 下载:
下载:  点击查看大图
点击查看大图全文HTML
--> --> --> 随着现代社会工业化的高速发展,天然水中砷的含量迅速增加[1]。水中砷污染已成为全社会亟待解决的重要环境问题之一[2]。目前,常用的除砷方法主要有混凝沉淀法、离子交换法、膜分离法、吸附法[3-6]等。混凝沉淀法中预氧化过程会增加后续污水处理的问题[3];离子交换法须采用特殊材料,工艺流程较繁琐[4];膜分离法易出现膜污染等问题,该法适用于处理量相对较少的砷污染水[5]。因此,吸附法在处理效率高、操作简单和适用性强等方面的优势越来越受到人们的重视[6]。多壁碳纳米管(CNTs)具有比表面积大、孔隙结构丰富、表面易于被修饰等特点,近年来在制备复合吸附材料方面已有诸多的应用[7]。IBRAHIM等[8]研究了深共熔溶剂(DESs)功能化CNTs对水中甲基橙的吸附性能;李德云等[9]在研究改性CNTs吸附水中亚甲基蓝过程时提到Cu/CuO改性CNTs对亚甲基蓝的吸附效果优于原始CNTs;杨爱丽等[10]在研究臭氧氧化改性CNTs对铀的吸附去除时提到,含氧量的增加会显著提高改性CNTs对铀的去除率。
目前,对水中砷去除的研究多集中在吸附剂的氧化改性以及对As(Ⅴ)的吸附去除上,对As(Ⅲ)的削减效果分析和去除机制等方面的研究相对较少[11]。本研究从活化CNTs管壁接枝点位、负载高活性基团的酰胺类材料的角度出发,采用强酸氧化和酰胺化等方法制备了酰胺化/氧化碳纳米管-聚苯胺(NMCNTs-PANI),并探究该复合材料对水中As(Ⅲ)的吸附性能。
1.1. 实验原料
