in producing higher quality food in agriculture and improving human health when used as animal models for various human diseases in biomedicine. Production of transgenic pigs, however, is a lengthy and inefficient process that hinders research using pig models. Recent applications of the CRISPR/Cas9 system for generating site-specific gene knockout/knockin models, including a knockout pig model, have significantly accelerated the animal model field. However, a knockin pig model containing a site-specific transgene insertion that can be passed on to its offspring remains lacking. Here, we describe for the first time the generation of a site-specific knockin pig model using a combination of CRISPR/Cas9 and somatic cell nuclear transfer. We also report a new genomic 'safe harbor' locus, named pH11, which enables stable and robust transgene expression. Our results indicate that our CRISPR/Cas9 knockin system allows highly efficient gene insertion at the pH11 locus of up to 54% using drug selection and 6% without drug selection. We successfully inserted a gene fragment larger than 9 kb at the pH11 locus using the CRISPR/Cas9 system. Our data also confirm that the gene inserted into the pH11 locus is highly expressed in cells, embryos and animals.
[本文引用: 1]
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
DOI:10.1007/s11248-012-9682-3URLPMID:23328918Magsci [本文引用: 1]
The feasibility of using the pig parotid secretory protein promoter to drive the β-glucanase transgene expression in mouse parotid glands was examined in this study. The parotid gland-specific vector expressing β-glucanase gene (GLU, from Paenibacillus polymyxa CP7) was constructed. Transgenic mice were produced by the pronuclear microinjection. Both PCR and Southern blot analysis showed that the mice carried the β-glucanase gene and the β-glucanase gene could be stably inherited. Furthermore, RT-PCR and northern blot analysis indicated that it was specifically expressed in the parotid. The β-glucanase activity in the saliva was found to be 0.18 U/mL. After feeding a diet containing 2 % β-glucan, the average daily gain of transgenic was significantly higher than non-transgenic mice. The crude protein and crude fat concentration in faeces of transgenic mice were significantly reduced compared with that of the non-transgenic mice. These results suggest that the successful expression of foreign β-glucanase in the animal parotid would offer a promising biological approach to reduce the anti-nutritional effect of β-glucans in feed.
DOI:10.1186/gb-2007-8-s1-s13URLPMID:18047690 [本文引用: 1]
Swine production has been an important part of our lives since the late Mesolithic or early Neolithic periods, and ranks number one in world meat production. Pig production also contributes to high-value-added medical markets in the form of pharmaceuticals, heart valves, and surgical materials. Genetic engineering, including the addition of exogenous genetic material or manipulation of the endogenous genome, holds great promise for changing pig phenotypes for agricultural and medical applications. Although the first transgenic pigs were described in 1985, poor survival of manipulated embryos; inefficiencies in the integration, transmission, and expression of transgenes; and expensive husbandry costs have impeded the widespread application of pig genetic engineering. Sequencing of the pig genome and advances in reproductive technologies have rejuvenated efforts to apply transgenesis to swine. Pigs provide a compelling new resource for the directed production of pharmaceutical proteins and the provision of cells, vascular grafts, and organs for xenotransplantation. Additionally, given remarkable similarities in the physiology and size of people and pigs, swine will increasingly provide large animal models of human disease where rodent models are insufficient. We review the challenges facing pig transgenesis and discuss the utility of transposases and recombinases for enhancing the success and sophistication of pig genetic engineering. 'The paradise of my fancy is one where pigs have wings.' (GK Chesterton).
DOI:10.1016/j.cell.2005.07.013URLPMID:16096065 [本文引用: 1]
Transposable elements have been routinely used for genetic manipulation in lower organisms, including generating transgenic animals and insertional mutagenesis. In contrast, the usage of transposons in mice and other vertebrate systems is still limited due to the lack of an efficient transposition system. We have tested the ability of piggyBac ( PB), a DNA transposon from the cabbage looper moth Trichoplusia ni, to transpose in mammalian systems. We show that PB elements carrying multiple genes can efficiently transpose in human and mouse cell lines and also in mice. PB permits the expression of the marker genes it carried. During germline transposition, PB could excise precisely from original insertion sites and transpose into the mouse genome at diverse locations, preferably transcription units. These data provide a first and critical step toward a highly efficient transposon system for a variety of genetic manipulations including transgenesis and insertional mutagenesis in mice and other vertebrates.
