冷欢, 杨清, 黄钢锋, 白丽萍


农业农村部沼气科学研究所, 四川 成都 610041
收稿日期:2019-12-07;修回日期:2020-04-03;网络出版日期:2020-06-15
基金项目:国家自然科学基金(31970066);中央级公益性科研院所基本科研业务费专项(Y2019XK23-02);成都农业科技中心地方财政专项资金(NASC2019TI01)
*通信作者:白丽萍, Tel:+86-28-85228239;E-mail:bailiping@caas.cn.
摘要:产甲烷古菌是一类极端厌氧的古菌域微生物,可以利用CO2、甲醇、乙酸等简单化合物产甲烷并获得能量。目前能够培养的氢营养型(CO2/H2)产甲烷古菌的种类较多,而且在三类产甲烷代谢类型中,氢营养型产甲烷途径的产能效率最高,并具有多种模式的特殊能量利用系统。近年来,随着质谱、光谱和晶体技术的发展与运用,人们对产甲烷代谢途径的研究进一步深入,尤其是对氢营养型产甲烷途径的生化机制有了新的认识,揭示了产甲烷古菌在能量极限条件下独特、高效的能量利用模式。本文从能量储存、代谢途径、蛋白功能与催化机制等方面概述产甲烷古菌利用CO2/H2产甲烷的详细过程,并对产甲烷古菌代谢途径的研究方向与技术发展进行展望。
关键词:产甲烷古菌CO2还原途径能量保存蛋白结构催化机制
Recent advances in hydrogenotrophic methanogenesis
Huan Leng, Qing Yang, Gangfeng Huang, Liping Bai
Biogas Institute of Ministry of Agriculture, Chengdu 610041, Sichuan Province, China
Received: 7 December 2019; Revised: 3 April 2020; Published online: 15 June 2020
*Corresponding author: Liping Bai, Tel:+86-28-85228239;E-mail:bailiping@caas.cn.
Foundation item: Supported by the National Natural Science Foundation of China (31970066), by the Central Public-Interest Scientific Institution Basal Research Fund (Y2019XK23-02) and by the The National Agricultural Science and Technology Center, Chengdu (NASC2019TI01)
Abstract: Methanogens are a group of extremely anaerobic microorganisms affiliated with the archaeal domain. They use simple compounds such as carbon dioxide, acetate and methyl compounds as substrate to grow. Hydrogenotrophic methanogens is the main type in all the cultured species. Among the three major methanogenic pathways, the hydrogenotrophic methanogenesis shows the highest energy productivity and includes a special energy metabolism system. In recent years, with the development and application of mass spectrum, spectroscopy and crystallization technology, the research on the methanogenic pathway has been further studied, especially in the biochemical mechanism of hydrogenotrophic methanogenesis, which revealing a unique and efficient way of energy conversation under extreme environment. Based on the massive research results, this review summarizes the details of hydrogenotrophic methanogenesis from the aspects of energy storage, metabolic pathways, enzyme structures and catalytic mechanism of enzymatic reactions. The prospect for the future research and technological development in the field of methanogenesis is also proposed in this review.
Keywords: methanogenshydrogenotrophic methanogenesisenergy conservationprotein structurecatalytic mechanism
产甲烷古菌是生命进化史上最古老的单细胞生命体[1],在生命起源、物种进化等研究领域起着至关重要的作用[2]。产甲烷古菌广泛存在于各种生境中,包括深海、湿地、地热温泉、动物瘤胃等,作为降解末端碳链的微生物参与地球化学元素循环[3-4]。产甲烷古菌利用简单化合物产甲烷,这一独特的生理现象使之成为生物能源、全球变暖等关键领域的研究对象[5]。因此,长期以来产甲烷古菌引起了科学界广泛的关注。国内外无数科研****致力于产甲烷古菌在系统进化、代谢途径、遗传体系构建等方面的研究。如德国Thauer、Gottschalk、美国Wolfe等团队多年来对CO2还原代谢途径进行了大量的研究工作[6-8];美国Kyzycki一直致力于甲基裂解途径关键酶蛋白的功能研究[9];美国Whiteman、Leigh等实验室建立了产甲烷古菌的遗传操作体系[10-11];在国内学术界中,产甲烷古菌的研究主要集中在基于宏基因组分析的系统发育、生态分布、分离培养等方面,而产甲烷代谢途径的研究相对较少。
1 产甲烷古菌的分类与生理特征 迄今为止,已发现的产甲烷古菌共有7个目,包括Methanomicrobiales、Methanobacteriales、Methanococcales、Methanocellales、Methanopyrales、Methanosarcinales以及近年发现的Methanomassiliicoccales,均属于广古菌门Euryarchaeota[3, 12]。最近发现一些深古菌门Ca. Bathyarchaeota及Ca. Verstraetearcheaota的古菌类群也具有产甲烷的能力[13]。产甲烷古菌的系统分类、形态特征、底物利用范围等在前人的综述中已有详尽的阐述,本文不再系统分析[12, 14-15]。
根据代谢底物类型将产甲烷古菌可分为三类:乙酸营养型、甲基营养型以及以CO2/H2为底物的氢营养型[2]。除了部分专性乙酸营养型与甲基营养型产甲烷古菌外,在目前已培养的模式菌株中,有3/4以上的产甲烷古菌属于利用CO2产甲烷的氢营养型[12]。相较于其他两类产甲烷途径,氢营养型产甲烷途径有更高的能量获取效率。在标准热力学状态下,1mol CO2还原产甲烷释放出131 kJ的能量,高于甲醇产甲烷(–106kJ)与乙酸产甲烷(–36kJ)释放的能量[2, 16]。CO2还原产甲烷代谢途径的复杂性、中间反应的多样性、能量储存模式的特殊性,使得氢营养型产甲烷古菌成为广泛研究的模式物种。
2 氢营养型产甲烷古菌及其生长特性 如上所述,大部分产甲烷古菌都能利用CO2/H2进行产甲烷生长。根据细胞膜结构的不同,将其分为两类:一类是细胞膜上含有细胞色素(Cytochrome)和甲烷吩嗪(Methanophenazine)的Methanosarcinales[16-17]。它们利用CO2/H2产甲烷过程的氢分压一般高于10Pa,代时一般大于10h,基本是中温菌;另一类氢营养型产甲烷古菌细胞膜上不含细胞色素和MP,只能以CO2/H2或甲酸为底物产甲烷,其代时最短可低至1h,可以忍耐环境中较低的氢分压[17]。
研究发现,有细胞色素的氢营养型产甲烷古菌在生长过程中表现出较高的ATP得率(得率约1.5,无细胞色素的类群得率约为0.5)[17-20]。当我们把CO2还原与ATP合成相偶联(4H2+CO2+ nADP+nPi=CH4+nATP+3H2O)时发现,若反应达到平衡,ATP得率n为0.5时,可推算出氢分压为2 Pa,而n为1时,氢分压增至30Pa[17, 21]。这一计算结果恰好解释了含细胞色素的产甲烷古菌具有较高的ATP得率和氢分压,也解释了为什么Methanosarcina在低氢分压环境下没有利用CO2/H2产甲烷的能力。产甲烷古菌在极低的ATP得率下,如何维持细胞的正常生命活动,能量代谢正常运转时接近热力学平衡的极限是什么,这些问题使产甲烷代谢途径的研究引起科学界的长期关注。
3 氢营养型产甲烷古菌能量代谢特征 产甲烷代谢是产甲烷古菌获取能量的唯一方式,其独特的能量保存方式清晰地体现在产甲烷代谢途径的各类酶反应中。其中大部分酶反应都是氧化还原、基团转移等,催化每一步反应的酶均是分子量庞大的复合体,并涉及到一系列含不同金属的辅因子[7, 22](详见第5部分)。产甲烷古菌巧妙地利用并保存反应过程中形成的ATP、膜内外离子梯度、还原力,并偶联于耗能反应,弥补ATP得率低的缺陷。
在产甲烷途径中,唯一能直接偶联ATP合成的反应只有Mtr催化的甲基转移反应。该复合体酶在催化过程中向膜外转移2个Na+并形成膜内外离子梯度,通过离子泵偶联膜上的ATP酶将离子梯度转化合成ATP[23-26](详见5.7)。此外,产甲烷古菌还利用膜蛋白催化过程偶联质子转运,形成膜内外质子梯度,这一电化学形式的能量既可用于ATP合成,也可用于推动耗能反应的发生。如催化甲烷生成的Hdr/Vht酶系向膜外转运2个H+,这一电化学梯度的能量用于Ech酶系合成还原力Fdred2–(+16 kJ/mol)[16, 27],详见5.10部分。在不含细胞色素的产甲烷古菌中,催化甲烷生成的Hdr/Mvh复合体位于胞质中,不能建立膜内外离子梯度[28]。因此,Hdr/Mvh采用特殊的黄素腺嘌呤二核苷酸(FAD)通过电子歧化方式合成还原的Fdred2–,用于CO2还原及其他代谢活动[29-30],详见5.9部分。
4 氢营养型甲烷途径概述 有无细胞色素的产甲烷古菌的CO2还原产甲烷代谢途径相似,只有部分酶促反应不同。CO2还原产甲烷途径以H2为电子供体,CO2通过一碳载体Methanofuran (MFR)、Tetrahydromethanopterin(H4MPT)、Coenzyme M (CoM)的传递并被逐级还原,最终形成甲烷,如图 1所示。每还原1分子CO2,需要消耗4分子H2,分别用于还原Fdox、F420以及CoM-S-S-CoB[2, 7, 22, 27]。产甲烷代谢途径涉及众多的酶促反应、一碳载体、金属辅酶。催化过程的酶主要包括脱氢酶、还原酶、转移酶。辅酶主要包括[Fe-S]簇、FAD、F420、F430、Tungstenpterin、Cobalamin、Cytochrome等。到目前为止,产甲烷途径的主要酶促反应已经基本明确,但部分酶蛋白的三维结构、催化机制等关键问题还需进一步的探索。CO2还原途径中各步酶促反应的详细过程在第5部分详细阐述。
 |
| 图 1 氢营养型产甲烷途径示意图[27, 31] Figure 1 Hydrogenotrophic methanogensis[27, 31]. MFR: methanofuran; H4MPT: tetrahydromethanopterin; CHO-MFR: formyl-MFR; CHO-H4MPT: formyl-H4MPT; CH≡H4MPT+: methenyl-H4MPT+; CH2=H4MPT: methylene-H4MPT; CH3-H4MPT: methyl-H4MPT; CoB-SH: coenzyme B; CoM-SH: coenzyme M; CoM-S-S-CoB: heterodisul?de; Fdred2–: reduced ferredoxin; Fdox: oxidized ferredoxin; F420: oxidized coenzyme F420; F420H2: reduced coenzyme F420; Fwd: tungsten-dependent formylmethanofuran dehydrogenase; Fmd: molybdenum-dependent formylmethanofuran dehydrogenase; Ftr: formyltransferase; Mch: methenyl-H4MPT+ cyclohydrolase; Frh: F420-reducing [NiFe]-hydrogenase; Mtd: F420-dependent methylenetetrahydromethanopterin dehydrogenase; Hmd: [Fe]-hydrogenase; Mer: F420-dependent methylene-H4MPT reductase; Mcr: methyl-coenzyme M reductase; Hdr-Mvh: heterodisulfide-reductase/[NiFe]-hydrogenase complex; Eha/Ehb: energy-converting [NiFe]-hydrogenases; Mtr: membrane-associated methyltransferase complex (MtrA-H). |
| 图选项 |
5 CO2还原途径酶促反应 5.1 Formylmethanofuran-脱氢酶(Fmd/Fwd) CO2还原途径起始于CO2的固定,催化该反应的酶复合体是甲酰基呋喃脱氢酶(Fmd或Fwd)[32-33],反应过程如公式(1)所示。
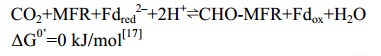 | 公式(1) |
Wagner等解析了M. wolfei的Fwd蛋白结构,结果显示Fwd是由6个亚基组成的同源四聚体Fwd(ABCDFG)4。四聚体结构中不仅有金属离子W、Zn、Fe等,还首次发现了多达46个[4Fe-4S]簇形成的长链电子传递体系(图 2-A)。数量众多的链式[Fe-S]簇不仅保证了CO2还原所需的低电势,也极有可能是古菌动态储存还原力的一种方式[38]。
 |
| 图 2 Fwd四聚体结构及催化机制示意图 Figure 2 Structure of Fwd tetrameric complex and the proposed catalytic mechanism. A: Crystal structure of the tetramer Fwd(ABCDFG)4 (PDB: 5T61[28]). The subunits and iron-sulfur cluster are shown with stick and cartoon model, respectively. B: Catalytic mechanism of Fwd. Tungsten accepts two electrons which delivered by [4Fe-4S] cluster, resulting W(Ⅳ). Then CO2 is reduced into formate with the two electrons from Fdred2– at the tungsten center. The formate is transferred into [ZnZn] center, where it reacts with MFR to form CHO-MFR. |