基准纯三氧化二砷(As2O3,99%);高锰酸钾(KMnO4)、硫酸(98%H2SO4)、氯化亚砜(SOCl2)、N-聚乙烯吡咯烷酮、三乙胺、乙二胺、N, N-二环己基碳二亚胺、聚苯胺(PANI)、丙酮均为分析纯。碳纳米管原粉(CNTs)购自苏州石墨烯科技有限公司,采用改性催化碳气相沉积法(CVD)制备。1.2. 实验装置
真空干燥箱(DZF-6050,上海福絮仪器厂);磁力加热搅拌器(HJ-4,江苏新瑞仪器厂);pH计(pHS-25型,上海仪电公司);原子荧光光度计(AFS-933,美国Thermo Fisher Scientific公司);扫描电子显微镜(TM3030,日立公司);比表面孔径分布测定仪(TRISTAR3000,美国MICROMERITICS公司);X射线衍射仪(D2 PHASER,德国布鲁克AXS公司);傅里叶变换红外光谱仪(EQUINOX 55,美国赛默飞公司);拉曼光谱仪(LABRAM ARAMIS,法国HORIBA公司)。实验采用恒温振荡器,实验装置及实物照片见图1。设置温度为25 ℃,转速为180 r·min?1,避光运行,振荡后静置,抽滤。
1.3. 实验方法
冰水浴中,称取1 g CNTs、3 g KMnO4加入到30 mL浓硫酸中,搅拌2 h;升温至40 ℃,搅拌反应4 h;置于70 ℃磁力搅拌器中搅拌0.3 h;加入适量FeCl3及FeSO4,溶解后抽滤,将所得固体烘干备用。取1 g上述烘干后的固体置于250 mL烧杯中,加入30 mL SOCl2、2 mL DMF和1 g N-聚乙烯吡咯烷酮,搅拌均匀,缓慢加入13 mL三乙胺、7 mL乙二胺,升温至80 ℃,搅拌0.5 h,加入适量N,N-二环己基碳二亚胺,超声0.5 h;置于摇床振荡,洗涤抽滤,干燥得到黑色固体[12-13]。