 ,1,*
,1,*Mapping and genetic analysis of QTLs for Fusarium head blight resistance to disease spread in Yangmai 16
HU Wen-Jing1, ZHANG Yong1, LU Cheng-Bin1, WANG Feng-Ju2, LIU Jin-Dong2, JIANG Zheng-Ning1, WANG Jin-Ping2, ZHU Zhan-Wang2, XU Xiao-Ting2, HAO Yuan-Feng2, HE Zhong-Hu2,3, GAO De-Rong,1,*通讯作者:
收稿日期:2019-07-22接受日期:2019-09-26网络出版日期:2019-10-09
| 基金资助: |
Received:2019-07-22Accepted:2019-09-26Online:2019-10-09
| Fund supported: |
作者简介 About authors
E-mail:huren2008@126.com。

摘要
关键词:
Abstract
Keywords:
PDF (2398KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
胡文静, 张勇, 陆成彬, 王凤菊, 刘金栋, 蒋正宁, 王金平, 朱展望, 徐小婷, 郝元峰, 何中虎, 高德荣. 小麦品种扬麦16赤霉病抗扩展QTL定位及分析[J]. 作物学报, 2020, 46(2): 157-165. doi:10.3724/SP.J.1006.2020.91048
HU Wen-Jing, ZHANG Yong, LU Cheng-Bin, WANG Feng-Ju, LIU Jin-Dong, JIANG Zheng-Ning, WANG Jin-Ping, ZHU Zhan-Wang, XU Xiao-Ting, HAO Yuan-Feng, HE Zhong-Hu, GAO De-Rong.
小麦赤霉病(Wheat scab, Fusarium head blight, FHB)是由禾谷镰刀菌(Fusarium graminearum)等引起的一种世界性真菌病害[1]。我国长江中下游麦区是赤霉病的常发区和重发区, 2012—2015年, 江苏省年均赤霉病发生面积约120万公顷[2]。近年来, 随着气候变暖和玉米秸秆还田, 小麦赤霉病发生愈来愈重, 正迅速扩展到黄淮麦区。据统计, 近10年, 河南省赤霉病年均发生面积达110万公顷左右, 其中2012年达333万公顷, 2016年达117万公顷[3]。
赤霉病的侵染时期主要是小麦开花期, 赤霉菌侵染小花后迅速在穗部扩展, 在小麦籽粒灌浆成熟过程中不断繁殖, 进而积累各种毒素, 例如脱氧雪腐镰孢菌烯醇(deoxynivalenol, DON)、雪腐镰孢菌烯醇(nivalenol, NIV)和玉米赤霉烯酮(zearalenol, ZEN), 严重影响小麦产量, 并对人畜健康造成巨大伤害, 成为粮食安全的主要威胁[4]。选育和种植抗病品种是应对赤霉病危害最经济、安全的途径。小麦赤霉病的抗性主要可以分为抗侵染(type I)、抗扩展(type II)、抗DON积累(type III)和籽粒抗性(type IV) 4种类型, 其中type II类型研究最深入[5]。多年来, 已报道超过250个抗赤霉病QTL, 覆盖小麦全部21条染色体[6], 但目前明确的小麦抗赤霉病基因只有7个, 其中Fhb1、Fhb2、Fhb3、Fhb6和Fhb7是抗扩展类型, Fhb4和Fhb5是抗侵染类型, 仅Fhb1已经被克隆[7,8]。苏麦3号是国际上研究和利用最广泛的赤霉病抗源, 望水白是已知的携带抗赤霉病位点最多的小麦品种之一[3]。它们均携带主效抗病基因Fhb1, 但两者综合农艺性状较差, 秆高易倒伏、穗数和穗粒数少、粒重低, 虽利用它们选育出一批抗性好的材料, 例如宁7840等, 但因其综合丰产性差未得到大面积应用[9]。据报道, 聚合Fhb1和其他抗赤霉病位点, 可累加抗性效应, 提高抗性水平[10,11]。扬麦系列品种一直是长江中下游麦区主体品种, 多数品种表现中抗赤霉病。扬麦16是近年来长江中下游推广面积最大的小麦品种, 利用其为亲本选育出优质丰产抗赤霉病的小麦品种如扬麦23和扬麦28等, 表明扬麦16在小麦遗传改良中可以作为稳定的抗赤亲本使用。经中国农业科学院开发的小麦功能基因标记检测, 扬麦16不含Fhb1[12,13], 可能存在其他主效抗赤霉病基因。因此, 挖掘扬麦16的抗赤霉病基因, 拓宽小麦赤霉病抗源, 对小麦抗赤霉病育种具有重要意义。本研究利用来自扬麦16/中麦895 的174份双单倍体家系, 经660K SNP芯片检测, 构建高密度遗传图谱, 结合多年表型鉴定结果, 挖掘扬麦16抗赤霉病QTL, 为开展标记辅助抗赤霉病育种提供技术支撑。
1 材料与方法
1.1 试验材料及田间试验
以扬麦16 (Yangmai 16, 简称YM16, 系谱: 扬91F138 (扬麦158选系)/扬90-30)为母本, 中麦895 (Zhongmai 895, 简称ZM895, 系谱: 周麦16/荔垦4号)为父本杂交, 利用玉米花粉诱导双单倍体培育174份DH家系。以苏麦3号(Sumai 3, 简称SM3)和周麦18 (Zhoumai 18, 简称ZM18)为抗赤霉病和感赤霉病对照。2016—2017年度(简称为2017年)于国际玉米小麦改良中心(International Maize and Wheat Improvement Center, CIMMYT)温室种植, 花盆直径20 cm, 每盆播种8粒, 最后定苗至5株, 2次重复, 参照CIMMYT的正常管理方法管理温室。2017—2018年度和2018—2019年度(简称为2018年和2019年)于江苏里下河地区农业科学研究所基地种植, 采用完全随机设计试验, 2行区, 每行30粒, 2次重复, 行长1.3 m, 行距0.3 m, 参照当地的正常栽培方法管理大田。1.2 赤霉病鉴定和表型数据处理
参照 Yu 等[14]方法制备赤霉菌孢子悬浮液(5000孢子 mL-1), 采用单花滴注法接种。于小麦开花初期, 用注射器吸取10 μL孢子液注射入麦穗中间小穗的一个小花中(顶部小穗开始自上而下第6个小穗), 接种每系15个穗子。温室单花滴注接种后套袋保湿3 d, 田间单花接种后立即采用人工弥雾保湿(每半小时田间弥雾喷水5 min, 6:00-18:00)。接种21 d后调查病小穗数, 计算病小穗率。病小穗率(percentage of scabbed spikelets, 简称PSS)作为赤霉病严重度的衡量指标[9]。病小穗率(%) = (病小穗数/总小穗数) ×100%。用 DPS 软件和IciMapping的ANOVA功能进行群体表型数据的相关分析、方差分析及遗传力的估算。1.3 分子标记的检测
取小麦幼苗, 采用CTAB法提取基因组DNA[15], 利用Affymetrix SNP技术检测平台(北京博奥生物有限公司)小麦660K SNP芯片分析亲本和群体, 利用Genomestudio v1.0软件进行多态性分析和整合(Illumina, http://www.illumina.com/)。将13个包括Rht-B1和Rht-D1[12]等重要农艺性状功能基因的相应KASP标记也整合入多态性标记中。1.4 遗传图谱构建和QTL定位
筛选出缺失率<10%, 最小等位基因频率>30%的多态性标记, 使用IciMapping v4.1 (http://www. isbreeding.net/)的BIN功能进行去冗余。根据SNP侧翼序列比对到中国春参考基因组的物理位置(IWGSC RefSeq v1.0, https://wheat-urgi.versailles. inra.fr/)和SNP在小麦660K整合图谱(赵光耀, 私人通讯) KJ-RIL遗传图谱中的位置[16], 挑选461个分布在小麦21条染色体的SNP作为Anchor标记, 利用IciMapping v4.1的MAP功能将标记分组, 利用MSTmap(http://mstmap.org/)的Kosambi功能对各组标记排序和计算遗传距离。利用JoinMap v4.0[17]校正遗传长度, 得到最终遗传图谱。利用MapChart 2.3 (https://www.wur.nl/en/show/Mapchart.htm)绘制遗传图谱。利用IciMapping v4.1的完备区间作图法(Inclusive composite interval mapping, ICIM)检测抗赤霉病QTL, LOD阈值设为2.5[18]。将同一染色体上检测到的峰值所在遗传位置之间距离小于10 cM的QTL视为同一个位点。QTL的命名方法为“Q”加性状缩写, 加单位名称缩写, 加QTL所在的染色体, 同一染色体上多个QTL用.1、.2、.3、……来表示。例如 QFhb.yaas-5A.1 代表 5A 染色体上检测出来的控制赤霉病抗扩展的第一个QTL。将在2个或者超过2个年份下能够检测到的QTL定义为“稳定QTL”, 将效应值大于10%的QTL定义为“具有较大表型贡献率的QTL”。为了与前人结果比较, 将连锁标记或者基因的序列与中国春参考基因组序列的EnsemblPlants数据库(http://plants.ensembl.org/)比对, 获得标记或者基因的物理位置[19]。2 结果与分析