| 图选项 |
Fwd的复合体结构由甲酸脱氢酶和金属水解酶两个催化核心组成[38]。其中,FwdB和FwdD共同组成甲酸脱氢酶的催化单位。FwdBD活性中心的W共价结合到嘌呤核苷酸的4个巯基端、Cys118及元素硫原子上,形成W-喋呤(tungstopterin)催化中心。FwdA的功能类似于金属水解酶,这类酶的显著特点是在由(α/β)8构成的一个TIM结构域入口处包含1个双锌的金属活性中心。FwdC在结构上与谷氨酸合成酶的C-末端相似,在整个复合体中起稳定结构的支撑功能。FwdF包含4个相似的Fd结合域,每一部分均携带2个[4Fe-4S]簇,以T字形分布。整个[4Fe-4S]簇、金属结合点、活性中心残基、金属原子结合模式都非常保守地分布在FwdBD与FwdA上。
FwdABCDFG复合体从催化功能上可以分为1个电子供应中心FwdFG和1个催化中心FwdABCD。首先,CO2通过一个狭长的疏水性气体通道(40 ? )到达FwdBD的W中心。深埋在蛋白内部的金属W中心与[4Fe-4S]簇相连,有效地转移CO2还原所需的电子。在W中心,CO2被还原成甲酸(E0’=–430 mV),还原反应所需的电子来自Fdred2– (E0’=–500 mV)。还原生成的甲酸通过FwdBD与FwdA之间的一个亲水通道(43 ? )转移到FwdA的双锌[ZnZn]中心,甲酸共价结合到甲烷呋喃MFR的氨基,最终生成CHO-MFR (图 2-B)。该甲酸通道将CO2还原的放能反应(ΔG0’=–15 kJ/mol)与HCHO转移的耗能反应(ΔG0’=+15 kJ/mol)偶联[39],这也体现了产甲烷古菌巧妙利用局部能量推动复杂反应的特点。
5.2 甲酰基转移酶Ftr 产甲烷途径的第二步反应,是在甲酰基转移酶Ftr的作用下将CHO-MFR的甲酰基转移到一碳载体H4MPT,形成N5-CHO-H4MPT[40]。反应过程如公式(2)所示。
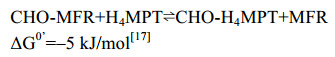 | 公式(2) |
Ftr的活性功能单位以二聚体或四聚体出现,这一推论在Ftr与底物CHO-MFR和H4MPT共结晶的结构中得以证实[47]。从M. kandleri Ftr的结构发现,底物CHO-MFR和H4MPT分别结合在不同的亚基上。CHO-MFR的结合中心位于两个亚基相互作用表面形成的狭长缝隙,主要靠疏水作用结合在蛋白上,而H4MPT松散地结合在单亚基中央β折叠片结构域的表面(图 3-A)。
 |
| 图 3 Ftr晶体结构及反催化反应中间态结构 Figure 3 Structure of Ftr and catalytic mechanism. A: Tetramer of Ftr with CHO-MFR and H4MPT (PDB: 2FHJ[47]). The substrates are shown with stick mode. Four subunits are shown with green, yellow, cyan, pink, respectively. B: Proposed catalytic mechanism of Ftr. The active formyl group of MFR is shown in red. |
| 图选项 |
Ftr四聚体结构显示该复合体由4个相同的亚基(32kDa)构成。活性中心Ser209与底物CHO-MFR的甲酰胺N、Glu245与CHO-MFR的甲酰胺O以及H4MPT喋呤环N3以氢键相连。这两个关键氨基酸在活性中心非常保守,且对Ftr的催化过程起重要作用[47]。Ftr催化甲酰基转移的过程类似包含三元中间态的SN2反应:Ser209与CHO-MFR的甲酰胺N之间形成的氢键、Glu245质子化的羧基COO–与甲酰胺O之间形成的氢键,都增加了甲酰基C的电负性,使其对H4MPT的N5进行亲核攻击,形成MFR-CHO/Ftr/H4MPT的三元中间态。随后,质子转移到MFR的氨基N,最终生成CHO-H4MPT,如图 3-B所示。
5.3 环化水解酶Mch 在产甲烷途径中,Mch催化CHO-H4MPT到CH≡H4MPT+的可逆缩合反应[48-49],反应过程如公式(3)所示。
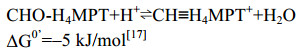 | 公式(3) |
 |
| 图 4 Mch晶体结构及催化反应中间态 Figure 4 Structure of homotrimer Mch and the catalytic intermediates. A: Trimer of Mch with CHO-H4MPT (PDB:4GVQ[52]). Substrate is shown with stick mode and the different subunits are shown with green, yellow, purple, respectively. B: catalytic intermediate state of Mch. The active methenyl group is shown in red. |
| 图选项 |
从活性中心底物结合状态可以推测Mch的催化机制[52]:首先,Arg183和Glu186捕获水分子对CH≡H4MPT+的C14发起亲核攻击,形成中间过渡态,同时Glu186成为能够获得质子的通用碱。质子化的Glu186贡献出从水分子获得的质子,并转移给邻近的H4MPT的N10。相对于接近Arg183的N5、N10通过氢键与Glu186相连,因此,N5成为最理想的离去基团。第二步反应中,C14与N10形成的局部环断裂,并形成N5-CHO-H4MPT(图 4-B)。
5.4 F420-dependent methylene-H4MPT脱氢酶(Mtd) Mtd催化氢负离子从F420H2转移到CH≡H4MPT+形成CH2=H4MPT[53-56](图 5-A)。反应过程如公式(4)所示。
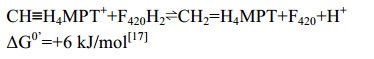 | 公式(4) |
 |
| 图 5 Mtd催化反应及其晶体结构与催化机制 Figure 5 The reaction catalyzed by homohexameric Mtd and its active site with binding substrate. A: The reverse reaction catalyzed by Mtd (PDB: 3IQF[60]). B: Crystal structure of Mtd hexamer. The substrates are shown as stick mode with green carbon. C: substrate binding site of Mtd and catalytic mechanism. H4MPT was show in green carbon and F420H2 with yellow carbon mode. |
| 图选项 |
M. kandleri的Mtd的蛋白晶体结构显示其不含任何辅基,由3个同源二聚体组成[57-58] (图 5-B)。高温古菌的蛋白通常在高盐环境下才能保持热稳定性,而Mtd在低盐浓度条件下已具有热稳定性。Mtd的复合体结构或许能解释这一反常行为:Mtd六聚体的亚基紧密互作,超过36%单亚基表面被包埋在六聚体内部。其次,Mtd每个残基之间的平均离子对是0.14,远高于中温蛋白和M. kandleri其他蛋白的平均值0.04,大量的残基离子对稳定了Mtd的表面[59]。
Mtd的底物结合中心位于单亚基α/β结合域与α-螺旋区组成的狭缝中[60]。两种底物CH≡H4MPT+与F420H2以面对面的方式结合在活性中心,F420H2的杂环部分与H4MPT的喋呤部分正面相对,二者的尾部直接延伸到狭缝的入口处。底物与蛋白氨基酸残基之间的结合主要依靠分子之间的范德华力,且底物的结合未引起蛋白结构构象的变化。如图 5-C所示,CH≡H4MPT+以re-face的方向面对F420H2的si-face方向,而且,F420H2的C5与CH≡H4MPT+的C14之间的距离仅2.6? ,因此氢负离子能够直接从F420H2的C5转移到CH≡H4MPT+的C14,形成CH2=H4MPT。
5.5 H2-forming methylene-H4MPT脱氢酶Hmd 细胞色素缺失的产甲烷古菌如果生长环境中Ni浓度过低,Mtd催化的正向反应被Ni诱导型蛋白Hmd替代。在低浓度Ni条件下,Hmd与Mtd的活性分别增加了6倍和4倍,而F420还原酶的活性降低了20倍[61-62]。同位素示踪实验发现Hmd催化氢分子的异裂后,直接转移氢负离子到底物CH≡H4MPT+上生成CH2=H4MPT[31, 63-65],Mtd则逆向氧化亚甲基获得电子并还原F420,为下一步反应提供还原力[60]。Afting等还发现在M.marburgensis中存在Hmd的同源蛋白HmdII与HmdIII (序列同源性为18.8%、17.6%)。但无酶活性的HmdII与HmdIII的表达并不受Ni的调控,其功能至今尚不明确[62]。Hmd催化的反应过程如公式(5)所示。
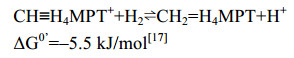 | 公式(5) |
 |
| 图 6 氢酶Hmd、辅因子结构及其催化机制 Figure 6 Homodimer of Hmd, cofactor structure and proposed catalytic mechanism of Hmd. A: Open (PDB: 6HAC[79]) and closed (PDB: 6HAV[79]) forms of homodimer Hmd. B: Chemical structure of FeGP cofactor. C: Heterolytic cleavage of hydrogen molecule at the Fe center and the transfer of the hydride and proton. |
| 图选项 |
该酶具有开放和闭合的两种构象,其特征为N末端结构域的刚体运动[79]。在开放构象中,FeGP的Fe中心与H4MPT的C14a之间的距离为9.3 ? ,不能直接发生反应(图 6-A)[80]。但通过结合CH2=H4MPT可以诱导闭合构象的形成,在闭合构象中,Fe中心与C14a之间的距离为3.8 ? [79],且活性位点裂隙中的大多数水分子被挤出。特别是结合在Fe中心的水配体由于封闭活性位点裂隙而被去除,使Fe中心产生了一个用于结合H2的空配位,同时吡啶环上的2-OH基团去质子化,由此激活Hmd。随后,H2进入活性中心并被捕捉到Fe的空配位上,异裂形成的Fe-H中间体将氢负离子转移至CH≡H4MPT+的C14a上生成CH2=H4MPT,质子被吡啶环上的去质子化2-OH接收,然后经质子传递链转移出催化活性中心(图 6-C)。闭合状态的酶-底物复合体晶体结构的QM/MM动力学模拟也支持了该预测模型[79]。
5.6 F420-reducing [NiFe]-氢酶Frh 大多数产甲烷古菌中存在Frh[81]。Frh以H2为电子供体,催化形成还原型的F420H2,此还原当量用于Mtd和Mer的催化反应。如公式(6)所示。
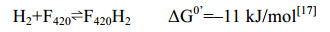 | 公式(6) |
 |
| 图 7 FrhABG蛋白结构以及近端Fe-S簇结构 Figure 7 Structure of FrhABG and special proximal [4Fe-4S] cluster in the active center. A: FrhABG monomer structure (PDB: 4OMF[84]). The hydrogen is cleavage into two protons and electrons at the active center of FrhA. Then electrons are transferred into FAD via [Fe-S] cluster to form FADH2. B: Mode of proximal [4Fe-4S] cluster, which bond to protein by cysteine and aspartate. |
| 图选项 |
在Frh的晶体结构中,FrhAGB的同源二聚体组合形成一个六聚体,底物氢分子和F420结合到位于蛋白六聚体表面的结合位点上,这一特点表明六聚体结构的核心区域可能不是Frh的催化中心[84]。然而,底物结合状态下的复合体结构尚未获得,Frh的详细催化机制也有待进一步的研究。
5.7 methylene-H4MPT还原酶Mer Mer广泛存在于产甲烷古菌和硫还原菌中,Mer在硫还原菌中参与乙酸氧化生成CO2的过程[87]。在产甲烷途径中,Mer催化CH2=H4MPT到CH3-H4MPT的可逆反应[88-89](图 8-A)。反应公式如公式(7)所示。
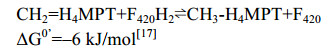 | 公式(7) |
 |
| 图 8 Mer催化的酶促反应及其蛋白结构 Figure 8 The reaction catalyzed by Mer and the tetramer structure of Mer. A: The reduction of methylene group with F420H2 catalyzed by Mer. B: Structure of Mer homotetramer (PDB: 1Z69[92]). C: Binding site of F420 which show with stick model in the active site. |
| 图选项 |
M. marburgensis、M. kandleri、M. barkeri等古菌的Mer蛋白结构相继被解析[90]。M. kandleri的Mer的晶体结构显示Mer是不包含任何辅基的单亚基蛋白,以同源二聚体或四聚体的形式存在[90] (图 8-B)。Mer属于细菌荧光素酶大家族(luciferase),该类酶蛋白以FMN、F420等为反应底物,TIM结合域是该类酶蛋白保守的底物结合中心[91]。Aufhammer等解析的Mer与F420结合的共结晶结构显示F420结合在TIM区域[92]。F420的核心嘧啶环通过Gly61、Val62、His36等氨基酸以微折叠的平面结构固定在结合中心(cis-face),F420侧链深入蛋白内部中,在结构上与F420依赖的乙醇脱氢酶相似[93] (图 8-C)。然而,目前Mer与底物H4MPT的结合还未报道。通过对M. barkeri的Mer的结构模型分析发现,TIM结合域与IR1、IR3结合域相互形成的狭缝空间可以结合H4MPT[90, 92]。Mer的催化机制也因底物结合状态的结构缺失而尚不明确。
5.8 H4MPT:CoM甲基转移酶Mtr 膜蛋白甲基转移酶MtrA-H复合体催化甲基从H4MPT转移到CoM[94-95]。反应过程如公式(8)所示。
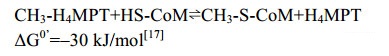 | 公式(8) |
目前,MtrA-H复合体的完整蛋白结构还未见报道。基于蛋白序列的结构预测显示,MtrC、MtrD以及MtrE是通过6–7个跨膜螺旋结合到膜上,MtrA、MtrB、MtrF和MtrG只有一个跨膜螺旋。MtrH与其他亚基结合锚定在膜的内侧,它是唯一缺乏跨膜疏水区的亚基,也是唯一能从Mtr复合体中分离出来的亚基(图 9-A)。纯化的MtrH能催化甲基从H4MPT转移到MtrA的钴胺素(cobalamin,维生素B12)的Co原子上[24, 105]。
 |