取0.5 g上述黑色固体分散在水溶液中,与50 mL含有2 g PANI的丙酮溶液混合,置于60 ℃水浴锅反应1 h,超声0.5 h;移至摇床振荡20 h,洗涤抽滤,110 ℃真空干燥箱烘干,得到NMCNTs-PANI。采用SEM观察表面特征;比表面积分析仪测定比表面积和孔径分布;X射线衍射仪(XRD)分析物相组成[14];傅里叶变换红外光谱仪(FT-IR)定性分析表面的化学键和官能团信息[15];拉曼光谱(Raman)分析材料的晶体结构[16]。
1.4. 分析方法
配置10 mg·L?1 As2O3溶液,取7份100 mL置于系列250 mL锥形瓶中,分别投加50 mg NCNTs/PANI,反应温度为25 ℃,设置不同的接触时间并置于振荡器中以180 r·min?1转速振荡,静置后过滤,用原子荧光光度计测定滤液中As(Ⅲ)浓度并计算吸附量。As(Ⅲ)初始浓度、初始pH、共存阴离子的考察过程与接触时间类似。溶液pH采用NaOH及HCl调节。吸附量、去除率依次按式(1)和式(2)计算。
式中:q为NMCNTs-PANI对溶液中As(Ⅲ)的吸附量,mg·g?1;η为去除率;C0为As(Ⅲ)初始浓度,mg·L?1;C为滤液中As(Ⅲ)浓度,mg·L?1;V为溶液体积,L;m为NMCNTs-PANI投加量,g。
2.1. 对NMCNTs-PANI的表征
图2是改性前后CNTs的SEM图。从图2(a)中可以看出,CNTs原粉是相互缠绕在一起的管状结构,其管壁无序、混乱;图2(b)和图2(c)显示,NMCNTs-PANI管状结构部分消失,管间缺陷增大,并出现聚集现象。该现象可能是因为引入某种活性剂或CNTs表面功能化导致CNTs结构受到破坏[17]。