2.1 亲本及DH群体的抗性表现
由表1和图1可以看出, 3年试验的PSS范围, 扬麦16是14.2%~23.3%, 中麦895是58.7%~69.7%, 抗病对照苏麦3号是5.2%~6.1%, 感病对照周麦18是60.2%~70.1%。群体的偏度和峰度绝对值均<1, 分布呈连续的正态分布, 可用于QTL定位研究[20]。Table 1
表1
表1扬麦16/中麦895 DH群体、亲本和对照的赤霉病严重度
Table 1
| 年份 Year | 亲本 Parents (%) | 对照 CK (%) | DH群体 DH population | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 扬麦16 Yangmai 16 | 中麦895 Zhongmai 895 | 苏麦3 Sumai 3 | 周麦18 Zhoumai 18 | 平均值 Mean (%) | 标准差 SD | 最大值 Max. (%) | 最小值 Min. (%) | 峰度 Kurt. | 偏度 Skew. | 遗传力 Hereditability | |
| 2017 | 14.2 A | 58.7 B | 5.2 A | 60.2 B | 29.1 | 18.9 | 83.7 | 3.4 | 0.2 | 0.9 | 0.67 |
| 2018 | 17.9 A | 65.0 B | 5.6 A | 62.3 B | 53.5 | 19.9 | 100.0 | 6.6 | -0.7 | -0.2 | 0.74 |
| 2019 | 23.3 A | 69.7 B | 6.1 A | 70.1 B | 49.5 | 18.1 | 89.8 | 10.3 | -0.5 | 0.1 | 0.82 |
新窗口打开|下载CSV
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1扬麦16/中麦895 DH群体赤霉病严重度频次分布(2017-2019)
横坐标代表病小穗率, 纵坐标代表相关病小穗率范围内的家系数目。
Fig. 1Frequency distributions of DH lines with different FHB severity (2017-2019)
The abscissa indicates the percentage of scabbed spikelets, and the ordinate indicates the line number.
3年试验结果均表明双亲赤霉病的病小穗率存在极显著的差异, DH群体赤霉病严重度的最小值和最大值之间差异明显, 存在超亲分离, 可以初步判断双亲都携带抗性基因且位点互补, 通过杂交可以得到超亲分离的株系。抗病和感病对照的病小穗率亦存在极显著差异。
2.2 遗传连锁图谱
利用660K SNP芯片, 过滤和筛选得到152,310个高质量多态性SNP标记, 经过去冗余分析后, 最终“扬麦16/中麦895”高密度遗传图谱的上图标记为14,480个。图谱覆盖小麦21条染色体, 长度为3681.7 cM, 密度为3.9个标记 cM-1。分布于小麦A、B和D染色体组的标记数分别为5036、7634和1810个, 连锁长度分别为1223.2、1035.6和1423.0 cM。其中, 标记密度最高的是1B染色体, 平均11.7个标记 cM-1, 标记密度最低的是7D染色体, 平均0.8个标记 cM-1。2.3 抗赤霉病QTL定位
共检测到6个抗赤霉病QTL, 分别位于2DL、3BL、4BS、4DS、5BL和6AS上。除QFhb.yaas-4BS外, 其他5个QTL均来自扬麦16。QFhb.yaas-4DS在3年中均被检测到, 抗性贡献率8.8%~13.7%, QFhb.yaas-6AS可在2年同时被检测到, 抗性贡献率9.5%~15.0%。这2个QTL是本研究检测到的稳定的抗赤霉病位点。QFhb.yaas-2DL和QFhb.yaas-3BL分别在2017和2019年被检测到, 抗性贡献率分别为10.5%和14.7%。QFhb.yaas-4BS和QFhb.yaas-5BL均仅在2018年被检测到, 抗性贡献率分别为8.3%和6.4% (表2和图2)。Table 2
表2
表2扬麦16/中麦895 DH 群体抗赤霉病QTL定位
Table 2
| QTL | 年份 Year | 物理位置 Physical position (Mb) | 遗传位置 Genetic position (cM) | 标记区间 Marker interval | LOD | 贡献率 PVE (%) | 加性效应# Add# |
|---|---|---|---|---|---|---|---|
| QFhb.yaas-2DL | 2017 | 512.7-532.0 | 49.6 | AX_111765614-AX_109738482 | 4.7 | 10.5 | -6.1 |
| QFhb.yaas-3BL | 2019 | 637.1-647.2 | 138.6 | AX_94528202-AX_109001202 | 8.9 | 14.7 | -6.9 |
| QFhb.yaas-4DS | 2017 | 27.1-33.1 | 61.9 | AX_109962849-AX_111071805 | 5.6 | 13.7 | -7.2 |
| 2018 | 18.3-19.3 | 54.7 | AX_111311200-AX_89421921 | 4.4 | 8.8 | -5.9 | |
| 2019 | 15.9-16.6 | 53.7 | AX_94558069-AX_95150762 | 6.2 | 9.5 | -5.6 | |
| QFhb.yaas-5BL | 2018 | 587.1-588.8 | 138.7 | AX_94646656-AX_109853998 | 3.2 | 6.4 | -5.0 |
| QFhb.yaas-6AS | 2018 | 12.0-12.4 | 64.9 | AX_109294414-AX_108848013 | 4.6 | 9.5 | -6.2 |
| 2019 | 12.0-12.4 | 65.0 | AX_109294414-AX_108848013 | 8.9 | 15.0 | -7.1 | |
| QFhb.yaas-4BS | 2018 | 82.7-91.3 | 14.7 | AX_110472183-AX_94859572 | 4.1 | 8.3 | 5.8 |
新窗口打开|下载CSV
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2扬麦16/中麦895 DH群体抗赤霉病QTL定位情况
连锁群右边是标记名称, 左边是遗传位置(CM)。红色、蓝色和绿色分别代
Fig. 2Mapped QTL for FHB resistance in the Yangmai 16/Zhongmai 895 DH population
Markers’ names are shown to the right of vertical axis, and their genetic positions are shown in cM to the left. Red, blue, and green represent 2017, 2018, and 2019, respectively. LOD values of QTL are shown on the right side.