| 图 9 MtrA-H复合体模型以及单亚基MtrA晶体结构 Figure 9 The complex model of membrane protein MtrA-H and the crystal structure of MtrA. A: MtrA-H packing model and the catalytic mechanism model. The methyl group is transferred into cobalt through MtrH, then it moved into MtrE, where the methyl-CoM formed. B: Crystal structure of MtrA with cofactor cobalamin (PDB: 5LAA[23]). Cobalt in cobalamin is shown with ball. |
| 图选项 |
MtrA包含一个辅因子钴胺素,Co原子以六配位的形式存在[23]。与其他含cobalamin的蛋白类似,MtrA由α/β折叠组成的Rossmann核心结构域组成,类钴啉环覆盖在Rossmann结构域的表面,而核苷酸尾部则深入α/β折叠区内部的疏水区域,通过氢键与氨基酸残基相结合。Co离子通过氢键与活性中心关键氨基酸His84的咪唑基N相连,氧化状态的cobamide(Ⅱ)与蛋白的结合状态呈base-off/His-on构型(图 9-B)。
研究发现,Mtr只有在cobamide(I)的状态下才有活性,且氧化状态的CoⅡ-cobalamin与CH3-CoⅢ-cobalamin都有一个轴向的配基,而未甲基化的CoⅠ-cobalamin却没有轴向配基。同位素追踪发现,该轴向配基是活性中心His84上的咪唑N[100]。因此,推测MtrA辅因子CoⅠ-cobalamin甲基化形成CH3-CoⅢ-cobalamin的过程引起MtrA蛋白构像的变化,使Co与His结合。随后CH3-CoⅢ-cobalamin的脱甲基化又使得蛋白构象逆转[23]。
紧接MtrA的催化产物,MtrE催化甲基从CH3-Co-MtrA转移到CoM,形成CH3-CoM,该反应高度依赖Na+[105]。这一预测主要基于两个原因:首先,MtrE有最大的细胞质区结构域,第一和第二跨膜螺旋之间的loop有62个氨基酸,且这一连接区包含一个保守的Zn结合位点(Asp26-XGlu28-X22-His51-X9-Glu61)[106]。目前所知的所有催化巯基烷基化的蛋白都具有保守的Zn结合位点。其次,MtrE包含一个含有天冬氨酸的跨膜螺旋。研究发现Asp对跨膜螺旋转运Na+至关重要[107]。
Mtr不仅能催化甲基从H4MPT转移到cobalamin的可逆过程(CH3-H4MPT+ Cob(Ⅰ)alamine?CH3-Cob(Ⅰ)alamine+H4MPT,ΔG0’= –15 kJ/mol),还能催化CH3-CoⅢ-MtrA到CoM的可逆反应(CH3-Cob(Ⅰ)alamine+HS-CoM?CH3-S- CoM+Cob(Ⅰ)alamine,ΔG0’=–15 kJ/mol)[95, 108]。甲基转移过程触发MtrA蛋白构型的改变,并将能量传递给MtrE促使Na+从膜内转移到膜外,形成膜内外电动势梯度,进而偶联ATP的合成,完成能量保存。
5.9 CH3-CoM还原酶Mcr 在已知的三条产甲烷代谢途径中,最后一步反应均为CH3-CoM还原生成甲烷,催化这一反应的还原酶是Mcr(图 10-A)。反应过程如公式(9)所示。
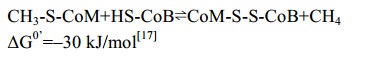 | 公式(9) |
 |
| 图 10 Mcr催化反应、晶体结构、活性位点及催化机制 Figure 10 The catalytic reaction, crystal structure, binding site and mechanism of Mcr. A: Methyl-CoM reduced into methane with CoB catalyzed by Mcr. B: Crystal structure of Mcr homodimer with cofactor F430/CoM/CoB (PDB: 5A8K[119]). The subunits and cofactor are shown in cartoon and stick model. C: Active site of Mcr with F430, CoM and CoB. Several post-transcriptional modified amino acids are arranged surrounding the active site. D: Hypothesized two type of catalytic mechanism of Mcr[122]. |
| 图选项 |
在甲烷氧化菌中,该酶也可以逆向催化甲烷的氧化[109]。Mcr在产甲烷古菌中是较为保守的蛋白,其序列同源性较高(61%–69%)[110]。因此,Mcr的编码基因也成为产甲烷古菌鉴定的标志基因[111]。
M.marburgensis、M. wolfeii、M.barkeri等古菌的Mcr的蛋白结构已获得解析[110, 112-113]。Mcr复合体是由α、β、γ三个亚基组成的同源二聚体(αβγ)2(图 10-B),每一个αβγ单位活性中心均存在含Ni的辅因子F430,EPR分析显示在完整催化过程中,Ni的氧还价态分为Ni(Ⅰ)、Ni(Ⅱ)、Ni(Ⅲ)三种[114-118]。Mcr单聚体不足以完成整个反应,由αβγα’或α’β’γ’α组成的催化单元形成直通蛋白表面的狭窄通道,F430结合在通道的底部,两个F430活性中心距离约为50? [112-113, 119]。F430通过与氨基酸残基形成氢键结合在活性中心,Ni的第五个配位与Gln147连接形成轴向基团,第六配位面向通道入口,也是催化反应过程中的关键配位。此外,底物CoM结合在F430的上方,CoM的巯基距离Ni约2.4? ,其磺酸基通过盐键、氢键与活性中心氨基酸残基结合。底物CoB主要通过盐键结合在狭窄通道的上半部分,这一狭窄通道也因为CoB的结合而处于完全封闭的状态[110, 119-120]。
Mcr的另一个显著特点是活性中心氨基酸残基的翻译后修饰[121]。在M.marburgensis的Mcr活性中心发现4个甲基化的氨基酸、1个巯基化的甘氨酸、以及1个醛基化的天冬氨酸(图 10-C)。翻译后修饰的氨基酸在所有的Mcr中并不保守,表明这些氨基酸残基的修饰对催化活性不是必需的,可能有助于提高催化效率以及活性中心的稳定性[119, 122-123]。
根据Mcr的结构分析预测其催化机制如下:辅因子F430的Ni(Ⅰ)攻击CH3-CoM产生CH3-Ni(Ⅲ)和CoM阴离子,电子从CoM转移到CH3-Ni(Ⅲ)形成CH3-Ni(Ⅱ)以及CoM-S自由基(radical),1个氢原子从CoB-SH转移到CH3-Ni(Ⅱ)生成甲烷(图 10-D)[124-125]。这种催化机制类似于cobalamin依赖的其他酶,催化过程中形成CH3-Co的中间态。通过DFT的计算预测Mcr的反应过程也可能基于自由基催化。另一种催化理论是Ni(Ⅰ)攻击CH3-S-CoM,生成甲基自由基和CoM-S-Ni(Ⅱ),随后,甲基自由基接受CoB-SH的氢原子生成甲烷。为避免甲基自由基的快速消旋,C-S的断裂与C-H的形成同时发生[122, 126-127]。
5.10 异二硫化合物还原酶/[NiFe]-氢酶复合体 在产甲烷途径的最后一步反应中,异二硫化合物被还原重新生成CoM与CoB后才能参与新的CoM转甲基反应[2]。在细胞色素缺失的产甲烷古菌中,CoM-S-S-CoB还原由异二硫化合物还原酶(Hdr)和[NiFe]-氢酶复合体(HdrABC-MvhAGD)完成,反应过程如公式(10c)所示。而在含细胞色素的产甲烷古菌中,由HdrDE-VhtACG复合体催化完成[128-129],反应过程如公式(10a–b)所示。不同的是HdrABC-MvhAGD复合体是细胞质蛋白,以黄素腺嘌呤二核苷酸(FAD)为基础的电子歧化完成氧化还原反应并偶联能量储存。而HdrDE-VhtACG复合体位于细胞膜上,通过电子载体MPH2和细胞色素传递电子,以形成质子泵的方式产生能量[17, 29-30]。CoM-S-S-CoB还原反应的不同体现了两类产甲烷古菌在能量保存方式上的差异。
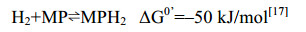 | 公式(10a) |
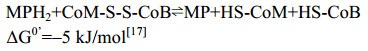 | 公式(10b) |
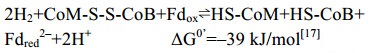 | 公式(10c) |
在生理条件下,铁氧还蛋白Fd的电动势E0’为–500 mV,而质子/氢分子电对2H+/H2的电势E0’为–400 mV,因此Fdox很难从电势较低的氢分子中得到电子。而在HdrABC-MvhAGD催化的还原反应中,氢分子裂解后产生的电子转移至电子载体FAD,通过FAD的转移作用,一半电子用于还原CoM- S-S-CoB (E0’为–140 mV),一半电子用于还原Fdox (E0’为–500 mV),这种现象称之为电子歧化[28, 30]。HdrABC-MvhAGD复合体采用电子歧化的方式还原CoM-S-S-CoB并获得还原力Fdred2–,直接用于产甲烷途径起始过程中CO2的固定,以及其他生物合成反应,以此偶联能量保存,如图 11所示。
 |
| 图 11 HdrABC-MvhAGD晶体结构及催化机制 Figure 11 Crystal structure of HdrABC-MvhAGD homodimer complex and its catalytic mechanism. The structure (PDB: 5ODR[28]) complex is shown with cartoon and surface model in different color, and the [Fe-S] cluster and FAD are shown with ball. MvhAGD, [NiFe]-hydrogenase, oxidize 2H2, producing 4H+ and 4 electrons. The electrons are transferred into FAD center via [Fe-S] cluster, where two electrons are used for ferredoxin reduction and another two electrons for CoM-S-S-CoB reduction. |
| 图选项 |
在含有细胞色素的产甲烷古菌中,HdrDE-VhtACG催化CoM-S-S-CoB的还原。虽然氢酶Vht与还原酶都位于细胞膜上,但并不形成紧密结合的复合体,在两者的催化过程中,甲烷吩嗪MP是氢酶与还原酶之间的电子传递物质[25, 130-131]。VhtACG催化氢气裂解产生的电子通过细胞色素传递给MP,生成还原型MPH2,MPH2通过自身二次氧化把获得的电子传递给还原酶HdrDE,用于还原CoM-S-S-CoB。该还原反应释放的能量与质子的膜内外转移(耗能反应)相偶联,由此形成膜内外质子梯度并用于ATP的合成,进而完成能量保存[16, 26-27]。
根据序列分析发现,在氢酶VhtACG复合体中,VhtA是[NiFe]-氢酶的大亚基,包含[NiFe]活性中心[132],VhtG是氢酶的小亚基。复合体中的[NiFe]活性中心面向细胞周质,因此催化氢分子裂解产生的质子直接释放到膜外[133-134]。而VhtC是包含细胞色素b的跨膜蛋白,其功能是作为从氢酶传递电子到还原酶HdrDE的电子通道。HdrDE复合体的HdrE是包含细胞色素的膜整合蛋白,而另一个亚基HdrD是通过蛋白结合作用锚定在HdrE上的胞质蛋白。亚基HdrE通过细胞色素再次氧化MPH2,并将获得的电子转移给HdrD用于CoM-S-S-CoB的还原[131, 135]。氢酶VhtACG与还原酶HdrDE的蛋白结构目前还尚未解析,其详细的催化机制也有待进一步的深入研究。
5.11 [NiFe]-氢酶Eha、Ehb、Ech 在细胞色素缺失的产甲烷途径中,Fdred2–产生于CoM-S-S-CoB还原反应的电子歧化过程,部分用于产甲烷途径起始CO2的固定,部分用于其他代谢途径。为补偿消耗的还原力,膜蛋白氢酶复合体EhaA-T、EhbA-Q也参与Fdred2–的生成[136]。这两类氢酶以氢分子(-414mv)为电子供体,催化Fdox的还原(–500mV),这一耗能反应由膜内外Na+梯度推动完成[137]。氢酶EhaA-T、EhbA-Q是无细胞色素产甲烷古菌中的两类同源蛋白,而在有细胞色素的产甲烷古菌中,膜蛋白复合体EchA-F代替Eha或者Ehb的功能[27]。反应过程如公式(11)所示。
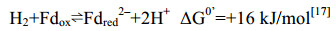 | 公式(11) |
EhaA-T与EhbA-Q、EchA-F具有同源性和相似的催化功能,由此推测其催化机制也类似。不同的是,在有细胞色素的产甲烷古菌中,推动Ech合成Fdred2–的不是Na+而是H+[16]。由于膜蛋白复合体的分离纯化和晶体生长难度极大,目前还没有获得完整的蛋白结构,其催化机制也尚不清楚。
6 展望 产甲烷古菌的研究始于1936年,Barker首次发现在微生物的作用下CO2还原产甲烷的现象,随后分离出第一株产甲烷古菌。随着Hungate厌氧培养方法的建立,越来越多的产甲烷古菌被发现,同时还发现了产甲烷古菌在不同底物条件下产甲烷的特殊生理现象[2]。经过长达半个世纪的探索,基本明确了不同的产甲烷途径,以及中间产物、酶复合体和能量代谢模式等。
在过去的20年中,产甲烷古菌的研究主要集中在产甲烷途径中的酶蛋白。基于对蛋白结构的解析,揭示了各种酶复合体的结构特征与催化机制[22]。然而,由于产甲烷古菌极端厌氧、难培养以及复合体蛋白难纯化等特点,部分产甲烷途径的蛋白结构(尤其是膜蛋白复合体)还尚未解析,这些蛋白的催化机制也没有完全确定。例如,偶联能量合成的膜蛋白MtrA-H复合体结构也亟待解析;复合体Hdr/Mvh电子歧化过程的详细机制尚不清楚,这需要更高分辨率的复合体结构去解析。此外,CH3-CoM还原酶Mcr的反应机制仍有很多疑问待解决,CoM-S-Ni(Ⅱ)中间体表明其催化机制可能是基于甲基自由基的反应,然而还需进一步验证[122]。目前预测的催化机制都是基于Mcr非活性状态的蛋白结构,因此,活性状态下的蛋白结构对催化机理的分析显得尤为重要,此外Mcr活性中心较多的转录后修饰氨基酸的功能仍待解析。
生化和光谱分析表明,产甲烷途径的酶蛋白在催化循环中构象变化很大,特别是HdrABC-MvhAGD、Mcr以及MtrA-H等复合体。将来,利用冷冻电子显微镜捕获催化反应中间体和酶蛋白构象变化,对产甲烷酶复合体的结构研究会发挥重要的作用。同时,构建完善稳定的古菌遗传体系也将会大量用于代谢途径调控机制、蛋白体外重组等多方面的研究中[11]。
此外,在针对产甲烷的生物能源应用研究中,产甲烷古菌广谱代谢底物和关键酶蛋白的研究也是未来重要的研究方向。本课题组正在研究的一株产甲烷古菌Methermicoccus shengliensis能够代谢甲氧基芳香化合物产甲烷,我们通过对新型底物特异的酶蛋白结构进行人工改造,可使其代谢众多结构类似的非特异底物,进而提高底物利用效率。另外,辅酶和辅基的生物合成途径对于全面了解酶蛋白催化机制起关键作用。更重要的是,在合成途径的研究基础上,通过合成生物学体外构建人工酶、代谢途径是未来代谢研究的重要方向,也是促进甲烷生产的新途径。
References
| [1] | Bapteste E, Brochier C, Boucher Y. Higher-level classification of the archaea:evolution of methanogenesis and methanogens. Archaea, 2005, 1: 353-363. DOI:10.1155/2005/859728 |
| [2] | |