改性前后CNTs物理性质见表1。可以看出,CNTs原粉、NMCNTs-PANI和吸附后材料的孔径为3.58~13.84 nm,说明样品为介孔材料;与CNTs原粉相比,NMCNTs-PANI和吸附后材料的比表面积稍有减小,而孔容和平均孔径均大大增加。H2SO4和KMnO4均具有强氧化性,可以氧化掉CNTs上部分管状结构,导致CNTs局部坍塌;或者少部分孔道在反应过程中被堵塞,碳管难以拉伸,从而减小NMCNTs-PANI和吸附后材料的比表面积;同时,也有部分管内杂质被溶解,疏通了CNTs孔道结构;或者使一些内部封闭的孔道被打开,导致NMCNTs-PANI和吸附后材料的孔容和平均孔径均增大[18]。
图3是改性前后CNTs的XRD图谱。从图3可以看出,改性前后表征CNTs晶面(002)的衍射角在26.0°处位置没发生变化[14];NMCNTs-PANI和吸附后材料衍射峰的强度均小于CNTs原粉,说明CNTs改性后结晶度变差。此外,NMCNTs-PANI中体现PANI的衍射特征峰在15.3°和22.6°凸起,可能原因是PANI大部分以无定性态存在[19]。
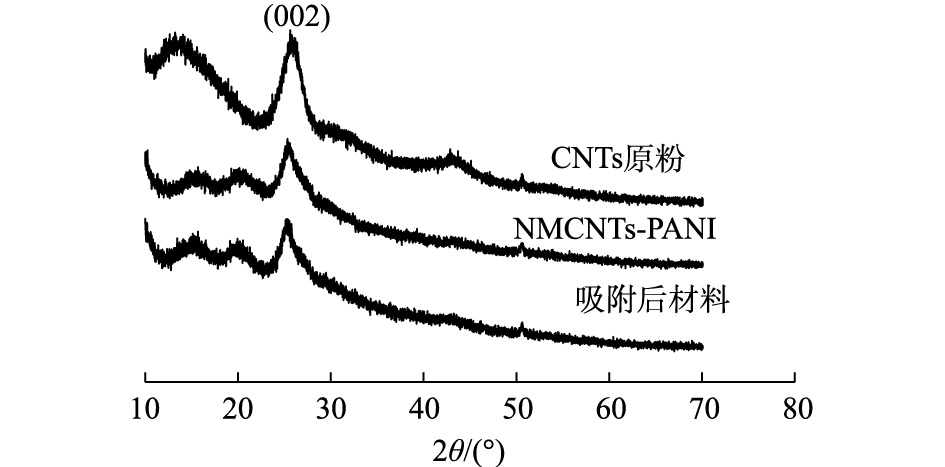
图4是改性前后CNTs的红外光谱图。可以看出,与CNTs原粉相比,NMCNTs-PANI和吸附后材料在500~4 000 cm?1出现多个吸收峰。其中NMCNTs-PANI的红外光谱中1 134 cm?1是O—H的振动峰[20];1 192 cm?1是环氧基C—O—C引起的伸缩振动峰[21];1 701 cm?1处的吸收峰为羰基C=O伸缩振动[22];1 257 cm?1处的红外吸收峰归属于C—O的伸缩振动[23];1 400 cm?1处是C—N键的伸缩振动特征峰,1 654 cm?1处是C=N的弯曲振动峰[24],以上说明制备过程中引入了羧基、羟基、环氧基等功能性基团;3 278 cm?1处归属于N—H的伸缩振动峰,该峰和C=O、C—N表明酰胺基团负载在材料表面;786 cm?1处的特征峰归属于Fe—O键的伸缩振动[25],这表明铁离子成功接枝在材料表面。