2.4 QTL效应分析
将在2个或者超过2个环境下能够检测到的QTL定义为“稳定QTL”, 本研究分别在3年和2年环境下均检测到QFhb.yaas-4DS和QFhb.yaas-6AS, 根据它们的基因型将174个家系分为4种类型, 用3年赤霉病严重度平均值进行类型间的差异分析。38个家系同时含有QFhb.yaas-4DS和QFhb.yaas-6AS, 赤霉病严重度范围9.9%~57.4% (平均32.3%), 多数家系PSS小于40%, 仅有8个家系PSS在40%~60%范围内; 28个家系只含有QFhb.yaas-6AS, 赤霉病严重度范围18.9%~78.8% (平均44.8%), 18个家系PSS小于50%, 10个家系PSS在50%~80%范围内; 有62个家系只含有QFhb.yaas-4DS, 赤霉病严重度范围18.6%~71.3% (平均43.2%), 41个家系PSS小于50%, 21个家系PSS在50%~80%范围内; 46个家系不含上述2个QTL, 赤霉病严重度范围26.1%~88.0% (平均54.3%), 多数家系PSS大于40%, 仅有6个家系PSS在20%~40%范围。聚合2个抗性位点的家系比含有1个抗性位点和不含有抗性位点的家系赤霉病严重度显著降低(表3)。Table 3
表3
表3扬麦16/中麦895 DH 群体中存在不同稳定赤霉病抗扩展QTL家系的赤霉病严重度情况
Table 3
| QTL组成 QTL combination | 家系数 Number of lines | 最小值 Minimum | 最大值 Maximum | 平均 Mean | 标准差 Standard deviation | 方差 Variance |
|---|---|---|---|---|---|---|
| None | 46 | 26.1 | 88.0 | 54.3 A | 13.7 | 187.6 |
| QFhb.yaas-4DS | 62 | 18.6 | 71.3 | 43.2 B | 13.1 | 172.0 |
| QFhb.yaas-6AS | 28 | 18.9 | 78.8 | 44.8 B | 16.4 | 269.6 |
| QFhb.yaas-4DS+QFhb.yaas-6AS | 38 | 9.9 | 57.4 | 32.3 C | 11.2 | 126.5 |
新窗口打开|下载CSV
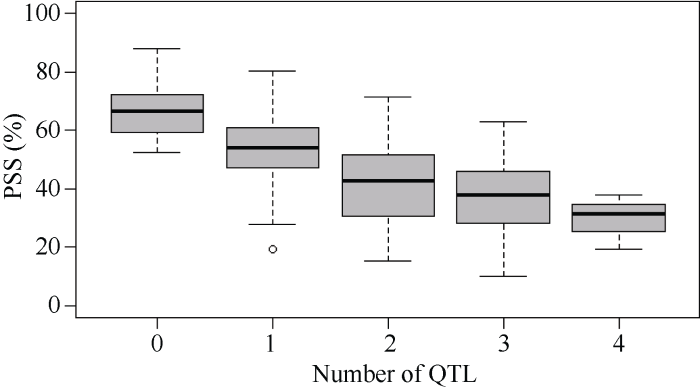
根据本研究中定位到的效应值大于10%的抗性QTL QFhb.yaas-2DL、QFhb.yaas-3BL、QFhb.yaas- 4DS 和QFhb.yaas-6AS将174个DH家系分为5种类型, 用3年PSS平均值进行类型间的差异分析。10个家系同时含有4个抗性QTL, 发病较轻, 平均PSS是30.5%; 46个家系含有3个抗性QTL, 平均PSS是36.9%; 65个家系含有2个抗性QTL, 平均PSS是41.8%; 46个家系只含有1个抗性QTL, 平均PSS是53.84%; 7个家系不含有任何一个上述4个抗性QTL, 平均PSS是67.1% (表4和图3)。
Table 4
表4
表4扬麦16/中麦895 DH群体中存在不同赤霉病抗扩展QTL家系的赤霉病严重度情况
Table 4
| QTL数 Number of QTL | 家系数 Number of lines | 平均PSS (100%)# Average of PSS (100%)# | 不同PSS范围内的家系数目 Number of lines in different PSS range | |||
|---|---|---|---|---|---|---|
| 0-25 | 25-50 | 50-75 | 75-100 | |||
| 0 | 7 | 67.1 A | 0 | 0 | 6 | 1 |
| 1 | 46 | 53.8 B | 1 | 15 | 28 | 2 |
| 2 | 65 | 41.8 C | 8 | 38 | 19 | 0 |
| 3 | 46 | 36.9 CD | 10 | 29 | 7 | 0 |
| 4 | 10 | 30.5 D | 2 | 8 | 0 | 0 |
新窗口打开|下载CSV
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3DH家系所含QTL数目与其平均赤霉病严重度的关系
PSS (percentage of scabbed spikelets)代表赤霉病严重度。
Fig. 3Relationship between DH lines with QTLs and FHB severity across environments
PSS (percentage of scabbed spikelets) indicates the FHB severity.