| [3] | Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, Castelle CJ, Butterfield CN, Hernsdorf AW, Amano Y, Ise K, Suzuki Y, Dudek N, Relman DA, Finstad KM, Amundson R, Thomas BC, Banfield JF. A new view of the tree of life. Nature Microbiology, 2016, 1: 1-6. |
| [4] | Vanwonterghem I, Evans PN, Parks DH, Jensen PD, Woodcroft BJ, Hugenholtz P, Tyson GW. Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota. Nature Microbiology, 2016, 1: 1-9. |
| [5] | Conrad R. The global methane cycle:recent advances in understanding the microbial processes involved. Environmental Microbiology Reports, 2009, 1(5): 285-292. DOI:10.1111/j.1758-2229.2009.00038.x |
| [6] | Thauer RK. My lifelong passion for biochemistry and anaerobic microorganisms. Annual Review of Microbiology, 2015, 69: 1-30. DOI:10.1146/annurev-micro-091014-104344 |
| [7] | Shima S, Warkentin E, Thauer RK, Ermler U. Structure and function of enzymes involved in the methanogenic pathway utilizing carbon dioxide and molecular hydrogen. Journal of Bioscience and Bioengineering, 2002, 93(6): 519-530. DOI:10.1016/S1389-1723(02)80232-8 |
| [8] | Dimarco AA, Bobik TA, Wolfe RS. Unusual coenzymes of methanogenesis. Annual Review of Biochemistry, 1990, 59: 355-394. DOI:10.1146/annurev.bi.59.070190.002035 |
| [9] | Ferguson DJ, Longstaff DG, Krzycki JA. Assay of methylotrophic methyltransferases from Methanogenic archaea. Methods Enzymology, 2011, 494: 139-158. DOI:10.1016/B978-0-12-385112-3.00008-1 |
| [10] | Moore BC, Leigh JA. Markerless mutagenesis in Methanococcus maripaludis demonstrates roles for alanine dehydrogenase, alanine racemase, and alanine permease. Journal of Bacteriology, 2005, 187(3): 972-979. DOI:10.1128/JB.187.3.972-979.2005 |
| [11] | Lyu Z, Chou CW, Shi H, Wang L, Ghebreab R, Phillips D, Yan Y, Duin EC, Whitman WB. Assembly of methyl coenzyme M reductase in the methanogenic archaeon Methanococcus maripaludis. Journal of Bacteriology, 2018, 200(7): 717-746. |
| [12] | Cheng L, Zheng ZZ, Wang C, Zhang H. Recent advances in methanogens. Microbiology China, 2016, 43(5): 1143-1164. (in Chinese) 承磊, 郑珍珍, 王聪, 张辉. 产甲烷古菌研究进展. 微生物学通报, 2016, 43(5): 1143-1164. |
| [13] | Evans PN, Parks DH, Chadwick GL, Robbins SJ, Orphan VJ, Golding SD, Tyson GW. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science, 2015, 350(6259): 434-438. DOI:10.1126/science.aac7745 |
| [14] | Fang XY, Li JB, Rui JP, Li XZ. Research progress in biochemical pathways of methanogenesis. Chinese Journal of Applied and Environmental Biology, 2015, 21(1): 1-9. (in Chinese) 方晓瑜, 李家宝, 芮俊鹏, 李香真. 产甲烷生化代谢途径研究进展. 应用与环境微生物学报, 2015, 21(1): 1-9. |
| [15] | Duan CH, Zhang CJ, Sun YH, Li M. Recent advances on the novel methanogens. Acta Microbiologica Sinica, 2019, 51(6): 981-995. (in Chinese) 段昌海, 张翠景, 孙艺华, 李猛. 新型产甲烷古菌研究进展. 微生物学报, 2019, 51(6): 981-995. |
| [16] | Welte C, Deppenmeier U. Bioenergetics and anaerobic respiratory chains of aceticlastic methanogens. Biochimica et Biophysica Acta, 2014, 1837(2014): 1130-1147. |
| [17] | Thauer RK, Kaster AK, Seedorf H, Buckel W, Hedderich R. Methanogenic archaea:ecologically relevant differences in energy conservation. Nature Reviews Microbiology, 2008, 6(8): 579-591. DOI:10.1038/nrmicro1931 |
| [18] | Weimer PJ, Zcikus JG. One carbon metabolism in methanogenie bacteria. Archives of Microbiology, 1978, 119: 49-57. DOI:10.1007/BF00407927 |
| [19] | Morii H, Koga Y, Nagai S. Energetic analysis of the growth of Methanobrevibacter arboriphilus A2 in hydrogen-limited continuous cultures. Biotechnology and Bioengineering, 1986, XXIX: 310-315. |
| [20] | Heijnen JJ, Dijken JPV. In search of a thermodynamic description of biomass yields for the chemotrophic growth of microorganisms. Biotechnology and Bioengineering, 1991, 39: 833-858. |
| [21] | Thauer RK, Jungermann K, Decker K. Energy conservation in chemotrophic anaerobic bacteria. Bacteriological Reviews, 1977, 41(1): 100-180. DOI:10.1128/MMBR.41.1.100-180.1977 |
| [22] | |
| [23] | Wagner T, Ermler U, Shima S. MtrA of the sodium ion pumping methyltransferase binds cobalamin in a unique mode. Scientific Reports, 2016, 6: 1-10. DOI:10.1038/s41598-016-0001-8 |
| [24] | Gottschalk G, Thauer RK. The Na+-translocating methyltransferase complex from methanogenic archaea. Biochimica et Biophysica Acta, 2001, 1505(1): 28-36. DOI:10.1016/S0005-2728(00)00274-7 |
| [25] | Deppenmeier U. The membrane-bound electron transport system of Methanosarcina species. Journal of Bioenergetics and Biomembranes, 2004, 36(1): 55-64. |
| [26] | Deppenmeier U, Müller V. Life close to the thermodynamic limit:how methanogenic archaea conserve energy. Results and Problems in Cell Differentiation, 2008, 45: 123-152. |
| [27] | Thauer RK, Kaster AK, Goenrich M, Schick M, Hiromoto T, Shima S. Hydrogenases from methanogenic archaea, nickel, a novel cofactor, and H2 storage. Annual Review of Biochemistry, 2010, 79: 507-536. DOI:10.1146/annurev.biochem.030508.152103 |
| [28] | Wagner T, Koch J, Ermler U, Shima S. Methanogenic heterodisulfide reductase (HdrABC-MvhAGD) uses two noncubane[4Fe-4S] clusters for reduction. Science, 2017, 357(6352): 699-703. DOI:10.1126/science.aan0425 |
| [29] | Buckel W, Thauer RK. Flavin-based electron bifurcation, a new mechanism of biological energy coupling. Chemical Reviews, 2018, 118(7): 3862-3886. DOI:10.1021/acs.chemrev.7b00707 |
| [30] | Buckel W, Thauer RK. Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation. Biochimica et Biophysica Acta, 2013, 1827(2013): 94-113. |
| [31] | Bai LP, Fujishiro T, Huang GF, Koch U, Takabayashi A, Yokono M, Tanaka A, Xu T, Hu XL, Ermler U, Shima S. Towards artificial methanogenesis:biosynthesis of the[Fe]-hydrogenase cofactor and characterization of the semisynthetic hydrogenase. Faraday Discussions, 2017, 198: 37-58. DOI:10.1039/C6FD00209A |
| [32] | Hochheimer A, Schmitz RA, Thauer RK, Hedderich R. The tungsten formylmethanofuran dehydrogenase from Methanobacterium thermoautotrophicum contains sequence motifs characteristic for enzymes containing molybdopterin dinucleotide. European Journal of Biochemistry, 1995, 234(3): 910-920. DOI:10.1111/j.1432-1033.1995.910_a.x |
| [33] | Bertram PA, Karrasch M, Schmitz RA, B? cher R, Albracht SPJ, Thauer RK. Formylmethanofuran dehydrogenases from methanogenic archaea substrate specificity, EPR properties and reversible inactivation by cyanide of the molybdenum or tungsten iron-sulfur proteins. European Journal of Biochemistry, 1994, 220: 477-484. DOI:10.1111/j.1432-1033.1994.tb18646.x |
| [34] | Hochheimer A, Linder D, Thauer RK, Hedderich R. The molybdenum formylmethanofuran dehydrogenase operon and the tungsten formylmethanofuran dehydrogenase operon from Methanobacterium thermoautotrophicum-structures and transcriptional regulation. European Journal of Biochemistry, 1996, 242(1): 156-162. DOI:10.1111/j.1432-1033.1996.0156r.x |
| [35] | Vorholt JA, Vaupel M, Thauer RK. A selenium-dependent and a selenium-independent formylmethanofuran dehydrogenase and their transcriptional regulation in the hyperthermophilic Methanopyrus kandleri. Molecular Microbiology, 1997, 23(5): 1033-1042. DOI:10.1046/j.1365-2958.1997.2931653.x |