吸附后材料的FT-IR图出现了位于582 cm?1处的金属配位体振动(Metal-N)[25],可能原因是水中As(Ⅲ)吸附于NMCNTs-PANI表面。
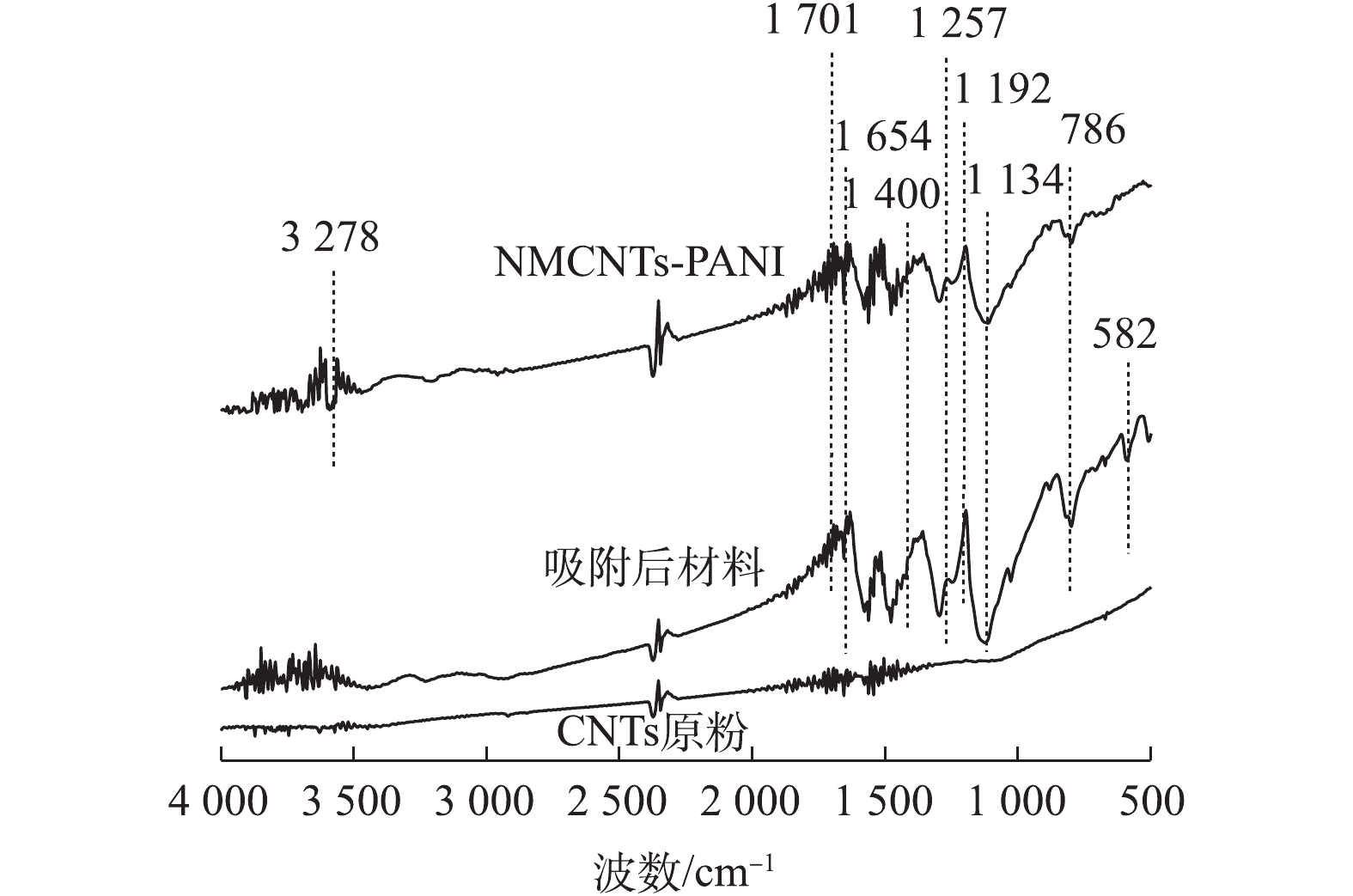
图5是改性前后CNTs的Raman光谱图。可以看出,改性前后CNTs均出现了2个典型特征峰,其中D峰位于1 340 cm?1左右,该缺陷峰反映的是碳材料中C—C的无序振动,峰值反映出CNTs结构的缺陷性和混乱程度;在1 580 cm?1左右的G峰是碳结构sp2的特征峰,用以说明CNTs的结晶程度,2个峰的强度比值ID/IG可以衡量CNTs结构缺陷密度,比值越低,结晶越完整[16]。计算表明,NMNCTs-PANI和吸附后材料的ID/IG分别为1.05、1.06,均高于CNTs原粉ID/IG(1.03),说明复合材料结构的无序性增加。综上所述,改性后CNTs的结构发生了变化,但XRD及Raman分析结果一致表明,CNTs的晶体结构并没有被破坏。

2.2. 静态吸附实验
图6为接触时间对吸附量的影响结果。当接触时间从5 min延至20 min时,吸附量从4 mg·g?1快速上升到13 mg·g?1;随后吸附量缓慢增加;在30 min左右达到吸附平衡。这是由于初始阶段NMCNTs-PANI表面高能量的活性位点迅速被占领,吸附速率较高,此时溶液中大部分As(Ⅲ)占据了表面活性位点;后期由低能量的活性位点继续吸附,吸附速率随之降低。综合考虑吸附速率,接触时间选择30 min。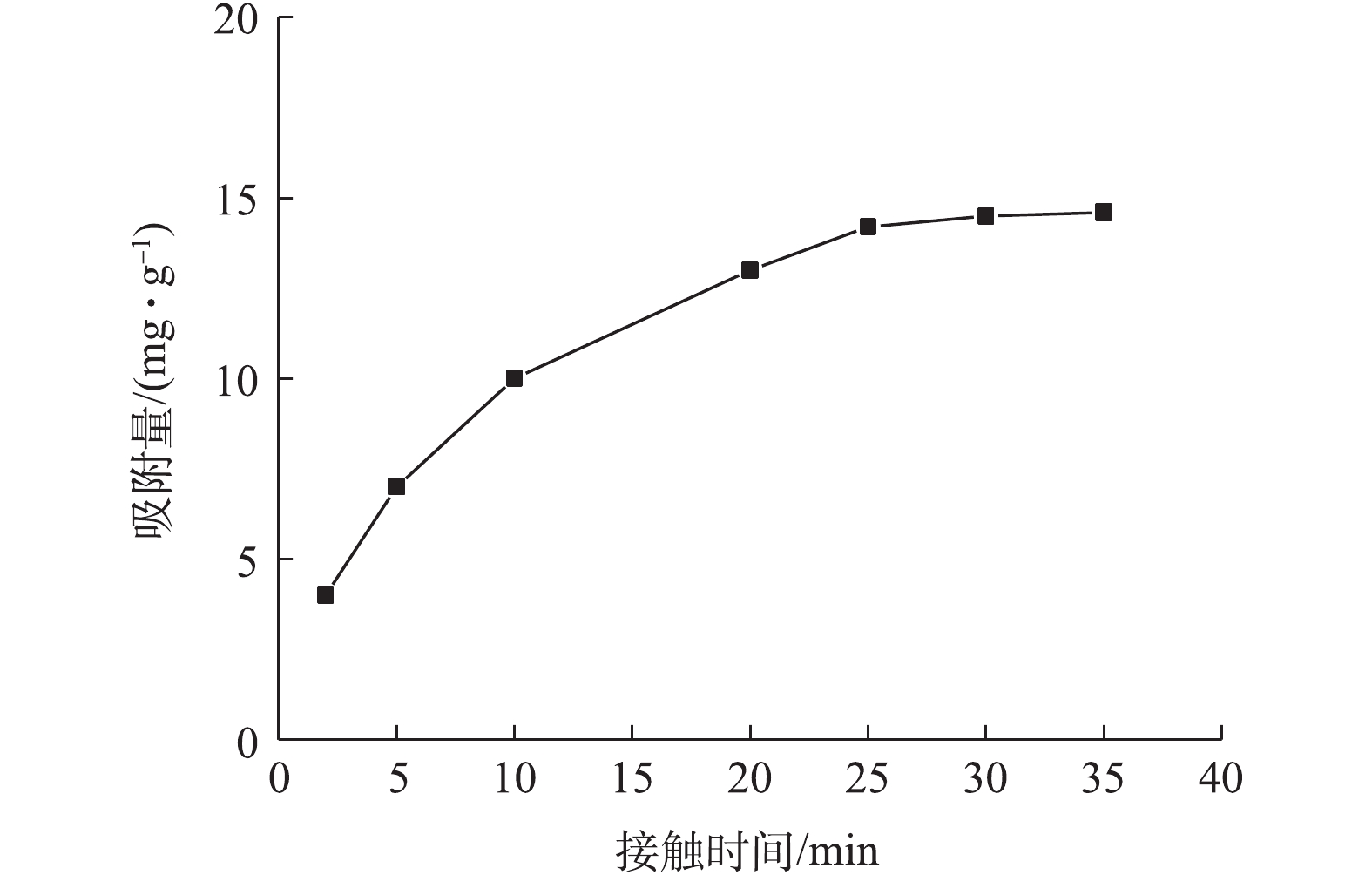
