 ,1,2, 杨小渝,1,2,*
,1,2, 杨小渝,1,2,*Integration and Optimization of Material Data Mining and Machine Learning Tools
Dong Jiayuan,1,2, Yang Xiaoyu,1,2,*通讯作者: * 杨小渝(E-mail:kxy@cnic.cn)
收稿日期:2020-03-25网络出版日期:2020-08-20
Received:2020-03-25Online:2020-08-20
作者简介 About authors

董家源,中国科学院计算机网络信息中心,在读硕士研究生,主要研究方向为材料信息学。
本文承担工作为:材料数据挖掘与机器学习工具以及相关算法的代码实现。
Dong Jiayuan is a master student at Computer Network Information Center of the Chinese Academy of Science. His main research interests are Materials informatics.
In this paper he undertakes the following tasks: code implementations of material data mining platform and related algorithms.
E-mail:

杨小渝,中国科学院计算机网络信息中心,研究员,主要研究方向为高通量材料计算,材料信息学。
本文承担工作为:想法思路的提出,材料数据挖掘与机器学习工具的架构设计、用户界面设计、相关算法的优化方法设计。
Prof. Xiaoyu Yang is a research fellow at Computer Network Information Center, the Chinese Academy of Sciences. His research interests include high-throughput materials simulation and materials informatics.
His work undertook in this paper includes: the proposal of the idea, system architectural design, user interface design, and associated processing logic design.
E-mail:

摘要
【目的】针对材料科学工作者开展机器学习工作门槛较高这一现状,本文基于MatCloud研发一个用户友好、自动化的材料数据挖掘与机器学习模块Auto-Mat。【方法】本文对MatMiner和scikit-learn中一些已有的获取数据的方法和机器学习算法进行了集成,并定义了数据字典以读取不同材料计算数据库的数据。同时,自主研发了一些特征筛选和处理方面的算法。【结果】能够提供一个具有可视化交互和展示界面的材料数据挖掘与机器学习模块,并将数据以统一的格式呈现。同时,自主研发的算法,对模型的性能均有一定提升。【局限】对于数据的获取,目前仅仅能获取到通过MatMiner API中的数据,相关代码的编写也完全和MatMiner API保持同步,因此可扩展性较差。而且,目前一些核心算法的执行速度有待提升。【结论】通过该模块与MatCloud的集成,用户可以“一站式”地读取Materials Project等几个主流数据库中的数据,并快速构建属于自己的材料数据挖掘与机器学习工作流程。并在最后通过2个案例的对比分析,说明了该模块对于降低用户开展材料数据挖掘与机器学习的使用门槛有着积极作用。
关键词:
Abstract
[Objective] Aiming at handling the current situation that there are high barriers impeding materials science researchers to take advantages of machine learning algorithms, this article focuses on developing a user-friendly and highly automated machine learning system for material data mining named Auto-Mat. [Methods] We have integrated some existing methods and machine learning algorithms in MatMiner and scikit-learn, and defined a data dictionary to read data from different material calculation databases. At the same time, we have developed some algorithms for feature selection and processing. [Results] It can provide the system with a visual interaction and display interface for data mining and machine learning modules under a unified data format. With the optimized algorithms, the performances of models are improved. [Limitations] For data acquisition, currently only the data in the MatMiner API can be obtained, and the writing of related code is also fully synchronized with the MatMiner API. So the scalability is poor. Moreover, at present, the execution speed of some core algorithms needs to be improved. [Conclusions] Through this system, users can read data from several mainstream databases such as Materials Project in one shot and quickly build their own material data mining workflow. In the end, a comparative analysis of two cases shows that our platform has a positive effect on reducing the barriers for users to use machine learning methods on material data mining.
Keywords:
PDF (10415KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
董家源, 杨小渝. 材料数据挖掘与机器学习工具的集成与优化. 数据与计算发展前沿[J], 2020, 2(4): 105-120 doi:10.11871/jfdc.issn.2096-742X.2020.04.009
Dong Jiayuan, Yang Xiaoyu.