| [36] | Schmitz RA, Albracht SPJ, Thauer RK. A molybdenum and a tungsten isoenzyme of formylmethanofuran dehydrogenase in the thermophilic archaeon Methanobacterium wolfei. European Journal of Biochemistry, 1992, 209: 1013-1018. DOI:10.1111/j.1432-1033.1992.tb17376.x |
| [37] | Hochheimer A, Hedderich R, Thauer RK. The formylmethanofuran dehydrogenase isoenzymes in Methanobacterium wolfei and Methanobacterium thermoautotrophicum:induction of the molybdenum isoenzyme by molybdate and constitutive synthesis of the tungsten isoenzyme. Archives of Microbiology, 1998, 170(5): 389-393. DOI:10.1007/s002030050658 |
| [38] | Wagner T, Ermler U, Shima S. The methanogenic CO2 reducing-and-fixing enzyme is bifunctional and contains 46[4Fe-4S] clusters. Science, 2016, 354(6308): 114-117. DOI:10.1126/science.aaf9284 |
| [39] | Bertram PA, Thauer RK. Thermodynamics of the formylmethanofuran dehydrogenase reaction in Methanobacterium thermoautotrophicum. European Journal of Biochemistry, 1994, 226(3): 811-818. DOI:10.1111/j.1432-1033.1994.t01-1-00811.x |
| [40] | Shima S, Weiss DS, Thauer RK. Formylmethanofuran:tetrahydromethanopterin formyltransferase (Ftr) from the hyperthermophilic Methanopyrus kandleri -Cloning, sequencing and functional expression of the ftr gene and one-step purification of the enzyme overproduced in Escherichia coli. European Journal of Biochemistry, 1995, 230(3): 906-913. DOI:10.1111/j.1432-1033.1995.tb20635.x |
| [41] | Shima S, Thauer RK, Michel H, Ermler U. Crystallization and preliminary X-ray diffraction studies of formylmethanofuran:tetrahydromethanopterin formyltransferase from Methanopyrus kandleri. Proteins:Structure Function and Genetics, 1996, 26(1): 118-120. DOI:10.1002/(SICI)1097-0134(199609)26:1<118::AID-PROT12>3.0.CO;2-J |
| [42] | Mamat B, Roth A, Grimm C, Ermler U, Tziatzios C, Schubert D, Thauer RK, Shima S. Crystal structures and enzymatic properties of three formyltransferases from archaea:environmental adaptation and evolutionary relationship. Protein Science, 2002, 11(9): 2168-2178. |
| [43] | Ermler U, Merckel MC, Thauer RK, Shima S. Formylmethanofuran:tetrahydromethanopterin formyltransferase from Methanopyrus kandleri-new insights into salt-dependence and thermostability. Structure, 1997, 5(5): 635-646. DOI:10.1016/S0969-2126(97)00219-0 |
| [44] | Kunow J, Shima S, Vorholt JA, Thauer RK. Primary structure and properties of the formyltransferase from the mesophilic Methanosarcina barkeri:comparison with the enzymes from thermophilic and hyperthermophilic methanogens. Archives of Microbiology, 1996, 165(2): 97-105. |
| [45] | Shima S, Thauer RK, Ermler U, Durchschlag H, Tziatzios C, Schubert D. A mutation affecting the association equilibrium of formyltransferase from the hyperthermophilic Methanopyrus kandleri and its influence on the enzyme's activity and thermostability. European Journal of Biochemistry, 2000, 267(22): 6619-6623. DOI:10.1046/j.1432-1327.2000.01756.x |
| [46] | Shima S, Tziatzios C, Schubert D, Fukada H, Takahashi K, Ermler U, Thauer RK. Lyotropic-salt-induced changes in monomer/dimer/tetramer association equilibrium of formyltransferase from the hyperthermophilic Methanopyrus kandleri in relation to the activity and thermostability of the enzyme. European Journal of Biochemistry, 1998, 258(1): 85-92. DOI:10.1046/j.1432-1327.1998.2580085.x |
| [47] | Acharya P, Warkentin E, Ermler U, Thauer RK, Shima S. The structure of formylmethanofuran:tetrahydromethanopterin formyltransferase in complex with its coenzymes. Journal of Molecular Biology, 2006, 357(3): 870-879. DOI:10.1016/j.jmb.2006.01.015 |
| [48] | Klein AR, Breitung J, Linder D, Stetter KO, Thauer RK. N5, N10-methenyltetrahydromethanopterin cyclohydrolase from the extremely thermophilic sulfate reducing Archaeoglobus fulgidus:comparison of its properties with those of the cyclohydrolase from the extremely thermophilic Methanopyrus kandleri. Archives of Microbiology, 1993, 159(3): 213-219. DOI:10.1007/BF00248474 |
| [49] | Brommelstroet BW, Hensgens CMH, Geerts WJ, Keltjens JT, Drift C, Vogels GD. Purification and properties of 5, 10-methenyltetrahydromethanopterin cyclohydrolase from Methanosarcina barkeri. Jounal of Bacteriology, 1990, 175: 564-571. |
| [50] | Grabarse W, Vaupel M, Vorholt JA, Shima S, Thauer RK, Wittershagen A, Bourenkov G, Bartunik HD, Ermler U. The crystal structure of methenyltetrahydromethanopterin cyclohydrolase from the hyperthermophilic archaeon Methanopyrus kandleri. Structure, 1999, 7(10): 1257-1268. |
| [51] | Vaupel M, Vorholt JA, Thauer RK. Overproduction and one-step purification of the N5, N10-methenyltetrahydromethanopterin cyclohydrolase (Mch) from the hyperthermophilic Methanopyrus kandleri. Extremophiles, 1998, 2(1): 15-22. DOI:10.1007/s007920050038 |
| [52] | Upadhyay V, Demmer U, Warkentin E, Moll J, Shima S, Ermler U. Structure and catalytic mechanism of N5, N10-methenyltetrahydromethanopterin cyclohydrolase. Biochemistry, 2012, 51(42): 8435-8443. DOI:10.1021/bi300777k |
| [53] | Klein AR, Thauer RK. Re-face specificity at C14a of methylenetetrahydromethanopterin and Si-face specificity at C5 of coenzyme F420 for coenzyme F420-dependent methylenetetrahydromethanopterin dehydrogenase from methanogenic archaea. European Journal of Biochemistry, 1995, 227(1/2): 169-174. |
| [54] | Klein AR, Berk H, Purwantini E, Daniels L, Thauer RK. Si-face stereospecificity at C5 of coenzyme F420 for F420-dependent glucose-6-phosphate dehydrogenase from Mycobacterium smegmatis and F420-dependent alcohol dehydrogenase from Methanoculleus thermophilicus. European Journal of Biochemistry, 1996, 239(1): 93-97. DOI:10.1111/j.1432-1033.1996.0093u.x |
| [55] | Smmelstroet B, Geerts WJ, Keltjens JT, Drift C, Vogels GD. Purification and properties of 5, 10-methylenetetrahydromethanopterin dehydrogenase and 5, 10-methylenetetrahydromethanopterin reductase, two coenzyme F420-dependent enzymes, from Methanosarcina barkeri. Biochimica et Biophysica Acta, 1991, 1079: 293-302. DOI:10.1016/0167-4838(91)90072-8 |
| [56] | Schworer B, Breitung J, Klein AR, Stetter KO, Thauer RK. Formylmethanofuran:tetrahydromethanopterin formyltransferase and N5, N10-methylenetetrahydromethanopterin dehydrogenase from the sulfate-reducing Archaeoglobus fulgidus:similarities with the enzymes from methanogenic Archaea. Archives of Microbiology, 1993, 159(3): 225-232. DOI:10.1007/BF00248476 |
| [57] | Klein AR, Thauer RK. Overexpression of the coenzyme-F420-dependent N5, N10-methylenetetrahydromethanopterin dehydrogenase gene from the hyperthermophilic Methanopyrus kandleri. European Journal of Biochemistry, 1997, 245(2): 386-391. DOI:10.1111/j.1432-1033.1997.t01-1-00386.x |
| [58] | Klein AR, Koch J, Stetter KO, Thauer RK. Two N5, N10-methylenetetrahydromethanopterin dehydrogenases in the extreme thermophile Methanopyrus kandleri:characterization of the coenzyme F420-dependent enzyme. Archives of Microbiology, 1993, 160(3): 186-192. |