3 讨论
3.1 QTL定位结果与已知抗赤霉病QTL/基因比较
在扬麦16/中麦895 DH群体中共检测到6个赤霉病抗扩展QTL, 其中QFhb.yaas-4DS和QFhb.yaas- 6AS在多年均可被检测到, 是来源于扬麦16的稳定QTL位点。根据比对结果, QFhb.yaas-4DS与Rht-D1紧密连锁, Srinivasachary等[21]报道Rht-D1附近确实存在赤霉病抗侵染QTL, 来源于Spark, Rht-D1b可显著降低小麦对赤霉病抗侵染能力, 但其与赤霉病抗扩展性不相关[22], Buerstmayr等[23]研究表明携带Rht-D1b位点的小麦品种易感染赤霉病, 表明较高的株高营造的穗部局部低湿度环境可以减少赤霉病孢子侵染小麦。本研究采用一年CIMMYT温室单花滴注然后套袋保湿, 两年扬州田间单花滴注试验采取弥雾保湿, 株高对穗部的微环境存在一定的影响, 从而影响发病的严重度。将2017—2019年DH群体株高数据平均数(未发表资料)与赤霉病严重度平均值进行相关性分析, 发现株高均值与赤霉病严重度均值相关系数-0.33 (P<0.01), 表明该群体赤霉病抗性与株高存在一定相关性。群体中携带QFhb.yaas-4DS抗病等位基因家系的平均株高显著高于携带感病等位基因家系的平均株高(P < 0.01), 表明该位点确实与株高显著相关。该位点为“一因多效”或者“紧密连锁”, 可通过后续增加温室环境下单花滴注试验进行验证。另据报道, 小麦粒重基因TaGW2-6A启动子区域的单倍型变异除了与粒重相关外, 还与株高显著相关[24], 本研究定位到了位于6AS染色体上的抗赤霉病QTL QFhb.yaas-6AS, DH群体2017—2019年的株高平均值(未发表资料)与赤霉病严重度平均值相关系数为-0.33 (P<0.01), 表明该位点的赤霉病抗性与株高也存在一定的关系。QFhb.yaas-6AS与来源于扬麦13的QFhbp-jaas. 6AS-1位置相近[6]。扬麦13的系谱是扬88-84//Maris Dove/扬麦3号, 扬麦3号是阿夫的红壳选系; 扬麦16的系谱是扬麦158优系(91F138)/扬90-30, 扬麦158的系谱是扬麦4号//St1472/506, 扬麦4号的系谱是南大2419/胜利麦1-3-2//阿夫选系。由于扬麦品种的亲本中存在共同的亲本阿夫, 且亲缘关系较近, 推测QFhb.yaas-6AS与QFhbp-jaas.6AS-1可能是相同抗病位点, 后续我们会继续研究上述材料, 以确定QFhb.yaas-6AS的来源。
其他QTL仅在1年被检测到, 其中QFhb.yaas- 2DL和QFhb.yaas-3BL的效应值较大, 来自扬麦16。QFhb.yaas-2DL可解释表型变异10.5%, 经两侧标记序列与中国春参考基因组序列比对发现, 其物理位置区间是512.7~532.0 Mb, 与已报道的Wuhan-1中检测到的2DL上抗赤霉病位点QFhs.crc-2D位置相近(513.1 Mb)[25], Wuhan-1与扬麦16均来自长江中下游麦区, 推测可能是相同的QTL。QFhb.yaas-3BL能解释表型变异14.7%, 经比对发现无同位置抗赤霉病QTL报道, 推测可能为新的抗赤霉病位点。小麦赤霉病抗性属于数量性状, 发病受环境影响很大, 还需增加多环境试验验证上述QTL的有效性和稳定性。
3.2 抗性改良与QTL/基因聚合
DH家系中仅存在单个抗赤霉病位点时, 多数家系的PSS在50%以上, 但当存在2位点或者2位点以上的QTL聚合时, 平均PSS明显降低。聚合包含4个QTL的家系具有最好的赤霉病抗性, 其中2个家系的PSS在0~25%区间, 达到高抗水平, 其余8个家系的PSS在25%~50%区间, 达到中抗水平, 表明多个抗病基因聚合能表现出较好的抗性。Jia等[10]报道Fhb1与其他赤霉病抗性QTL/基因的聚合能有效提高小麦的赤霉病抗扩展和抗侵染水平。Arruda等[11]研究表明, 聚合多个3B上抗性位点能使赤霉病抗性水平提高。因此, 将多个抗病基因聚合到同一品种(系)中, 对于提高赤霉病抗性具有重要意义。3.3 抗赤霉病基因的利用
我国长江中下游麦区是赤霉病的常发区和重发区, 国内外广泛使用的主效抗病基因Fhb1区段与不利农艺性状存在连锁[26], 因此单纯利用苏麦3号和望水白背景的Fhb1不足以解决该麦区赤霉病的危害。关于这一点程顺和等[27]早有阐述, 并提出抗赤霉病育种的第2条路线, 即选用丰产、中感-中抗材料杂交, 后代注重丰产, 兼顾抗赤性等选择, 选育出扬麦158、扬麦11、扬麦16等一批抗赤性与丰产性相结合的大面积种植品种。扬麦系列多数品种的赤霉病抗性好且遗传力较强正在被大范围用作育种亲本, 更有价值的是其抗病基因不是Fhb1, 需要进一步发掘其中的主效赤霉病抗性QTL并开发相应的标记, 为扬麦抗赤霉病基因的高效利用奠定基础。由于多数抗病扬麦品种不具Fhb1, 将Fhb1导入扬麦背景, 提高抗赤霉病等级, 不失为解决长江中下游麦区赤霉病危害的有效途径, 如扬16-157 (含Fhb1), 其在2017—2019年度国家小麦良种重大联合攻关试验中表现高抗赤霉病, 与苏麦3号相当, 2017—2018年品比试验中产量为6325.5 kg hm-2, 比对照扬麦20增产7.0%, 2018—2019年品比试验中产量为6886.5 kg hm-2, 比对照增产4.5%。4 结论
扬麦16中抗赤霉病, 具有主效QTL, 其中4个QTL具有较大的表型贡献率, 2个QTL多年被检测到, QTL聚合能显著降低小麦赤霉病严重度。扬麦系列小麦品种QTL的发掘可以为小麦抗赤霉病改良提供优异基因资源。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1111/pbr.2009.128.issue-1URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
DOI:10.3864/j.issn.0578-1752.2014.18.012URL [本文引用: 1]

脱氧雪腐镰刀菌烯醇(DON),又称呕吐毒素,是由小麦赤霉病菌禾谷镰刀菌复合群(Fusariumgraminearum species complex)产生的单端孢霉烯族毒素,毒素在小麦籽粒中累积。作为B型单端孢霉烯族毒素,DON可以引起呕吐、拒食、腹泻、出血甚至死亡, 对猪的危害尤其严重。近年来,小麦赤霉病在世界各地高频率流行,尤其在中国长江中下游小麦生产区以及黄淮部分小麦产区、美国中西部小麦主产区,导致小麦中DON毒素严重超标,并引发了严重的食品安全问题。本文对国内外小麦中DON毒素的污染现状、产毒镰刀菌种类及其化学型的分布及其趋势、毒素产生的调控机制以及小麦中DON毒素的防控途径进行了论述,希望有助于镰刀菌毒素污染小麦质量安全的风险评估、监管以及科学处置。
DOI:10.3864/j.issn.0578-1752.2014.18.012URL [本文引用: 1]
脱氧雪腐镰刀菌烯醇(DON),又称呕吐毒素,是由小麦赤霉病菌禾谷镰刀菌复合群(Fusariumgraminearum species complex)产生的单端孢霉烯族毒素,毒素在小麦籽粒中累积。作为B型单端孢霉烯族毒素,DON可以引起呕吐、拒食、腹泻、出血甚至死亡, 对猪的危害尤其严重。近年来,小麦赤霉病在世界各地高频率流行,尤其在中国长江中下游小麦生产区以及黄淮部分小麦产区、美国中西部小麦主产区,导致小麦中DON毒素严重超标,并引发了严重的食品安全问题。本文对国内外小麦中DON毒素的污染现状、产毒镰刀菌种类及其化学型的分布及其趋势、毒素产生的调控机制以及小麦中DON毒素的防控途径进行了论述,希望有助于镰刀菌毒素污染小麦质量安全的风险评估、监管以及科学处置。
DOI:10.1007/s00122-018-3213-4URLPMID:30327846 [本文引用: 1]
A novel QTL for FHB resistance was mapped on wheat 7DL, being effective in multiple genetic backgrounds and environments, and comparable to Fhb1 in effect magnitude. Fusarium head blight (FHB) is one of the major fungal diseases affecting wheat production in many countries. The wheat line AQ24788-83 (AQ) possesses FHB resistance. The American wheat cultivar Luke is FHB susceptible. A Luke?