开放科学标识码(OSID)

引言
对于材料科学研究人员而言,由于材料数据的稀缺和不易获取、特征向量的构建过程较为困难,以及最终构建的模型往往难以在较短的时间内取得一个比较好的拟合精度,想要开展材料数据挖掘与机器学习的相关工作,有较高的门槛。为了解决这些问题,我们提出并研发了一种能自动寻找供材料数据挖掘所需的特征变量的算法,能自动进行超参数调节使模型达到一个较好拟合精度的算法,以及能获取不同来源的材料计算数据方法和技术。通过对这些算法的研发和一些数据获取方法的集成,系统降低材料数据挖掘的门槛,使得研发人员可以更快地构建一个拟合精度较高的高可用模型。同时系统还可提供一些用于结果展示的可视化模块,方便研发人员使用。在工具集成方面,我们基于高通量材料计算与数据管理云平台MatCloud[28,29],通过集成一些主流的材料计算数据库的API[20, 26],实现了对于一些材料计算数据库的访问,如Materials Project[1],The Materials Data Facility[2]等,使得用户可以一键获取到这些数据库中的数据,同时还可以进行拼接等操作,使得同时使用这些数据成为了可能;其次是对于特征向量的获取方法,用户想要获取存在于繁杂的文献当中的描述材料结构的特征因子非常困难,我们也是通过集成的方法,整合了一些文献中的特征向量构建方法供用户使用;最后是关于特征的选择、模型的选择、以及模型超参数的优化方面,我们提出和集成了几种算法并做了一些对比试验,简化了材料数据挖掘与机器学习的流程。
在最后的部分,本文引入了两个案例,讲述了用户如何通过使用我们的模块,加速他们的工作流程,尤其是数据的获取和特征向量的构建,以及模型的选择和参数设置这几个方面。
综上所述,本文基于MatCloud平台,着眼于研发一个操作简便的材料数据挖掘与机器学习模块Auto-Mat,使得用户在基于MatCloud的交互式界面仅通过鼠标点击的方式,即可完成数据获取、特征提取、模型训练等一系列流程,并且不要求用户具有机器学习和材料学等领域的背景知识。
1 架构设计
图1给出了本文的数据挖掘模块Auto-Mat的架构设计图。该模块在架构设计上共分为三个部分:(1)前端用户交互界面,负责在数据导入界面和模型训练界面提供一个图形化的接口,接受用户的输入参数。同时也负责为用户在前端提供一个可视化的模型下载接口。由HTML5和AngularJS编写;(2)后台处理模块,负责对用户输入的参数进行预处理,以及调用数据读取、特征提取、模型训练等相应脚本,并完成将训练结果等数据对前端页面的回传;(3)脚本模块,用来执行实际的数据挖掘功能,包括通过调用MatMiner API获取数据、提取特征,通过调用scikit-learn工具包进行模型的训练,以及相关自动化特征筛选和自动化超参数优化算法的实现。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1整体架构
Fig.1Overall architecture
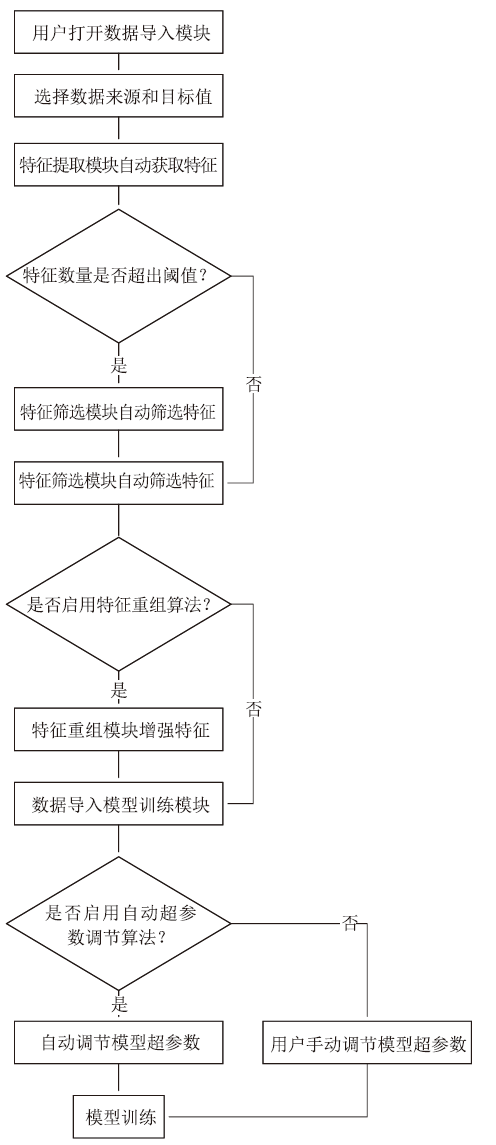
图2给出了用户通过该模块构建的机器学习任务的主要工作流程。主要步骤为:(1)用户通过数据导入模块,选择数据的来源与待预测的目标值,该阶段获取的数据只包括原始的结构信息;(2)系统自动以原始的结构信息作为输入,尽可能多地提取特征;(3)如果特征数量较多,启用我们的特征筛选方法,只留下对目标值地预测贡献度较高的特征;(4)根据用户的选择,决定是否启用特征重组算法,来增强模型的性能;(5)模型训练阶段,根据用户的选择,使用用户输入的超参数或者是启用我们的算法自动选择超参数;(6)开始模型训练。