| [59] | Hagemeier CH, Shima S, Thauer RK, Bourenkov G, Bartunik HD, Ermler U. Coenzyme F420-dependent methylenetetrahydro-methanopterin dehydrogenase (Mtd) from Methanopyrus kandleri:a methanogenic enzyme with an unusual quarternary structure. Journal of Molecular Biology, 2003, 332(5): 1047-1057. DOI:10.1016/S0022-2836(03)00949-5 |
| [60] | Ceh K, Demmer U, Warkentin E, Moll J, Thauer RK, Shima S, Ermler U. Structural basis of the hydride transfer mechanism in F420-dependent methylenetetrahydromethanopterin dehydrogenase. Biochemistry, 2009, 48(42): 10098-10105. DOI:10.1021/bi901104d |
| [61] | Afting C, Hochheimer A, Thauer RK. Function of H2-forming methylenetetrahydromethanopterin dehydrogenase from Methanobacterium thermoautotrophicum in coenzyme F420 reduction with H2. Archives of Microbiology, 1998, 169(3): 206-210. DOI:10.1007/s002030050562 |
| [62] | Afting C, Kremmer E, Brucker C, Hochheimer A, Thauer RK. Regulation of the synthesis of H2-forming methylenetetrahydromethanopterin dehydrogenase (Hmd) and of HmdII and HmdIII in Methanothermobacter marburgensis. Archives of Microbiology, 2000, 174(4): 225-232. DOI:10.1007/s002030000197 |
| [63] | Klein AR, Fernandez VM, Thauer RK. H2-forming N5, N10-methylenetetrahydromethanopterin dehydrogenase:mechanism of H2 formation analyzed using hydrogen isotopes. FEBS Letters, 1995, 368(2): 203-206. DOI:10.1016/0014-5793(95)00642-M |
| [64] | Hartmann GC, Santamaria E, Fernandez VM, Thauer RK. Studies on the catalytic mechanism of H2-forming methylenetetrahydromethanopterin dehydrogenase:para-ortho H2 conversion rates in H2O and D2O. Journal of Biological Inorganic Chemistry, 1996, 1(5): 446-450. DOI:10.1007/s007750050077 |
| [65] | Klein AR, Hartmann GC, Thauer RK. Hydrogen isotope effects in the reactions catalyzed by H2-forming N5, N10-methylenetetrahydromethanopterin dehydrogenase from methanogenic Archaea. European Journal of Biochemistry, 1995, 233(1): 372-376. DOI:10.1111/j.1432-1033.1995.372_1.x |
| [66] | Shima S, Pilak O, Vogt S, Schick M, Stagni MS, Meyer-Klaucke W, Warkentin E, Thauer RK, Ermler U. The crystal structure of[Fe]-hydrogenase reveals the geometry of the active site. Science, 2008, 321(5888): 572-575. DOI:10.1126/science.1158978 |
| [67] | Schick M, Xie XL, Ataka K, Kahnt J, Linne U, Shima S. Biosynthesis of the iron-guanylylpyridinol cofactor of [Fe]-hydrogenase in methanogenic archaea as elucidated by stable-isotope labeling. Journal of the American Chemical Society, 2012, 134(6): 3271-3280. DOI:10.1021/ja211594m |
| [68] | Hiromoto T, Ataka K, Pilak O, Vogt S, Stagni MS, Meyer-Klaucke W, Warkentin E, Thauer, R K, Shima S, Ermler U. The crystal structure of C176A mutated[Fe]-hydrogenase suggests an acyl-iron ligation in the active site iron complex. FEBS Letters, 2009, 583(3): 585-590. DOI:10.1016/j.febslet.2009.01.017 |
| [69] | Fujishiro T, Bai LP, Xu T, Xie XL, Schick M, Kahnt J, Rother M, Hu XL, Ermler U, Shima S. Identification of HcgC as a SAM-Dependent Pyridinol Methyltransferase in [Fe]-Hydrogenase Cofactor Biosynthesis. Angewandte Chemie International Edition, 2016, 55(33): 9648-9651. DOI:10.1002/anie.201604352 |
| [70] | Fujishiro T, Kahnt J, Ermler U, Shima S. Protein-pyridinol thioester precursor for biosynthesis of the organometallic acyl-iron ligand in [Fe]-hydrogenase cofactor. Nature Communications, 2015, 6(6895): 1-8. |
| [71] | Bai LP, Wagner T, Xu T, Hu XL, Ermler U, Shima S. A water-bridged H-bonding network contributes to the catalysis of the SAM-dependent C-methyltransferase HcgC. Angewandte Chemie International Edition, 2017, 56(36): 10806-10809. DOI:10.1002/anie.201705605 |
| [72] | Shima S, Thauer RK. A third type of hydrogenase catalyzing H2 activation. The Chemical Record, 2007, 7(1): 37-46. |
| [73] | Lubitz W, Ogata H, Rudiger O, Reijerse E. Hydrogenases. Chemical Reviews, 2014, 114(8): 4081-4148. DOI:10.1021/cr4005814 |
| [74] | Hidese R, Ataka K, Bill E, Shima S. CuI and H2O2 inactivate and FeⅡ inhibits[Fe]-hydrogenase at very low concentrations. Chembiochem, 2015, 16(13): 1861-1865. DOI:10.1002/cbic.201500318 |
| [75] | Shima S, Ataka K. Isocyanides inhibit[Fe]-hydrogenase with very high affinity. FEBS Letters, 2011, 585(2011): 353-356. |
| [76] | Tamura H, Salomone-Stagni M, Fujishiro T, Warkentin E, Meyer-Klaucke W, Ermler U, Shima S. Crystal structures of[Fe]-hydrogenase in complex with inhibitory isocyanides:implications for the H2-activation site. Angewandte Chemie International Edition, 2013, 52(37): 9656-9659. DOI:10.1002/anie.201305089 |
| [77] | Lyon EJ, Shima S, Buurman G, Chowdhuri S, Batschauer A, Steinbach K, Thauer RK. UV-A/blue-light inactivation of the 'metal-free' hydrogenase (Hmd) from methanogenic archaea. European Journal of Biochemistry, 2004, 271(1): 195-204. DOI:10.1046/j.1432-1033.2003.03920.x |
| [78] | Buurman G, Shima S, Thauer RK. The metal-free hydrogenase from methanogenic archaea:evidence for a bound cofactor. FEBS Letters, 2000, 485(2/3): 200-204. |
| [79] | Huang GF, Wagner T, Wodrich MD, Ataka K, Bill E, Ermler U, Hu X, Shima S. The atomic-resolution crystal structure of activated[Fe]-hydrogenase. Nature Catalysis, 2019, 2(6): 537-543. DOI:10.1038/s41929-019-0289-4 |
| [80] | Hiromoto T, Warkentin E, Moll J, Ermler U, Shima S. The crystal structure of an[Fe]-hydrogenase-substrate complex reveals the framework for H2 activation. Angewandte Chemie International Edition, 2009, 48(35): 6457-6460. DOI:10.1002/anie.200902695 |
| [81] | Alex LA, Reeve JN, Orme-Johnson WH, Walsh CT. Cloning, sequence determination, and expression of the genes encoding the subunits of the nickel-containing 8-hydroxy-5-deazaflavin reducing hydrogenase from Methanobacterium thermoautotrophicum delta H. Biochemistry, 1990, 29(31): 7237-7244. DOI:10.1021/bi00483a011 |
| [82] | Vaupel M, Thauer RK. Two F420-reducing hydrogenases in Methanosarcina barkeri. Archives of Microbiology, 1998, 169(3): 201-205. |
| [83] | Bingemann R, Klein A. Conversion of the central[4Fe-4S] cluster into a[3Fe-4S] cluster leads to reduced hydrogen-uptake activity of the F420-reducing hydrogenase of Methanococcus voltae. European Journal of Biochemistry, 2000, 267: 6612-6618. DOI:10.1046/j.1432-1327.2000.01755.x |
| [84] | Vitt S, Ma K, Warkentin E, Moll J, Pierik AJ, Shima S, Ermler U. The F420-reducing[NiFe]-hydrogenase complex from Methanothermobacter marburgensis, the first X-ray structure of a group 3 family member. Journal of Molecular Biology, 2014, 426(15): 2813-2826. DOI:10.1016/j.jmb.2014.05.024 |
| [85] | Mills DJ, Vitt S, Strauss M, Shima S, Vonck J. De novo modeling of the F420-reducing[NiFe]-hydrogenase from a methanogenic archaeon by cryo-electron microscopy. Elife, 2013, 2: 1-21. |
| [86] | Thauer RK. Biochemistry of methanogenesis:a tribute to Marjory Stephenson. Microbiology, 1998, 144: 2377-2406. DOI:10.1099/00221287-144-9-2377 |
| [87] | Thauer RK. Sulfate-reducing bacteria, Biotechnology Handbook. New York: Springer, 1995: 33-48. |