×?AQ population consisting of 1652 advanced recombinant inbred lines (RILs) was developed, from which 272 RILs were randomly sampled and used to construct a linkage map. Another 154 RILs were selected for homogeneity in plant height (PH) and flowering date (FD). This selection strategy was adopted to reduce possible confounding effects on FHB assessment due to variation in PH and FD. The 272 and 154 RILs were genotyped applying simple sequence repeat (SSR), diversity arrays technology (DArT) and single-nucleotide polymorphism (SNP) markers. The two sets of RILs were evaluated for FHB resistance applying point inoculation in greenhouses; the 154 RILs were also evaluated applying spray inoculation in multiple field environments. The linkage map consisted of 2088 SSR, DArT, and SNP markers. A FHB resistance quantitative trait locus (QTL), designated as QFhb.cau-7DL, was detected on chromosome arm 7DL; this QTL was closely linked to the SSR marker gwm428 ( http://www.wheat.pw.usda.gov/ggpages/SSR/ ). QFhb.cau-7DL was significantly effective (α?=?0.01) in every test trial, and its effectiveness was validated using three additional wheat crosses. Sumai 3 (donor wheat of the FHB resistance gene Fhb1) was used in one of these crosses. QFhb.cau-7DL was comparable to Fhb1 in effect magnitude, providing a great potential for improving FHB resistance.
DOI:10.3389/fpls.2018.00573URLPMID:29780395 [本文引用: 2]
Fusarium head blight (FHB) is a destructive wheat disease present throughout the world, and host resistance is an effective and economical strategy used to control FHB. Lack of adequate resistance resource is still a main bottleneck for FHB genetics and wheat breeding research. The synthetic-derived bread wheat line C615, which does not carry the Fhb1 gene, is a promising source of FHB resistance for breeding. A population of 198 recombinant inbred lines (RILs) produced by crossing C615 with the susceptible cultivar Yangmai 13 was evaluated for FHB response using point and spray inoculations. As the disease phenotype is frequently complicated by other agronomic traits, we used both traditional and multivariate conditional QTL mapping approaches to investigate the genetic relationships (at the individual QTL level) between FHB resistance and plant height (PH), spike compactness (SC), and days to flowering (FD). A linkage map was constructed from 3,901 polymorphic SNP markers, which covered 2,549.2 cM. Traditional and conditional QTL mapping analyses found 13 and 22 QTL for FHB, respectively; 10 were identified by both methods. Among these 10, three QTL from C615 were detected in multiple years; these QTL were located on chromosomes 2AL, 2DS, and 2DL. Conditional QTL mapping analysis indicated that, at the QTL level, SC strongly influenced FHB in point inoculation; whereas PH and SC contributed more to FHB than did FD in spray inoculation. The three stable QTL (QFhbs-jaas.2AL, QFhbp-jaas.2DS, and QFhbp-jaas.2DL) for FHB were partly affected by or were independent of the three agronomic traits. The QTL detected in this study improve our understanding of the genetic relationships between FHB response and related traits at the QTL level and provide useful information for marker-assisted selection for the improvement of FHB resistance in breeding.
DOI:10.1038/s41588-019-0546-0URLPMID:31873299 [本文引用: 1]
We conducted a large-scale genome-wide association study evaluation of 683?common bean accessions, including landraces and breeding lines, grown over 3?years and in four environments across China, ranging in latitude from 18.23° to 45.75° N, with different planting dates and abiotic or biotic stresses. A total of 505?loci were associated with yield components, of which seed size, flowering time and harvest maturity traits were stable across years and environments. Some loci aligned with candidate genes controlling these traits. Yield components were observed to have strong associations with a gene-rich region on the long arm of chromosome?1. Manipulation of seed size, through selection of seed length versus seed width and height, was deemed possible, providing a genome-based means to select for important yield components. This study shows that evaluation of large germplasm collections across north-south geographic clines is useful in the detection of marker associations that determine grain yield in pulses.
