摘要/Abstract
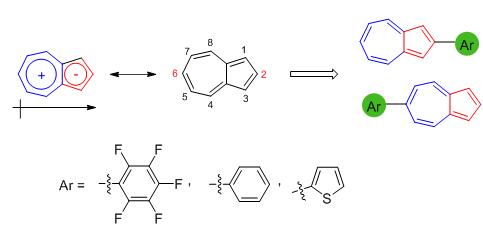
设计合成了薁2/6-位芳基取代的六个模型化合物1~6,化合物1~3和4~6分别为薁2-位和6-位取代的化合物,其取代基顺序均分别为五氟苯、苯和α-噻吩.对化合物的紫外吸收光谱、荧光光谱、电化学以及质子响应等物理化学性质进行了研究,并结合密度泛函理论(DFT)计算研究了2/6-位不同取代芳基对于薁衍生物的基本物理化学性质的影响.吸收光谱研究表明,在薁的2/6-位引入不同取代芳基均可以使其S0→S2跃迁吸收峰红移(Δλ=6~68 nm),2/6-位引入给电子的噻吩基团发生明显红移(Δλ=68/48 nm),其中2-位引入给电子噻吩基团红移更加明显(Δλ=68 nm).荧光光谱研究表明,在薁6-位引入强拉电子的五氟苯,所得化合物4荧光强度最强(?F=0.082);质子化后,同样含有五氟苯基的化合物1-H+的荧光强度最强((?F=0.359).电化学和DFT理论计算表明,在薁2/6-位引入拉电子的五氟苯基可显著降低分子的最高已占分子轨道(HOMO)和最低空分子轨道(LUMO)能级(1和4的ΔEHOMO/ΔELUMO分别为-0.23/-0.18和-0.20/-0.15 eV).这些研究结果为基于薁的有机功能分子的设计合成及性质研究提供了有效依据.
关键词: 薁, 芳基取代, 质子响应
Six 2/6-aryl substituted azulene derivatives 1~6 were designed and synthesised. Compounds 1~3 and 4~6 are 2-and 6-substituted derivatives, respectively, where the arly substituents were pentafluorobenzene, benzene and α-thiophene. The UV-Vis spectra, fluorescence spectra, electrochemical properties and proton-responsive properties of 1~6 were studied. To investigate the molecular sturcture, absorption spectra and energy levels of compounds 1~6, density functional theory (DFT) calculations were carried out. In comparison with the UV-Vis spectra of azulene, the absorption of S0→S2 transition of 1~6 showed red-shift (Δλ=6~68 nm). Owing to the strong electron-donating ability of α-thiophene group, remarkable bathochromic shifts of 3 and 6 (Δλ=68 and 48 nm, respectively) were obseved. The fluorescence spectra revealed that 4 (?F=0.082) has the highest fluorescence quantum yield of 1~6, while 1-H+ (?F=0.359) has the highest fluorescence quantum yield of the protonated compounds 1-H+~6-H+, benefiting from the electron-withdrawing pentafluorophenyl group of 1 and 1-H+. Moreover, the electrochemical analysis and DFT calculations demonstated that the introduction of electron-withdrawing pentafluorophenyl unit in the 2/6-positon of azulene can significantly lower the energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). In comparison with the HOMO/LUMO energy levels of azulene, those of 1 and 4 shift downward with ΔEHOMO/ΔELUMO of -0.23/-0.18 and -0.20/-0.15 eV, respectively. The investigations of physical/chemical properties of 2/6-aryl substituted azulene derivatives will provide valuable insights for developing azulene-based organic functional molecules.
Key words: azulene, aryl substitution, proton response
PDF全文下载地址:
点我下载PDF