| [88] | Ma K, Thauer RK. Purification and properties of N5, N10-methylenetetrahydromethanopterin reductase from Methanobacterium thermoautotrophicum (strain Marburg). Biotechnology and Bioengineering, 1990, 191(1): 187-193. |
| [89] | Ma K, Linder D, Stetter KO, Thauer RK. Purification and properties of N5, N10-methylenetetrahydromethanopterin reductase (coenzyme F420-dependent) from the extreme thermophile Methanopyrus kandleri. Archives of Microbiology, 1991, 155(6): 593-600. DOI:10.1007/BF00245355 |
| [90] | Shima S, Warkentin E, Grabarse W, Sordel M, Wicke M, Thauer RK, Ermler U. Structure of coenzyme F420 dependent methylenetetrahydromethanopterin reductase from two methanogenic archaea. Journal of Molecular Biology, 2000, 300(4): 935-950. |
| [91] | Fisher AJ, Thompson TB, Thoden JB, Baldwin TO, Rayment I. The 1.5-? resolution crystal structure of bacterial luciferase in low salt conditions. Journal of Biology Chemistry, 1996, 271(36): 21956-21968. DOI:10.1074/jbc.271.36.21956 |
| [92] | Aufhammer SW, Warkentin E, Ermler U, Hagemeier CH, Thauer RK, Shima S. Crystal structure of methylenetetrahydromethanopterin reductase (Mer) in complex with coenzyme F420:architecture of the F420/FMN binding site of enzymes within the nonprolyl cis-peptide containing bacterial luciferase family. Protein Science, 2005, 14(7): 1840-1849. DOI:10.1110/ps.041289805 |
| [93] | Aufhammer SW, Warkentin E, Berk H, Shima S, Thauer RK, Ermler U. Coenzyme binding in F420-dependent secondary alcohol dehydrogenase, a member of the bacterial luciferase family. Structure, 2004, 12(3): 361-370. |
| [94] | Fischer R, Gartner P, Yeliseev A, Thauer RK. N5-methyltetrahydromethanopterin:coenzyme M methyltransferase in methanogenic archaebacteria is a membrane protein. Archives of Microbiology, 1992, 158(3): 208-217. DOI:10.1007/BF00290817 |
| [95] | Weiss DS, Gartner P, Thauer RK. The energetics and sodiumion dependence of N5-methyltetrahydromethanopterin:coenzyme M methyltransferase studied with cob(Ⅰ)alamin as methyl acceptor and methylcob(Ⅲ)alamin as methyl donor. European Journal of Biochemistry, 1994, 226(3): 799-809. DOI:10.1111/j.1432-1033.1994.00799.x |
| [96] | Müller V, Blaut M, Gottschalk G. The transmembrane electrochemical gradient of Na+ as driving force for methanol oxidation in Methanosarcina barkeri. European Journal of Biochemistry, 1988, 172(3): 601-606. |
| [97] | Peinemann S, Blaut M, Gottschalk G. ATP synthesis coupled to methane formation from methyl-CoM and H2 catalyzed by vesicles of the methanogenic bacterial strain G?1. European Journal of Biochemistry, 1989, 186(1): 175-180. |
| [98] | Becher B, Müller V, Gottschalk G. N5-methyl-tetrahydromethanopterin:coenzyme M methyltransferase of Methanosarcina strain G? 1 is an Na(+)-translocating membrane protein. Journal of Bacteriology, 1992, 174(23): 7656-7660. DOI:10.1128/JB.174.23.7656-7660.1992 |
| [99] | Lienard T, Becher B, Marschall M, Bowien S, Gottschalk G. Sodium ion translocation by N5-methyltetrahydromethanopterin:coenzyme M methyltransferase from Methanosarcina mazei G? 1 reconstituted in ether lipid liposomes. European Journal of Biochemistry, 1996, 239(3): 857-864. DOI:10.1111/j.1432-1033.1996.0857u.x |
| [100] | Harms U, Thauer RK. Identification of the active site histidine in the corrinoid protein MtrA of the energy-conserving methyltransferase complex from Methanobacterium thermoautotrophicum. European Journal of Biochemistry, 1997, 250(3): 783-788. DOI:10.1111/j.1432-1033.1997.00783.x |
| [101] | Harms U, Weiss DS, Gartner P, Linder D, Thauer RK. The energy conserving N5-Methyltetrahydromethanopterin:coenzyme M methyltransferase complex from Methanobacterium thermoautotrophicum is composed of eight different subunits. European Journal of Biochemistry, 1995, 228(3): 640-648. DOI:10.1111/j.1432-1033.1995.0640m.x |
| [102] | Gartner P, Ecker A, Fischer R, Linder D, Fuchs G, Thauer RK. Purification and properties of N5-methyltetrahydromethanopterin:coenzyme M methyltransferase from Methanobacterium thermoautotrophicum. European Journal of Biochemistry, 1993, 213(1): 537-545. |
| [103] | Harms U, Thauer RK. Methylcobalamin:coenzyme M methyltransferase isoenzymes MtaA and MtbA from Methanosarcina barkeri-cloning, sequencing and differential transcription of the encoding genes, and functional overexpression of the mtaA gene in Escherichia coli. European Journal of Biochemistry, 1996, 235(3): 653-659. DOI:10.1111/j.1432-1033.1996.00653.x |
| [104] | Harmer U, Thauer RK. The corrinoid-containing 23-kDa subunit MtrA of the energy-conserving N5-methyltetrahydromethanopterin:coenzyme M methyltransferase complex from Methanobacterium thernionutotrophicum-EPR spectroscopic evidence for a histidine residue as a cobalt ligand of the cobamide. European Journal of Biochemistry, 1996, 241: 149-154. DOI:10.1111/j.1432-1033.1996.0149t.x |
| [105] | Hippler B, Thauer RK. The energy conserving methyltetrahydromethanopterin:coenzyme M methyltransferase complex from methanogenic archaea:function of the subunit MtrH. FEBS Letters, 1999, 449(2-3): 165-168. DOI:10.1016/S0014-5793(99)00429-9 |
| [106] | Vallee BL, Auld DS. Active-site zinc ligands and activated H2O of zinc enzymes. Proceedings of the National Academy of Sciences, 1990, 89: 220-224. |
| [107] | Berardino MD, Dimroth P. Aspartate 203 of the oxaloacetate decarboxylase β-subunit catalyses both the chemical and vectorial reaction of the Na+ pump. The EMBO Journal, 1996, 15(8): 1842-1849. DOI:10.1002/j.1460-2075.1996.tb00534.x |
| [108] | Gartner P, Weiss DS, Harms U, Thauer RK. N5-methyltetrahydromethanopterin:coenzyme M methyltransferase from Methanobacterium thermoautotrophicum. European Journal of Biochemistry, 1994, 226(2): 465-472. |
| [109] | Friedmann HC, Klein A, Thauer RK. Structure and function of the nickel porphinoid, coenzyme F430 and of its enzyme, methyl coenzyme M reductase. FEMS Microbiology Reviews, 1990, 7(3): 339-348. |
| [110] | Grabarse WG, Mahlert F, Shima S, Thauer RK, Ermler U. Comparison of three methyl-coenzyme M reductases from phylogenetically distant organisms:unusual amino acid modification, conservation and adaptation. Journal of Molecular Biology, 2000, 303(2): 329-344. |
| [111] | Evans PN, Boyd JA, Leu AO, Woodcroft BJ, Parks DH, Hugenholtz P, Tyson GW. An evolving view of methane metabolism in the Archaea. Nature Revivews Microbiology, 2019, 17: 219-232. DOI:10.1038/s41579-018-0136-7 |
| [112] | Ermler U, Grabarse W, Shima S, Goubeaud M, Thauer RK. Crystal structure of methyl coenzyme M reductase:the key enzyme of biological methane formation. Science, 1997, 278(5342): 1457-1462. DOI:10.1126/science.278.5342.1457 |
| [113] | Grabarse W, Mahlert F, Duin EC, Goubeaud M, Shima S, Thauer RK, Lamzin V, Ermler U. On the mechanism of biological methane formation:structural evidence for conformational changes in methyl-coenzyme M reductase upon substrate binding. Journal of Molecular Biology, 2001, 309(1): 315-330. DOI:10.1006/jmbi.2001.4647 |