DOI:10.1038/s41588-019-0546-0URLPMID:31873299 [本文引用: 1]
We conducted a large-scale genome-wide association study evaluation of 683?common bean accessions, including landraces and breeding lines, grown over 3?years and in four environments across China, ranging in latitude from 18.23° to 45.75° N, with different planting dates and abiotic or biotic stresses. A total of 505?loci were associated with yield components, of which seed size, flowering time and harvest maturity traits were stable across years and environments. Some loci aligned with candidate genes controlling these traits. Yield components were observed to have strong associations with a gene-rich region on the long arm of chromosome?1. Manipulation of seed size, through selection of seed length versus seed width and height, was deemed possible, providing a genome-based means to select for important yield components. This study shows that evaluation of large germplasm collections across north-south geographic clines is useful in the detection of marker associations that determine grain yield in pulses.
DOI:10.1270/jsbbs.15124URLPMID:27436944 [本文引用: 2]
Molecular markers associated with known quantitative trait loci (QTLs) for type 2 resistance to Fusarium head blight (FHB) in bi-parental mapping population usually have more than two alleles in breeding populations. Therefore, understanding the association of each allele with FHB response is particularly important to marker-assisted enhancement of FHB resistance. In this paper, we evaluated FHB severities of 192 wheat accessions including landraces and commercial varieties in three field growing seasons, and genotyped this panel with 364 genome-wide informative molecular markers. Among them, 11 markers showed reproducible marker-trait association (p &lt; 0.05) in at least two experiments using a mixed model. More than two alleles were identified per significant marker locus. These alleles were classified into favorable, unfavorable and neutral alleles according to the normalized genotypic values. The distributions of effective alleles at these loci in each wheat accession were characterized. Mean FHB severities increased with decreased number of favorable alleles at the reproducible loci. Chinese wheat landraces and Japanese accessions have more favorable alleles at the majority of the reproducible marker loci. FHB resistance levels of varieties can be greatly improved by introduction of these favorable alleles and removal of unfavorable alleles simultaneously at these QTL-linked marker loci.
[本文引用: 2]
DOI:10.3835/plantgenome2016.02.0021URLPMID:27902807 [本文引用: 2]
Potato ( L.) breeders consider a large number of traits during cultivar development and progress in conventional breeding can be slow. There is accumulating evidence that some of these traits, such as yield, are affected by a large number of genes with small individual effects. Recently, significant efforts have been applied to the development of genomic resources to improve potato breeding, culminating in a draft genome sequence and the identification of a large number of single nucleotide polymorphisms (SNPs). The availability of these genome-wide SNPs is a prerequisite for implementing genomic selection for improvement of polygenic traits such as yield. In this review, we investigate opportunities for the application of genomic selection to potato, including novel breeding program designs. We have considered a number of factors that will influence this process, including the autotetraploid and heterozygous genetic nature of potato, the rate of decay of linkage disequilibrium, the number of required markers, the design of a reference population, and trait heritability. Based on estimates of the effective population size derived from a potato breeding program, we have calculated the expected accuracy of genomic selection for four key traits of varying heritability and propose that it will be reasonably accurate. We compared the expected genetic gain from genomic selection with the expected gain from phenotypic and pedigree selection, and found that genetic gain can be substantially improved by using genomic selection.
DOI:10.1007/s00122-016-2743-xURLPMID:27306516 [本文引用: 2]
We developed and validated a robust marker toolkit for high-throughput and cost-effective screening of a large number of functional genes in wheat. Functional markers (FMs) are the most valuable markers for crop breeding programs, and high-throughput genotyping for FMs could provide an excellent opportunity to effectively practice marker-assisted selection while breeding cultivars. Here we developed and validated kompetitive allele-specific PCR (KASP) assays for genes that underpin economically important traits in bread wheat including adaptability, grain yield, quality, and biotic and abiotic stress resistances. In total, 70 KASP assays either developed in this study or obtained from public databases were validated for reliability in application. The validation of KASP assays were conducted by (a) comparing the assays with available gel-based PCR markers on 23 diverse wheat accessions, (b) validation of the derived allelic information using phenotypes of a panel comprised of 300 diverse cultivars from China and 13 other countries, and (c) additional testing, where possible, of the assays in four segregating populations. All KASP assays being reported were significantly associated with the relevant phenotypes in the cultivars panel and bi-parental populations, thus revealing potential application in wheat breeding programs. The results revealed 45 times superiority of the KASP assays in speed than gel-based PCR markers. KASP has recently emerged as single-plex high-throughput genotyping technology; this is the first report on high-throughput screening of a large number of functional genes in a major crop. Such assays could greatly accelerate the characterization of crossing parents and advanced lines for marker-assisted selection and can complement the inflexible, high-density SNP arrays. Our results offer a robust and reliable molecular marker toolkit that can contribute towards maximizing genetic gains in wheat breeding programs.
DOI:10.3724/SP.J.1006.2018.00505URL [本文引用: 1]
赤霉病已上升为黄淮冬麦区的主要病害, 提高小麦品种对赤霉病的抗性成为该麦区主要的育种目标之一。宁麦9号、生选6号、建阳798、建阳84、苏麦3号和宁麦13均携带Fhb1基因, 对赤霉病表现中抗水平以上。本研究以这6个品种(系)为供体, 分别与高感赤霉病的周麦16矮败小麦近等基因系杂交和回交, 构建6个回交群体。利用Fhb1基因的KASP标记在回交后代中进行基因型分析, 分别选择携带和不携带Fhb1基因的可育株, 对后代株系进行单花滴注接种鉴定和田间病圃自然鉴定。回交后代携带Fhb1家系整体抗性达到中感, 比不携带Fhb1家系的平均病小穗数低4.2 (P < 0.01), 平均病情指数低4.0, 比轮回亲本周麦16的平均病小穗数和病情指数分别低8.1 (P < 0.01)和28.4 (P < 0.01)。不同供体品种(系)回交后代在赤霉病抗性上表现出明显差异, 以生选6号为供体的回交后代家系抗性表现最好。本研究表明, 利用Fhb1基因分子标记辅助选择技术能够有效地提高黄淮冬麦区小麦品种的赤霉病抗性水平。
DOI:10.3724/SP.J.1006.2018.00505URL [本文引用: 1]
赤霉病已上升为黄淮冬麦区的主要病害, 提高小麦品种对赤霉病的抗性成为该麦区主要的育种目标之一。宁麦9号、生选6号、建阳798、建阳84、苏麦3号和宁麦13均携带Fhb1基因, 对赤霉病表现中抗水平以上。本研究以这6个品种(系)为供体, 分别与高感赤霉病的周麦16矮败小麦近等基因系杂交和回交, 构建6个回交群体。利用Fhb1基因的KASP标记在回交后代中进行基因型分析, 分别选择携带和不携带Fhb1基因的可育株, 对后代株系进行单花滴注接种鉴定和田间病圃自然鉴定。回交后代携带Fhb1家系整体抗性达到中感, 比不携带Fhb1家系的平均病小穗数低4.2 (P < 0.01), 平均病情指数低4.0, 比轮回亲本周麦16的平均病小穗数和病情指数分别低8.1 (P < 0.01)和28.4 (P < 0.01)。不同供体品种(系)回交后代在赤霉病抗性上表现出明显差异, 以生选6号为供体的回交后代家系抗性表现最好。本研究表明, 利用Fhb1基因分子标记辅助选择技术能够有效地提高黄淮冬麦区小麦品种的赤霉病抗性水平。
DOI:10.1007/s00122-006-0297-zURL [本文引用: 1]
The major quantitative trait locus (QTL) on 3BS from Sumai 3 and its derivatives has been used as a major source of resistance to Fusarium head blight (FHB) worldwide, but resistance genes from other sources are necessary to avoid complete dependence on a single source of resistance. Fifty-nine Asian wheat landraces and cultivars differing in the levels of FHB resistance were evaluated for type II FHB resistance and for genetic diversity on the basis of amplified fragment length polymorphism (AFLP) and simple sequence repeats (SSRs). Genetic relationships among these wheat accessions estimated by cluster analysis of molecular marker data were consistent with their geographic distribution and pedigrees. Chinese resistant landraces had broader genetic diversity than that of accessions from southwestern Japan. The haplotype pattern of the SSR markers that linked to FHB resistance quantitative trait loci (QTLs) on chromosomes 3BS, 5AS and 6BS of Sumai 3 suggested that only a few lines derived from Sumai 3 may carry all the putative QTLs from Sumai 3. About half of the accessions might have one or two FHB resistance QTLs from Sumai 3. Some accessions with a high level of resistance, may carry different FHB resistance loci or alleles from those in Sumai 3, and are worth further investigation. SSR data also clearly suggested that FHB resistance QTLs on 3BS, 5AS, and 6BS of Sumai 3 were derived from Chinese landrace Taiwan Xiaomai.