将数据代入准一级动力学方程和准二级动力学方程进行拟合,准一级动力学方程[26]和准二级动力学方程[27]见式(3)和式(4)。
式中:t为吸附过程某一时刻,min;qe为平衡吸附量,mg·g?1;qt为t时刻吸附量,mg·g?1;k1为一级动力学速率常数,min?1;k2为二级动力学速率常数,g·(mg·min)?1。
图7为采用不同动力学方程对动力学实验数据的拟合结果,拟合参数见表2。由图7可知,准一级动力学方程和准二级动力学方程均能较好描述NMCNTs-PANI吸附As(Ⅲ)的动力学过程。但表2显示准二级动力学方程R2为0.994,准一级动力学方程R2为0.983,故准二级动力学方程更为符合NMCNTs-PANI吸附As(Ⅲ)的动力学过程,即NMCNTs-PANI吸附水中As(Ⅲ)主要以化学吸附为主[28]。吸附过程中物理吸附主要是分子间作用力;化学吸附可能涉及离子交换或NMCNTs-PANI上活性基团与As(Ⅲ)形成化学键的吸附[28],尤其是改性材料表面所带有的酰胺基团,具有较高反应活性,可与As(Ⅲ)发生结合[29]。
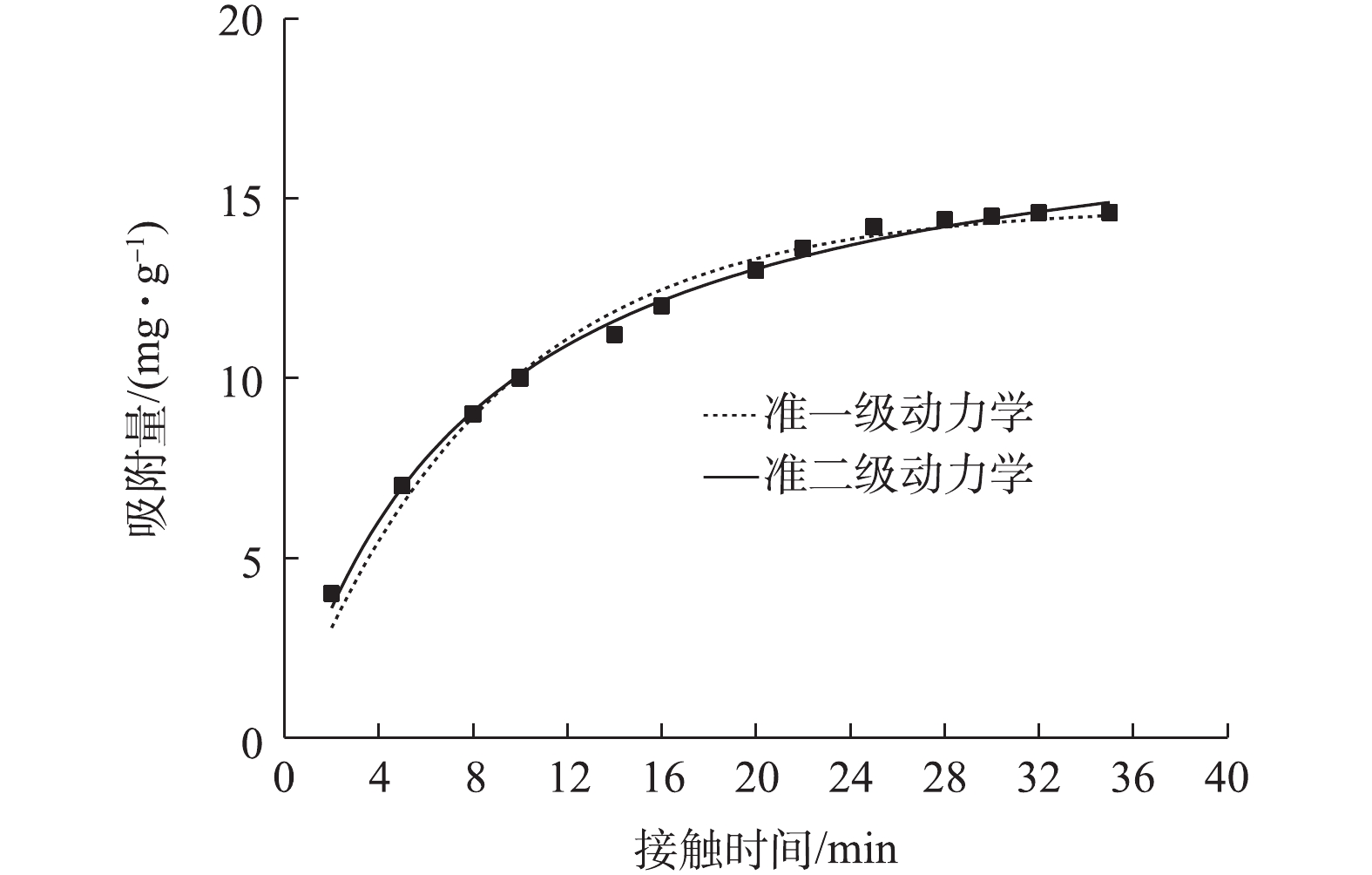
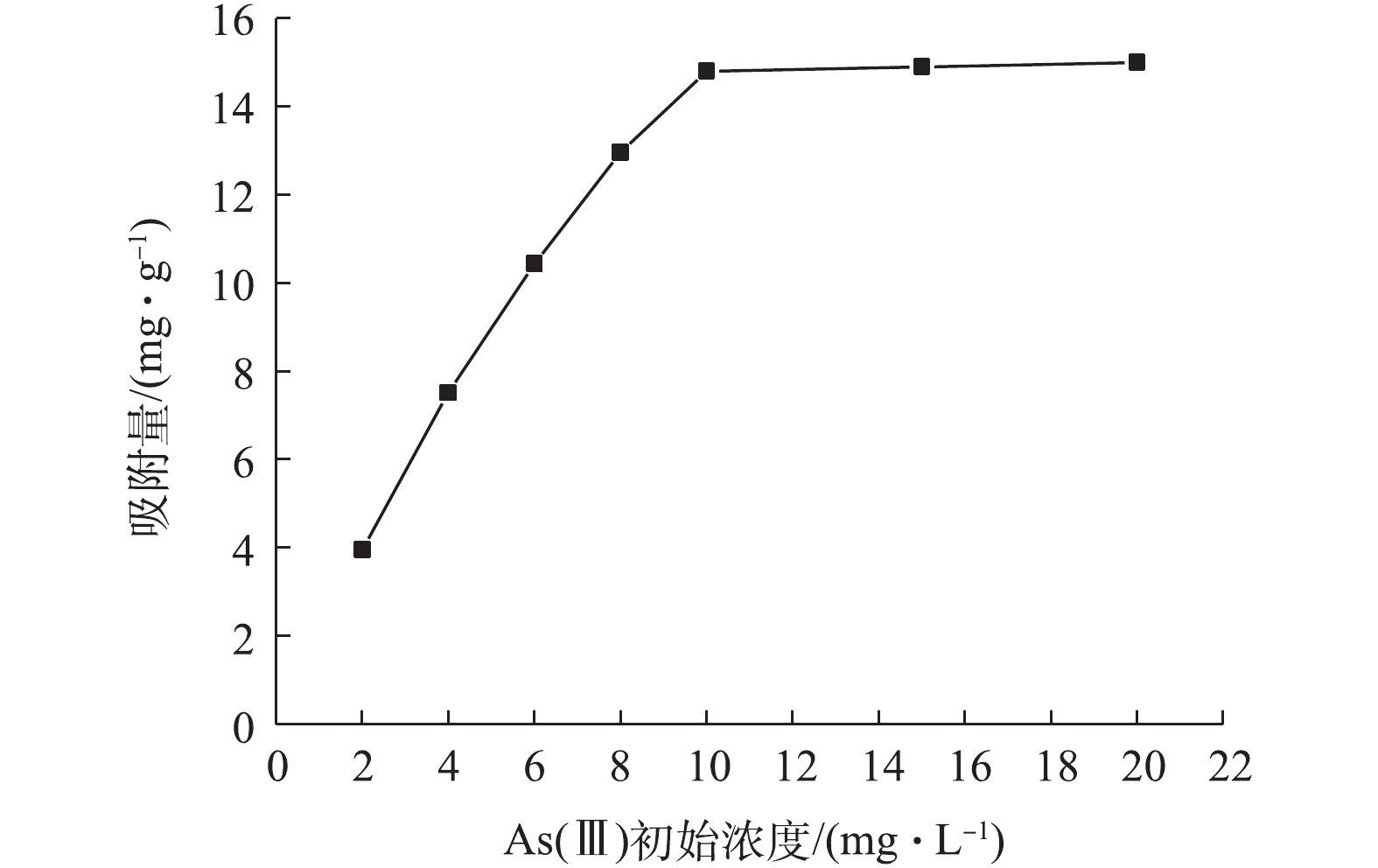
图8为As(Ⅲ)初始浓度对吸附量的影响结果。由图8可知,As(Ⅲ)初始浓度从1 mg·L?1增加到8 mg·L?1时,吸附量从1.98 mg·g?1增加到12.96 mg·g?1;当As(Ⅲ)初始浓度达到10 mg·L?1以上时,NMCNTs-PANI对As(Ⅲ)吸附量几乎不再增加。