DOI:10.1093/nar/gkr764URLPMID:3239208 [本文引用: 1]
Abstract The development of technologies that allow the stable delivery of large genomic DNA fragments in mammalian systems is important for genetic studies as well as for applications in gene therapy. DNA transposons have emerged as flexible and efficient molecular vehicles to mediate stable cargo transfer. However, the ability to carry DNA fragments >10 kb is limited in most DNA transposons. Here, we show that the DNA transposon piggyBac can mobilize 100-kb DNA fragments in mouse embryonic stem (ES) cells, making it the only known transposon with such a large cargo capacity. The integrity of the cargo is maintained during transposition, the copy number can be controlled and the inserted giant transposons express the genomic cargo. Furthermore, these 100-kb transposons can also be excised from the genome without leaving a footprint. The development of piggyBac as a large cargo vector will facilitate a wider range of genetic and genomic applications. The Author(s) 2011. Published by Oxford University Press.
DOI:10.1073/pnas.0606979103URLPMID:17005721 [本文引用: 1]
A nonviral vector for highly efficient site-specific integration would be desirable for many applications in transgenesis, including gene therapy. In this study we directly compared the genomic integration efficiencies of piggyBac, hyperactive Sleeping Beauty (SB11), Tol2, and Mos1 in four mammalian cell lines. piggyBac demonstrated significantly higher transposition activity in all cell lines whereas Mos1 had no activity. Furthermore, piggyBac transposase coupled to the GAL4 DNA-binding domain retains transposition activity whereas similarly manipulated gene products of Tol2 and SB11 were inactive. The high transposition activity of piggyBac and the flexibility for molecular modification of its transposase suggest the possibility of using it routinely for mammalian transgenesis.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/s41598-017-02785-yURLPMID:28596588 [本文引用: 2]
Abstract Genetically modified pigs have important roles in agriculture and biomedicine. However, genome-specific knock-in techniques in pigs are still in their infancy and optimal strategies have not been extensively investigated. In this study, we performed electroporation to introduce a targeting donor vector (a non-linearized vector that did not contain a promoter or selectable marker) into Porcine Foetal Fibroblasts (PFFs) along with a CRISPR/Cas9 vector. After optimization, the efficiency of the EGFP site-specific knock-in could reach up to 29.6% at the pRosa26 locus in PFFs. Next, we used the EGFP reporter PFFs to address two key conditions in the process of achieving transgenic pigs, the limiting dilution method and the strategy to evaluate the safety and feasibility of the knock-in locus. This study demonstrates that we establish an efficient procedures for the exogenous gene knock-in technique and creates a platform to efficiently generate promoter-less and selectable marker-free transgenic PFFs through the CRISPR/Cas9 system. This study should contribute to the generation of promoter-less and selectable marker-free transgenic pigs and it may provide insights into sophisticated site-specific genome engineering techniques for additional species.
DOI:10.1186/s12896-016-0234-4URLPMID:4715285 [本文引用: 1]
The CRISPR/Cas9 system is increasingly used for gene inactivation in mouse zygotes, but homology-directed mutagenesis and use of inbred embryos are less established. In particular,Rosa26knock-in alleles for the insertion of transgenes in a genomic ‘safe harbor’ site, have not been produced. Here we applied CRISPR/Cas9 for the knock-in of 8–1102kb inserts intoRosa26of C57BL/6 zygotes. We found that 10–2002% of live pups derived from microinjected zygotes were founder mutants, without apparent off-target effects, and up to 5002% knock-in embryos were recovered upon coinjection of Cas9 mRNA and protein. Using this approach, we established a new mouse line for the Cre/loxP-dependent expression of Cas9. Altogether, our protocols and resources support the fast and direct generation of newRosa26knock-in alleles and of Cas9-mediated in vivo gene editing in the widely used C57BL/6 inbred strain. The online version of this article (doi:10.1186/s12896-016-0234-4) contains supplementary material, which is available to authorized users.
DOI:10.1038/srep24360URLPMID:4827023 [本文引用: 1]
Recent advances in our ability to design DNA binding factors with specificity for desired sequences have resulted in a revolution in genetic engineering, enabling directed changes to the genome to be made relatively easily. Technologies that facilitate specific and precise genome editing, such as knock-in, are critical for determining the functions of genes and for understanding fundamental biological processes. The CRISPR/Cas9 system has recently emerged as a powerful tool for functional genomic studies in mammals. Rosa26 gene can encode a non-essential nuclear RNA in almost all organizations, and become a hot point of exogenous gene insertion. Here, we describe efficient, precise CRISPR/Cas9-mediated Integration using a donor vector with tGFP sequence targeted in the sheep genomic Rosa26 locus. We succeeded in integrating with high efficiency an exogenous tGFP (turboGFP) gene into targeted genes in frame. Due to its simplicity, design flexibility, and high efficiency, we propose that CRISPR/Cas9-mediated knock-in will become a standard method for the generation transgenic sheep.