DOI:10.1385/0-89603-254-x:9URLPMID:8118521 [本文引用: 1]
DOI:10.1038/s41598-019-56977-9URLPMID:31892747 [本文引用: 1]
The bacterial and fungal communities from the olive (Olea europaea L.) root systems have not yet been simultaneously studied. We show in this work that microbial communities from the olive root endosphere are less diverse than those from the rhizosphere. But more relevant was to unveil that olive belowground communities are mainly shaped by the genotype of the cultivar when growing under the same environmental, pedological and agronomic conditions. Furthermore, Actinophytocola, Streptomyces and Pseudonocardia are the most abundant bacterial genera in the olive root endosphere, Actinophytocola being the most prevalent genus by far. In contrast, Gp6, Gp4, Rhizobium and Sphingomonas are the main genera in the olive rhizosphere. Canalisporium, Aspergillus, Minimelanolocus and Macrophomina are the main fungal genera present in the olive root system. Interestingly enough, a large number of as yet unclassified fungal sequences (class level) were detected in the rhizosphere. From the belowground microbial profiles here reported, it can be concluded that the genus Actinophytocola may play an important role in olive adaptation to environmental stresses. Moreover, the huge unknown fungal diversity here uncovered suggests that fungi with important ecological function and biotechnological potential are yet to be identified.
DOI:10.1111/j.1365-313X.1993.00739.xURL [本文引用: 1]
DOI:10.3724/SP.J.1006.2009.00239URL [本文引用: 1]
结合分子标记和表型数据的QTL作图已成为数量性状遗传分析的常规方法。复合区间作图是近10多年来广泛应用的一种QTL定位方法,但它在算法上有一些缺陷,致使QTL效应可能会被侧连标记区间之外的标记变量吸收,同时不同的背景标记选择方法对作图结果的影响较大,并且难以推广到上位型互作QTL的定位。针对这些问题,笔者提出完备区间作图方法。本文介绍了该方法的遗传和统计原理,并通过一个大麦加倍单倍体群体说明其在定位加性QTL和加性×加性互作QTL中的应用。完备区间作图包含两个步骤:首先利用所有标记的信息,通过逐步回归选择重要的标记变量并估计其效应;然后利用逐步回归得到的线性模型校正表型数据,通过一维扫描定位加(显)性效应QTL,通过二维扫描定位上位型互作QTL。这种作图策略简化了复合区间作图中控制背景遗传变异的过程,提高了对QTL的检测功效。
DOI:10.3724/SP.J.1006.2009.00239URL [本文引用: 1]
结合分子标记和表型数据的QTL作图已成为数量性状遗传分析的常规方法。复合区间作图是近10多年来广泛应用的一种QTL定位方法,但它在算法上有一些缺陷,致使QTL效应可能会被侧连标记区间之外的标记变量吸收,同时不同的背景标记选择方法对作图结果的影响较大,并且难以推广到上位型互作QTL的定位。针对这些问题,笔者提出完备区间作图方法。本文介绍了该方法的遗传和统计原理,并通过一个大麦加倍单倍体群体说明其在定位加性QTL和加性×加性互作QTL中的应用。完备区间作图包含两个步骤:首先利用所有标记的信息,通过逐步回归选择重要的标记变量并估计其效应;然后利用逐步回归得到的线性模型校正表型数据,通过一维扫描定位加(显)性效应QTL,通过二维扫描定位上位型互作QTL。这种作图策略简化了复合区间作图中控制背景遗传变异的过程,提高了对QTL的检测功效。
DOI:10.1007/s00122-015-2634-6URLPMID:26602234 [本文引用: 1]
Fifty-six QTL for flour color-related traits and polyphenol oxidase activity were identified using a genome-wide linkage mapping of data from a RIL population derived from a Gaocheng 8901/Zhoumai 16 cross.
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s00122-008-0742-2URL [本文引用: 1]
Fusarium head blight (FHB) is an important disease of wheat worldwide. The cultivar Spark is more resistant than most other UK winter wheat varieties but the genetic basis for this is not known. A mapping population from a cross between Spark and the FHB susceptible variety Rialto was used to identify quantitative trait loci (QTL) associated with resistance. QTL analysis across environments revealed nine QTL for FHB resistance and four QTL for plant height (PH). One FHB QTL was coincident with the Rht-1D locus and accounted for up to 51% of the phenotypic variance. The enhanced FHB susceptibility associated with Rht-D1b is not an effect of PH per se as other QTL for height segregating in this population have no influence on susceptibility. Experiments with near-isogenic lines supported the association between susceptibility and the Rht-D1b allele conferring the semi-dwarf habit. Our results demonstrate that lines carrying the Rht-1Db semi-dwarfing allele are compromised in resistance to initial infection (type I resistance) while being unaffected in resistance to spread within the spike (type II resistance).