| [114] | Mahlert F, Grabarse W, Kahnt J, Thauer RK, Duin EC. The nickel enzyme methyl-coenzyme M reductase from methanogenic archaea:in vitro interconversions among the EPR detectable MCR-red1 and MCR-red2 states. Journal of Biological Inorganic Chemistry, 2002, 7(1-2): 101-112. DOI:10.1007/s007750100270 |
| [115] | Finazzo C, Harmer J, Jaun B, Duin EC, Mahlert F, Thauer RK, Doorslaer VS, Schweiger A. Coenzyme B induced coordination of coenzyme M via its thiol group to Ni(Ⅰ) of F430 in active methyl-coenzyme M reductase. Journal of American Chemical Society, 2003, 125(17): 4988-4989. DOI:10.1021/ja0344314 |
| [116] | Finazzo C, Harmer J, Jaun B, Duin EC, Mahlert F, Thauer RK, Doorslaer VS, Schweiger A. Characterization of the MCRred2 form of methyl-coenzyme M reductase:a pulse EPR and ENDOR study. Journal of Biological Inorganic Chemistry, 2003, 8(5): 586-593. DOI:10.1007/s00775-003-0450-y |
| [117] | Duin EC, Signor L, Piskorski R, Mahlert F, Clay MD, Goenrich M, Thauer RK, Jaun B, Johnson MK. Spectroscopic investigation of the nickel-containing porphinoid cofactor F430. Comparison of the free cofactor in the +1, +2 and +3 oxidation states with the cofactor bound to methyl-coenzyme M reductase in the silent, red and ox forms. Journal of Biological Inorganic Chemistry, 2004, 9(4): 563-576. |
| [118] | Goenrich M, Mahlert F, Duin EC, Bauer C, Jaun B, Thauer RK. Probing the reactivity of Ni in the active site of methyl-coenzyme M reductase with substrate analogues. Journal of Biological Inorganic Chemistry, 2004, 9(6): 691-705. DOI:10.1007/s00775-004-0552-1 |
| [119] | Wagner T, Kahnt J, Ermler U, Shima S. Didehydroaspartate modification in methyl-coenzyme M reductase catalyzing methane formation. Angewandte Chemie International Edition, 2016, 55(36): 10630-10633. DOI:10.1002/anie.201603882 |
| [120] | Shima S, Krueger M, Weinert T, Demmer U, Kahnt J, Thauer RK, Ermler U. Structure of a methyl-coenzyme M reductase from Black Sea mats that oxidize methane anaerobically. Nature, 2012, 481(7379): 98-101. DOI:10.1038/nature10663 |
| [121] | Kahnt J, Buchenau B, Mahlert F, Kruger M, Shima S, Thauer RK. Post-translational modifications in the active site region of methyl-coenzyme M reductase from methanogenic and methanotrophic archaea. FEBS Journal, 2007, 274(18): 4913-4921. DOI:10.1111/j.1742-4658.2007.06016.x |
| [122] | Thauer RK. Methyl (alkyl)-coenzyme M reductases:nickel F430-containing enzymes involved in anaerobic methane formation and in anaerobic oxidation of methane or of short chain alkanes. Biochemistry, 2019: 1-23. |
| [123] | Selmer T, Kahnt J, Goubeaud M, Shima S, Grabarse W, Ermler U, Thauer RK. The biosynthesis of methylated amino acids in the active site region of methyl-coenzyme M reductase. Journal of Biological Chemistry, 2000, 275(6): 3755-3760. DOI:10.1074/jbc.275.6.3755 |
| [124] | Harmer J, Finazzo C, Piskorski R, Bauer C, Jaun B, Duin EC, Goenrich M, Thauer RK, Van Doorslaer S, Schweiger A. Spin density and coenzyme M coordination geometry of the ox1 form of methyl-coenzyme M reductase:a pulse EPR study. Journal of American Chemical Society, 2005, 127(50): 17744-17755. DOI:10.1021/ja053794w |
| [125] | Harmer J, Finazzo C, Piskorski R, Ebner S, Duin EC, Goenrich M, Thauer RK, Reiher M, Schweiger A, Hinderberger D, Jaun B. A nickel hydride complex in the active site of methyl-coenzyme M reductase:implications for the catalytic cycle. Journal of American Chemical Society, 2008, 130(33): 10907-10920. DOI:10.1021/ja710949e |
| [126] | Wongnate T, Sliwa D, Ginovska B, Smith D, Wolf MW, Lehnert N, Raugei S, Ragsdale SW. The radical mechanism of biological methane synthesis by methylcoenzyme M reductase. Science, 2016, 352(6288): 953-958. DOI:10.1126/science.aaf0616 |
| [127] | Pelmenschikov V, Blomberg MRA, Siegbahn PEM, Crabtree RH. A mechanism from quantum chemical studies for methane formation in methanogenesis. Journal of American Chemical Society, 2002, 124: 4039-4049. DOI:10.1021/ja011664r |
| [128] | Setzke E, Hedderich R, Heiden S, Thauer RK. H2:heterodisulfide oxidoreductase complex from Methanobacterium thermoautotrophicum composition and properties. European Journal of Biochemistry, 1994, 220(1): 139-148. DOI:10.1111/j.1432-1033.1994.tb18608.x |
| [129] | Kaster AK, Moll J, Parey K, Thauer RK. Coupling of ferredoxin and heterodisulfide reduction via electron bifurcation in hydrogenotrophic methanogenic archaea. Proceedings of the National Academy of Sciences, 2011, 108(7): 2981-2986. DOI:10.1073/pnas.1016761108 |
| [130] | Heiden S, Hedderich R, Setzke E, Thauer RK. Purification of a cytochrome b containing H2:heterodisulfide oxidoreductase complex from membranes of Methanosarcina barkeri. European Journal of Biochemistry, 1993, 213(1): 529-535. DOI:10.1111/j.1432-1033.1993.tb17791.x |
| [131] | Heiden S, Hedderich R, Setzke E, Thauer RK. Purification of a two-subunit cytochrome-b-containing heterodisulfide reductase from methanol-grown Methanosarcina barkeri. European Journal of Biochemistry, 1994, 221(2): 855-861. DOI:10.1111/j.1432-1033.1994.tb18800.x |
| [132] | Deppenmeier U, Blaut M, Schmidt B, Gottschalk G. Purification and properties of a F420-nonreactive, membrane-bound hydrogenase from Methanosarcina strain G?1. Archives of Microbiology, 1992, 157(6): 505-511. |
| [133] | Deppenmeier U, Blaut M, Lentes S, Herzberg C, Gottschalk G. Analysis of the vhoGAC and vhtGAC operons from Methanosarcina mazei strain G?1, both encoding a membrane-bound hydrogenase and a cytochrome b. European Journal of Biochemistry, 1995, 227(1/2): 261-269. |
| [134] | Deppenmeier U. Different structure and expression of the operons encoding the membrane-bound hydrogenases from Methanosarcina mazei G?1. Archives of Microbiology, 1995, 164(5): 370-376. DOI:10.1007/BF02529985 |
| [135] | Kunkel A, Vaupel M, Heim S, Thauer RK, Hedderich R. Heterodisulfide reductase from methanol-grown cells of Methanosarcina barkeri is not a flavoenzyme. European Journal of Biochemistry, 1997, 244(1): 226-234. DOI:10.1111/j.1432-1033.1997.00226.x |
| [136] | Tersteegen A, Hedderich R. Methanobacterium thermoautotrophicum encodes two multisubunit membrane-bound[NiFe] hydrogenases. European Journal of Biochemistry, 1999, 264: 930-943. DOI:10.1046/j.1432-1327.1999.00692.x |
| [137] | Bott M, Thauer RK. Proton-motive-force-driven formation of CO from CO2 and H2 in methanogenic bacteria. European Journal of Biochemistry, 1987, 168(2): 407-412. DOI:10.1111/j.1432-1033.1987.tb13434.x |
| [138] | Kunkel A, Vorholt JA, Thauer RK, Hedderich R. An Escherichia coli hydrogenase-3-type hydrogenase in methanogenic archaea. European Journal of Biochemistry, 1998, 252(3): 467-476. DOI:10.1046/j.1432-1327.1998.2520467.x |
| [139] | Meuer J, Bartoschek S, Koch J, Kunkel A, Hedderich R. Purification and catalytic properties of ech hydrogenase from Methanosarcina barkeri. European Journal of Biochemistry, 1999, 265(1): 325-335. DOI:10.1046/j.1432-1327.1999.00738.x |