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2机器学习任务流水线
Fig.2Machine learning task pipeline
2 材料数据挖掘工具详细介绍
2.1 数据导入模块
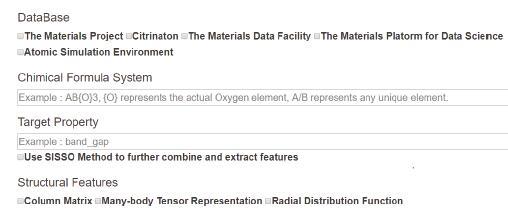
2.1.1 用户交互界面图3是数据导入模块的前端交互界面展示图。用户可以通过选择数据库、化合物体系、目标值来决定数据的选取。我们在界面设计上借鉴了Citrination [6]的search data界面,主要在于通过用户的输入来筛选数据集的逻辑。Citrination中用户可以输入的信息包括:化学式、数据库筛选、目标性质选择。而我们的筛选方式和Citrination最大的不同在于,我们支持选择一类化合物体系,而Citrination只支持一个特定的化学式。
与此同时,我们目前集成了几个材料信息学领域很经典的特征,包括库伦矩阵[8]、MBTR[9]等,可以供熟悉材料领域的研究者们选择。
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3数据导入模块的人机交互界面
Fig.3The interactive interface of the data import module
2.1.2 数据处理
依托于后端集成的Materials Project、Aflow、The Materials Data Facility、ASE等丰富的材料数据,我们的用户可以在这个界面内实现“一站式”的数据读取。我们在后端对数据进行了统一化的处理,使得我们可以把不同来源的数据“拼接”到一起来使用。这一点对于材料科学的研究者而言将会非常有帮助。
材料科学中面临的一个比较大的问题就是,因为计量单位、命名规范、数据格式等因素的不统一,使得数据在汇总的过程中存在着很大的困难。为此我们设计了一个统一的规范,并通过定义一个JSON格式的语义文本,将所有的规范以“key-value”键值对的形式存储在这个语义文本当中。
举例说明,在Materials Project和The Materials DataFacility两个数据库中,对于带隙band_gap这一性质的命名和单位都不同,因此在我们的语义文本中定义了如表1所示的片段。对于band_gap的命名,在Materials Project和MDF数据库中分别命名为’band_gap’和’Band Gap’,我们统一命名为’BandGap’;对于band_gap的单位,在MaterialsProject和MDF数据库中分别采取了以eV为单位和以mV为单位,我们统一规定使用eV作为单位,即需要对一些数据库的数据进行单位进制上的转化。
Table 1
表1
表1语义文本片段
Table 1
| 语义文本中关于band_gap的片段 |
|---|
| {‘Property_Name’: { ‘BandGap’: { { ‘MaterialsProject’: ‘band_gap’, ‘MDF’: ‘Band Gap’ } }, ‘Units’: { ‘BandGap’: { { ‘MaterialsProject’: ‘eV’, ‘MDF’: ‘mV’, ‘OurStandard’: ‘eV’ } } } } |