URLPMID:26756580 [本文引用: 1]
The porcine pluripotent cells that can generate germline chimeras have not been developed. The Oct4 promoter-based fluorescent reporter system, which can be used to monitor pluripotency, is an important tool to generate authentic porcine pluripotent cells. In this study, we established a porcine Oct4 reporter system, wherein the endogenous Oct4 promoter directly controls red fluorescent protein (RFP). 2A-tdTomato sequence was inserted to replace the stop codon of the porcine Oct4 gene by homogenous recombination (HR). Thus, the fluorescence can accurately show the activation of endogenous Oct4. Porcine fetal fibroblast (PFF) lines with knock-in (KI) of the tdTomato gene in the downstream of endogenous Oct4 promoter were achieved using the CRISPR/CAS9 system. Transgenic PFFs were used as donor cells for somatic cell nuclear transfer (SCNT). Strong RFP expression was detected in the blastocysts and genital ridges of SCNT fetuses but not in other tissues. Two viable transgenic piglets were also produced by SCNT. Reprogramming of fibroblasts from the fetuses and piglets by another round of SCNT resulted in tdTomato reactivation in reconstructed blastocysts. Result indicated that a KI porcine reporter system to monitor the pluripotent status of cells was successfully developed.
.
[本文引用: 1]
.
[本文引用: 1]
DOI:10.1186/1472-6750-12-84URLPMID:23140586 [本文引用: 2]
Background Somatic cell nuclear transfer (SCNT) is currently the most efficient and precise method to generate genetically tailored pig models for biomedical research. However, the efficiency of this approach is crucially dependent on the source of nuclear donor cells. In this study, we evaluate the potential of primary porcine kidney cells (PKCs) as cell source for SCNT, including their proliferation capacity, transfection efficiency, and capacity to support full term development of SCNT embryos after additive gene transfer or homologous recombination. Results PKCs could be maintained in culture with stable karyotype for up to 71 passages, whereas porcine fetal fibroblasts (PFFs) and porcine ear fibroblasts (PEFs) could be hardly passaged more than 20 times. Compared with PFFs and PEFs, PKCs exhibited a higher proliferation rate and resulted in a 2-fold higher blastocyst rate after SCNT and in vitro cultivation. Among the four transfection methods tested with a GFP expression plasmid, best results were obtained with the NucleofectorTM technology, resulting in transfection efficiencies of 70% to 89% with high fluorescence intensity, low cytotoxicity, good cell proliferation, and almost no morphological signs of cell stress. Usage of genetically modified PKCs in SCNT resulted in approximately 150 piglets carrying at least one of 18 different transgenes. Several of those pigs originated from PKCs that underwent homologous recombination and antibiotic selection before SCNT. Conclusion The high proliferation capacity of PKCs facilitates the introduction of precise and complex genetic modifications in vitro. PKCs are thus a valuable cell source for the generation of porcine biomedical models by SCNT.
DOI:10.1089/clo.2007.0021URLPMID:18154513 [本文引用: 4]
Abstract Porcine embryonic fibroblasts (PEF) are important as donor cells for nuclear transfer for generation of genetically modified pigs. In this study, we determined an optimal protocol for transfection of PEF with the Amaxa Nucleofection system, which directly transfers DNA into the nucleus of cells, and compared its efficiency with conventional lipofection and electroporation. Cell survival and transfection efficiency were assessed using dye-exclusion assay and a green fluorescent protein (GFP) reporter construct, respectively. Our optimized nucleofection parameters yielded survival rates above 60%. Under these conditions, FACS analysis demonstrated that 79% of surviving cells exhibited transgene expression 48 h after nucleofection when program U23 was used. This efficiency was higher than that of transfection of PEFs with electroporation (ca. 3-53%) or lipofection (ca. 3-8%). Transfected cells could be expanded as stably transgene-expressing clones over a month. When porcine nuclear transfer (NT) was performed using stable transformant expressing GFP as a donor cell, 5-6% of reconstituted embryos developed to blastocysts, from which 30-50% of embryos exhibited NT-embryo-derived green fluorescence. Under the conditions evaluated, nucleofection exhibited higher efficiency than conventional electroporation and lipofection, and may be a useful alternative for generation of genetically engineered pigs through nuclear transfer.