这是因为随着As(Ⅲ)浓度的增加,NMCNTs-PANI与As(Ⅲ)之间的碰撞机率增大,导致吸附量不断增高;同时NMCNTs-PANI表面吸附位点数量有限,吸附位点被占据后不能继续吸附溶液中As(Ⅲ),NMCNTs-PANI逐渐达到吸附饱和状态。改性后CNTs表面官能团数量大大增加,水中As(Ⅲ)可以与羟基、羧基、酰胺键等官能团发生络合,极大地促进了NMCNTs-PANI对As(Ⅲ)的吸附。
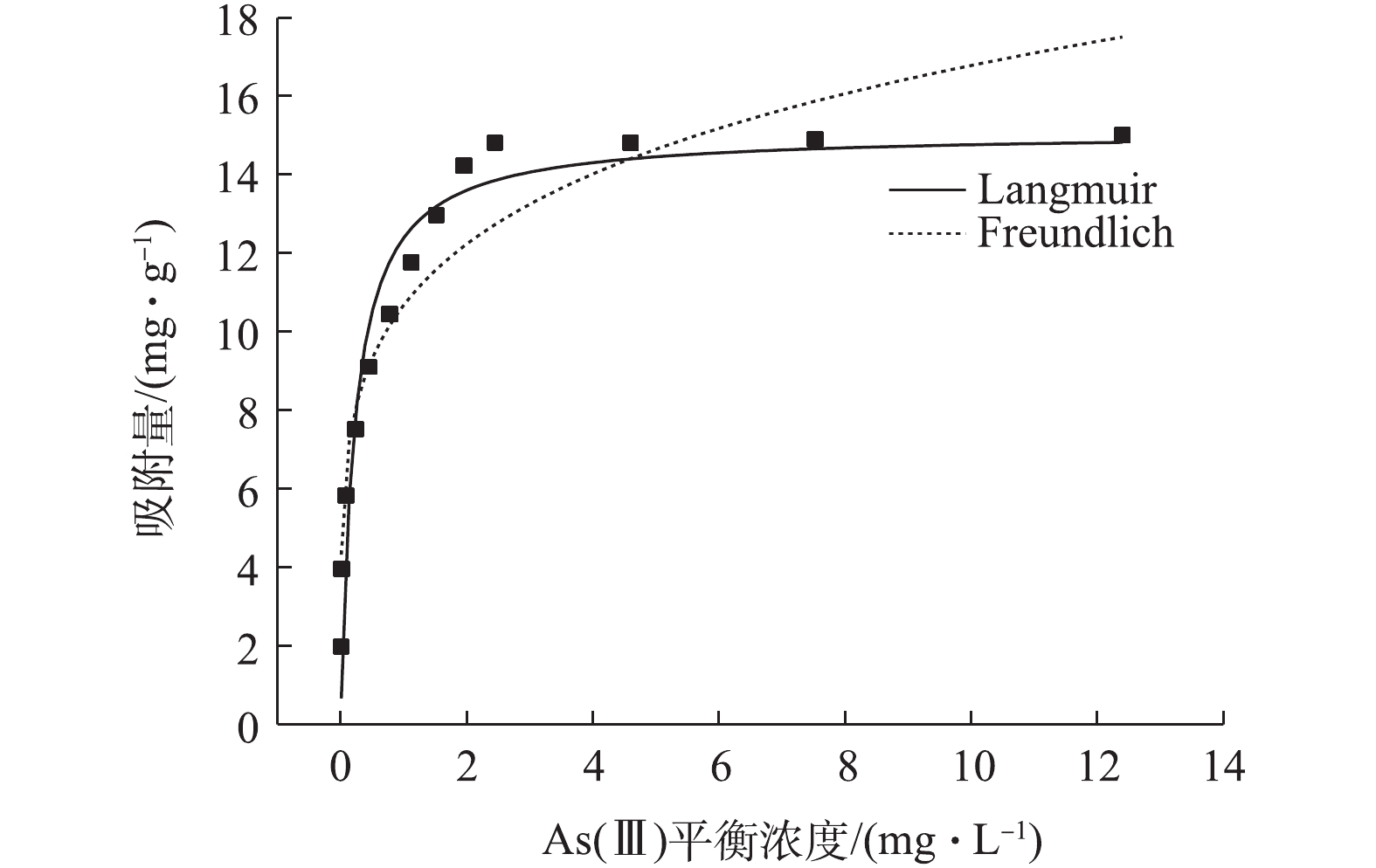
将数据代入Langmuir等温线方程和Freundlich等温线方程进行拟合,前者常用于描述单分子层吸附;后则多用于描述复杂不均匀表面的多分子层吸附[30-31]。Langmuir方程[30]和Freundlich方程[31]见式(5)和式(6)。
式中:Ce为平衡浓度,mg·L?1;qe为平衡吸附量,mg·L?1;qm为饱和吸附量,mg·g?1;kL为Langmuir平衡常数,L·mg?1;kF为Freundlich常数mg·g?1;n为吸附强度。
图9是利用Langmuir、Freundlich等温线对实验数据的拟合结果,拟合参数见表3。由图9可知,Langmuir、Freundlich等温线均能较好描述NMCNTs-PANI吸附水中As(Ⅲ)过程,但表3显示,前者R2大于后者,说明Langmuir等温线能更好地描述NMCNTs-PANI吸附水中As(Ⅲ)过程。实验中NMCNTs-PANI对10 mg·L?1 As(Ⅲ)饱和吸附量为14.80 mg·g?1。