DOI:10.1007/s00122-008-0930-0URLPMID:19034409 [本文引用: 1]
Fusarium head blight (FHB) is an important disease of wheat worldwide. Soissons is one of the most resistant varieties grown in UK. The current study was undertaken to identify QTL for FHB resistance in Soissons and to determine whether the semi-dwarfing alleles Rht-B1b and Rht-D1b have a similar influence on susceptibility to FHB. A Soissons (Rht-B1b; Rht-D1a) x Orvantis (Rht-B1a; Rht-D1b) doubled haploid (DH) population was assessed for FHB resistance in three trials. Soissons contributed a single, stable major FHB QTL linked to the Rht-D1 locus. In contrast, the Rht-B1b allele (contributed by Soissons) conferred no negative effect on FHB resistance, even conferring a very minor positive effect in one trial. The influence of the Rht-B1b and Rht-D1b alleles on FHB resistance was further investigated using both Mercia and Maris Huntsman near-isogenic lines. Under high disease pressure both Rht-B1b and Rht-D1b significantly decreased Type 1 resistance (resistance to initial infection). However, whilst Rht-D1b has no effect on Type 2 resistance (resistance to spread of the fungus within the spike), Rht-B1b significantly increased Type 2 resistance. Our study demonstrates that the choice of semi-dwarfing gene used in plant breeding programmes may be a significant consideration where resistance to FHB is an important breeding target.
DOI:10.1111/pbr.2009.128.issue-1URL [本文引用: 1]
DOI:10.1371/journal.pone.0129400URLPMID:26076351 [本文引用: 1]
TaGW2 is an orthologue of rice gene OsGW2, which encodes E3 RING ubiquitin ligase and controls the grain size in rice. In wheat, three copies of TaGW2 have been identified and mapped on wheat homoeologous group 6 viz. TaGW2-6A, TaGW2-6B and TaGW2-6D. In the present study, using as many as 207 Indian wheat genotypes, we identified four SNPs including two novel SNPs (SNP-988 and SNP-494) in the promoter sequence of TaGW2-6A. All the four SNPs were G/A or A/G substitutions (transitions). Out of the four SNPs, SNP-494 was causal, since it was found associated with grain weight. The mean TGW (41.1 g) of genotypes with the allele SNP-494_A was significantly higher than mean TGW (38.6 g) of genotypes with the allele SNP-494_G. SNP-494 also regulates the expression of TaGW2-6A so that the wheat genotypes with SNP-494_G have higher expression and lower TGW and the genotypes with SNP-494_A have lower expression but higher TGW. Besides, SNP-494 was also found associated with grain length-width ratio, awn length, spike length, grain protein content, peduncle length and plant height. This suggested that gene TaGW2-6A not only controls grain size, but also controls other agronomic traits. In the promoter region, SNP-494 was present in 'CGCG' motif that plays an important role in Ca2+/calmodulin mediated regulation of genes. A user-friendly CAPS marker was also developed to identify the desirable allele of causal SNP (SNP-494) for use in marker-assisted selection for improvement of grain weight in wheat. Using four SNPs, five haplotypes were identified; of these, Hap_5 (G_A_G_A) was found to be a desirable haplotype having significantly higher grain weight (41.13g) relative to other four haplotypes (36.33-39.16 g).
DOI:10.1139/g03-033URLPMID:12897863 [本文引用: 1]
Fusarium head blight of wheat is an extremely damaging disease, causing severe losses in seed yield and quality. The objective of the current study was to examine and characterize alternate sources of resistance to Fusarium head blight (FHB). Ninety-one F1-derived doubled haploid lines from the cross Triticum aestivum 'Wuhan-1' x Triticum aestivum 'Maringa' were examined for disease reaction to Fusarium graminearum by single-floret injection in replicated greenhouse trials and by spray inoculation in replicated field trials. Field and greenhouse experiments were also used to collect agronomic and spike morphology characteristics. Seed samples from field plots were used for deoxynivalenol (DON) determination. A total of 328 polymorphic microsatellite loci were used to construct a genetic linkage map in this population and together these data were used to identify QTL controlling FHB resistance, accumulation of DON, and agronomic and spike morphology traits. The analysis identified QTL for different types of FHB resistance in four intervals on chromosomes 2DL, 3BS, and 4B. The QTLs on 4B and 3BS proximal to the centromere are novel and not reported elsewhere. QTL controlling accumulation of DON independent of FHB resistance were located on chromosomes 2DS and 5AS. Lines carrying FHB resistance alleles on 2DL and 3BS showed a 32% decrease in disease spread after single-floret injection. Lines carrying FHB resistance alleles on 3BS and 4B showed a 27% decrease from the mean in field infection. Finally, lines carrying favourable alleles on 3BS and 5AS, showed a 17% reduction in DON accumulation. The results support a polygenic and quantitative mode of inheritance and report novel FHB resistance loci. The data also suggest that resistance to FHB infection and DON accumulation may be controlled, in part, by independent loci and (or) genes.
DOI:10.3724/SP.J.1006.2018.00473URL [本文引用: 1]
提高赤霉病抗性已成为我国小麦主产区的重要育种目标之一。Fhb1是抗性最强且最稳定的抗赤霉病基因, 阐明其在我国小麦育种中的应用及传递路径, 对抗赤霉病育种有重要意义。本研究通过分析229份小麦品种(系) Fhb1区段内PFT (pore-forming toxin-like)、HC (HCBT-like defense response protein)和His (histidine-rich calcium-binding protein)基因的多样性与赤霉病抗性的关系, 发现PFT-I/His-I为抗病单倍型。基因检测和系谱分析表明, 中国小麦品种所含Fhb1至少有2个来源, 分别为苏麦3号和宁麦9号, 并以后者为主。本研究开发的诊断性标记PFT-CAPS和His-InDel可有效用于Fhb1的分子标记辅助育种。
DOI:10.3724/SP.J.1006.2018.00473URL [本文引用: 1]
提高赤霉病抗性已成为我国小麦主产区的重要育种目标之一。Fhb1是抗性最强且最稳定的抗赤霉病基因, 阐明其在我国小麦育种中的应用及传递路径, 对抗赤霉病育种有重要意义。本研究通过分析229份小麦品种(系) Fhb1区段内PFT (pore-forming toxin-like)、HC (HCBT-like defense response protein)和His (histidine-rich calcium-binding protein)基因的多样性与赤霉病抗性的关系, 发现PFT-I/His-I为抗病单倍型。基因检测和系谱分析表明, 中国小麦品种所含Fhb1至少有2个来源, 分别为苏麦3号和宁麦9号, 并以后者为主。本研究开发的诊断性标记PFT-CAPS和His-InDel可有效用于Fhb1的分子标记辅助育种。
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}