DOI:10.1007/s11248-009-9345-1URLPMID:19937273 [本文引用: 6]
Development of a transgenic porcine biomedical research model requires effective delivery of DNA into the donor cell followed by selection of genetically modified somatic cell lines to be used for nuclear transfer. The objective of the current study was 2-fold: (1) to compare the effectiveness of a single 102ms pulse of different voltages (V; 100, 150, 200, 250, 300, 350) and multiple 102ms pulses (1, 2, 3, 4 or 5) at 30002V for delivery and expression of super-coiled GFP vector in surviving cells of three fetal fibroblast cell lines, and (2) to determine the ability of these electroporation parameters to produce stably transfected fibroblast colonies following G418 selection. Cell line ( P 02<020.001) and voltage ( P 02<020.001) affected DNA delivery into the cell as assessed by GFP expression while survival at 2402h was affected by voltage ( P 02<020.001) and not by cell line ( P 02=020.797). Using a single pulse while increasing voltage resulted in the percentage of GFP expressing cells increasing from 3.202±020.8% to 43.002±023.4% while survival decreased from 90.502±028.0% to 44.802±022.0%. The number of pulses at 30002V significantly affected survival ( P 02<020.001) and GFP expression ( P 02<020.001). Survival steadily decreased following 1–5 pulses from 63.202±026.3% to 3.002±020.3% with GFP expression of surviving cells increasing from 35.602±022.67% to 71.402±026.1%. Electroporation of a selectable marker at a 1:1 copy number ratio to a co-electroporated transgene resulted in 83% of G418 resistant colonies also being PCR positive for the secondary transgene. These electroporation conditions, specifically, three 102ms pulses of 30002V to 20002μL of 102×0210 6 02cells/mL in the presence of 12.502μg DNA/mL effectively introduced DNA into somatic cells. The utilization of these conditions produced numerous transgenic fibroblast colonies following G418 selection that when used for somatic cell nuclear transfer resulted in the production of live offspring.
[本文引用: 1]
DOI:10.3389/fncir.2014.00041URLPMID:4018551 [本文引用: 1]
The zebrafish has various advantages as a model organism to analyze the structure and function of neural circuits but efficient viruses or other tools for fast gene transfer are lacking. We show that transgenes can be introduced directly into the adult zebrafish brain by herpes simplex type I viruses (HSV-1) or electroporation. We developed a new procedure to target electroporation to defined brain areas and identified promoters that produced strong long-term expression. The fast workflow of electroporation was exploited to express multiple channelrhodopsin-2 variants and genetically encoded calcium indicators in telencephalic neurons for measurements of neuronal activity and synaptic connectivity. The results demonstrate that HSV-1 and targeted electroporation are efficient tools for gene delivery into the zebrafish brain, similar to adeno-associated viruses and lentiviruses in other species. These methods fill an important gap in the spectrum of molecular tools for zebrafish and are likely to have a wide range of applications.
DOI:10.2220/biomedres.39.95URLPMID:29669988 [本文引用: 2]
Abstract Recently, gene-editing using the clustered regularly interspaced short palindromic repeats (CRISPR)/ CRISPR-associated protein 9 (Cas9) technique has attempted to utilize fibroblasts of livestock animals for somatic cell nuclear transfer. In this study, we establish the procedure for preparing skin fibroblast clones whose genes were edited by the CRISPR/Cas9 technique. After isolating fibroblasts from earlobes of Japanese Black cattle, subsequent collagenase-digestion and extensive wash procedures enabled us to avoid contamination of fungi. Electroporation using NEPA21, rather than lipofection using commercially available liposome reagents, allowed us to perform more efficient transfection of plasmid constructs. Although bovine ear-derived fibroblasts were not able to proliferate in single cell cultures in Dulbecco's modified Eagle medium containing 10% fetal calf serum, supplementation with insulin-transferrin-selenium mixture, human recombinant epidermal growth factor, or human recombinant basic fibroblast growth factor promoted proliferation of the cells, even in a single cell culture. Taking advantage of our established protocol, we eventually obtained eight ear-derived fibroblast clones with a recessive mutation in the isoleucyl-tRNA synthetase gene corrected by the CRISPR/Cas9 technique.
[本文引用: 3]
.