有研究发现,除部分研究[32]针对含As(Ⅲ)地下水利用生物催化合成施氏矿物对As(Ⅲ)的饱和吸附量为67.70 mg·g?1外,大多数研究[33-35]中吸附剂对水中As(Ⅲ)的饱和吸附量为1.78~10.90 mg·g?1,如Fe-Mn双氧化物改性的硅藻土(1.68 mg·g?1)、针铁矿(0.38 mg·g?1)和赤铁矿(0.26 mg·g?1)。因此,相比其他研究的结果,本研究所用吸附剂的吸附性能更优。
溶液pH可影响溶液中As(Ⅲ)的存在形态及吸附剂表面官能团的性能[36]。图10为初始pH对As(Ⅲ)吸附量的影响结果。由图10可知,在较低pH(3.3~4.2)条件下,As(Ⅲ)吸附量从9.87 mg·g?1升至11.76 mg·g?1;在pH为4.2~6.5的弱酸性环境中,As(Ⅲ)的吸附量下降至9.92 mg·g?1;当pH升高至6.5~8.6时,吸附量达到最大值,为14.82 mg·g?1,此时,去除率为74.1%;继续提高pH,吸附量则再次降低。有研究[37]表明,在溶液pH<9.2时,As(Ⅲ)主要以中性H3AsO3形态存在(
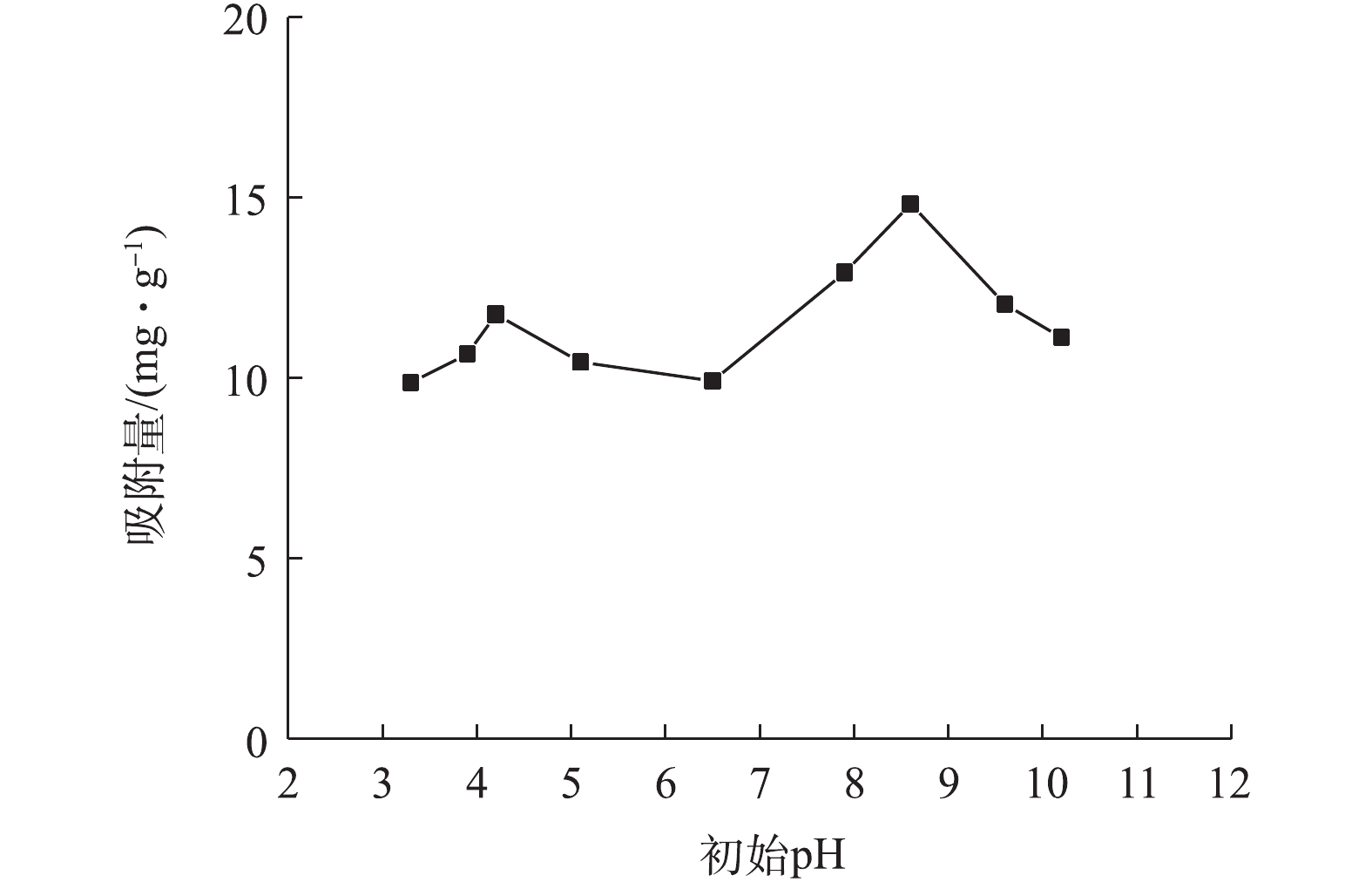
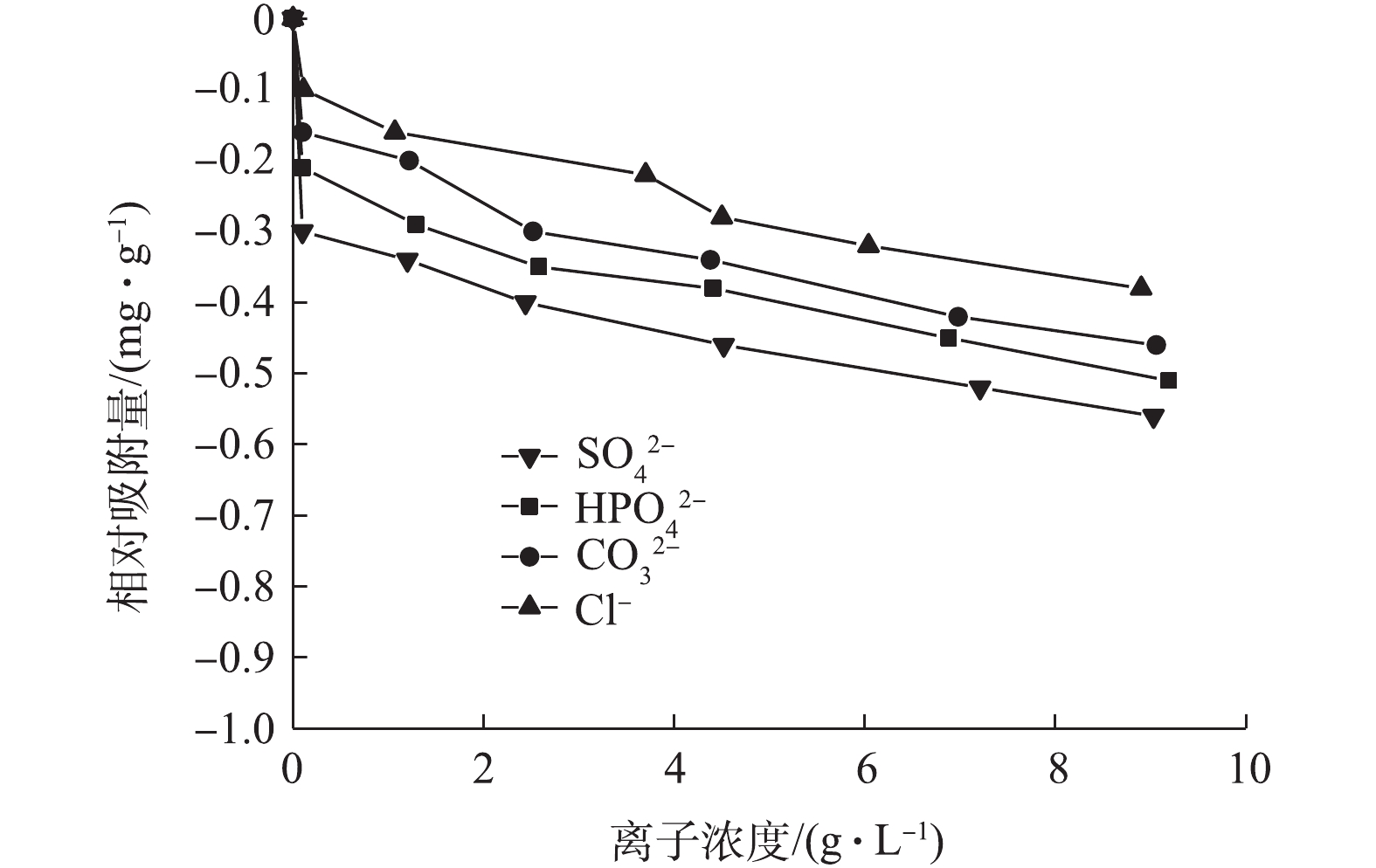
图11为共存阴离子对吸附量的影响结果。将Cl?、
2)动力学特征符合准一级动力学和准二级动力学方程;吸附等温线符合Langmuir模型,饱和吸附容量为14.80 mg·g?1。吸附动力学和等温线研究结果表明,NMCNTs-PANI吸附水中As(Ⅲ)的过程以化学吸附为主。
3)酰胺化/氧化碳纳米管-聚苯胺具有较佳的吸附性能,当微污染水中As(Ⅲ)浓度不超过饮用水标准50倍时,采用0.5 g·L?1 NMCNTs/PANI吸附处理可达到饮用水标准。此研究为解决饮用水中低浓度三价砷的污染问题提供了新方法。
参考文献