[本文引用: 3]
DOI:10.1021/bp060029sURLPMID:16889402 [本文引用: 1]
A plasmid expressing the -galactosidase enzyme was used to transfect Vero cells in order to evaluate the efficiency of a liposome-mediated transfection by circular and linear DNA. The results obtained showed a low rate of transfection by linear DNA:liposome complexes. To explore whether the structure of the complexes was interfering with the transfection, atomic force microscopy (AFM) was used. It has confirmed the difference between the linear and circular condensates: whereas the circular DNA:liposome complexes presented compact spherical or cylindrical structures of about 100-800 nm, the linear DNA showed pearl necklace-like structures, with pearls varying from 250 to 400 nm. On the basis of the theory proposed by Kuhn et al. (1999), low concentrations of cationic amphihile were used to neutralize or reverse the DNA charge in order to improve the transfection efficiency of the linear DNA. Using this method, we were able to obtain the expression of the transgene without an associated toxicity observed with the linear DNA liposome delivery.
DOI:10.1101/gr.161638.113URLPMID:24179142Magsci [本文引用: 1]
Sequence-specific nucleases like TALENs and the CRISPR/Cas9 system have greatly expanded the genome editing possibilities in model organisms such as zebrafish. Both systems have recently been used to create knock-out alleles with great efficiency, and TALENs have also been successfully employed in knock-in of DNA cassettes at defined loci via homologous recombination (HR). Here we report CRISPR/Cas9-mediated knock-in of DNA cassettes into the zebrafish genome at a very high rate by homology-independent double-strand break (DSB) repair pathways. After co-injection of a donor plasmid with a short guide RNA (sgRNA) and Cas9 nuclease mRNA, concurrent cleavage of donor plasmid DNA and the selected chromosomal integration site resulted in efficient targeted integration of donor DNA. We successfully employed this approach to convert eGFP into Gal4 transgenic lines, and the same plasmids and sgRNAs can be applied in any species where eGFP lines were generated as part of enhancer and gene trap screens. In addition, we show the possibility of easily targeting DNA integration at endogenous loci, thus greatly facilitating the creation of reporter and loss-of-function alleles. Due to its simplicity, flexibility, and very high efficiency, our method greatly expands the repertoire for genome editing in zebrafish and can be readily adapted to many other organisms.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[J].
[本文引用: 1]
[本文引用: 1]
DOI:10.1371/journal.pone.0019986URLPMID:3094386 [本文引用: 1]
Multiple genetic modifications in pigs can essentially benefit research on agriculture, human disease and xenotransplantation. Most multi-transgenic pigs have been produced by complex and time-consuming breeding programs using multiple single-transgenic pigs. This study explored the feasibility of producing multi-transgenic pigs using the viral 2A peptide in the light of previous research indicating that it can be utilized for multi-gene transfer in gene therapy and somatic cell reprogramming. A 2A peptide-based double-promoter expression vector that mediated the expression of four fluorescent proteins was constructed and transfected into primary porcine fetal fibroblasts. Cell colonies (54.3%) formed under G418 selection co-expressed the four fluorescent proteins at uniformly high levels. The reconstructed embryos, which were obtained by somatic cell nuclear transfer and confirmed to express the four fluorescent proteins evenly, were transplanted into seven recipient gilts. Eleven piglets were delivered by two gilts, and seven of them co-expressed the four fluorescent proteins at equivalently high levels in various tissues. The fluorescence intensities were directly observed at the nose, hoof and tongue using goggles. The results suggest that the strategy of combining the 2A peptide and double promoters efficiently mediates the co-expression of the four fluorescent proteins in pigs and is hence a promising methodology to generate multi-transgenic pigs by a single nuclear transfer.
[本文引用: 1]
DOI:10.1038/ncomms6560URLPMID:4263139 [本文引用: 1]
Abstract Genome engineering using programmable nucleases enables homologous recombination (HR)-mediated gene knock-in. However, the labour used to construct targeting vectors containing homology arms and difficulties in inducing HR in some cell type and organisms represent technical hurdles for the application of HR-mediated knock-in technology. Here, we introduce an alternative strategy for gene knock-in using transcription activator-like effector nucleases (TALENs) and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated 9 (Cas9) mediated by microhomology-mediated end-joining, termed the PITCh (Precise Integration into Target Chromosome) system. TALEN-mediated PITCh, termed TAL-PITCh, enables efficient integration of exogenous donor DNA in human cells and animals, including silkworms and frogs. We further demonstrate that CRISPR/Cas9-mediated PITCh, termed CRIS-PITCh, can be applied in human cells without carrying the plasmid backbone sequence. Thus, our PITCh-ing strategies will be useful for a variety of applications, not only in cultured cells, but also in various organisms, including invertebrates and vertebrates.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}