新窗口打开|下载CSV
2.1.3 特征提取
为了让没有领域知识的人,仅给定化合物的晶体描述[4](一般为CIF 文件,Crystallographic Information File)就能自动获取到关键材料特征,我们分为2 步:(1)用户给出化合物的CIF 文件,我们尽可能多的获取该化合物的特征,形成基础特征库;(2)系统自动筛选出与目标值相关度最高,而自身相关性最低的特征变量。本章节先介绍特征提取的基本方法。
目前主流的特征提取方法主要分为以下几种[10]:
(1)结构(Structure)特征,利用晶胞的形状和晶胞的总体结构,提取描述晶体结构的特征,比如键长的最大值、空间群编号等;
(2)组分(Composition)特征,根据化学式构成,提取元素相关的特征;
(3)原子位置(Sites)特征,基于原子位置和坐标的分布,提取的特征,该类特征向量的长度可能会与原子个数有关。
2.1.3.1 结构特征
晶胞的结构特征,一般包括全局的键长、键角、电荷作用等;以及描述间接结构信息的编码方式,一般有库伦矩阵、径向分布函数[12]、Ewald能量、结构异质性[13]、多体张量表示等。
下面主要介绍关于库伦矩阵的详细内容:库伦矩阵是一个比较经典的结构特征,在Goh[15]等人的工作中提到,很多使用传统或是深度学习方法构建“结构-性能”模型的文章中都会尝试使用库伦矩阵作为输入特征。
晶胞的结构特征需要具有平移、旋转、翻转不变的特性[16],而库伦矩阵就具备这一特性。库伦矩阵是一个n×n的矩阵,其中n为晶胞内的原子个数,而矩阵中的每一项为:
其中,Zi表示第i个原子的核电荷数,Ri表示第i个原子的位置。因此,非对角线位置的值是对自由原子势能的近似,描述了两个原子之间的相互作用;而对角线上的值则是对原子对之间的库伦排斥的近似,描述了了其自身的原子能量。
然而库伦矩阵在实际使用中依然会遇到两个问题:(1)当输入的数据集中各个化合物之间原子个数不一致时,构建的库伦矩阵的大小也不一致;(2)库伦矩阵中的原子顺序是不确定的,因此当交换库伦矩阵的行和列后,该矩阵依然对应于同一个晶胞的表征。
解决第一个问题的方法是,可以通过向晶胞中增加“虚无原子”(invisible atoms) [8],将所有的晶胞扩充为d个原子(d为数据集中原子数最多的晶胞的原子个数)。第二个问题属于物理学领域内的一大难题,即相似结构的检验。但是在这里我们可以将该问题简化,我们不需要检验相似结构,只需要保证库伦矩阵中的原子顺序固定不变即可。我们在构建库伦矩阵之前先对原子做一个排序,不同元素的原子按照其在元素周期表的顺序排序,相同元素的原子按照其距离晶胞质心的距离排序。这样就保证了库伦矩阵中原子顺序的唯一性。
2.1.3.2 组分特征
组分特征,顾名思义,根据化合物的原子组成成分,使用化学式提取得到的特征。在这里可以分为两种:(1)从化学式中的每一种元素分别提取的性质,如,化学式中该原子的个数、电负性(electronegativity)、原子(在元素周期表中的)序号、原子质量、原子半径、平均离子半径、最大氧化态、最小氧化态、在元素周期表中的行号和主族号等;(2)除此之外,还有一些基于整个化学式提取的特征,包括原子轨道、能带中心、内聚能、电子亲和力、电负性差、每一种原子的比例、Miedema模型、化学计量学统计信息、磁性过渡金属比例、化学价轨道等。
2.1.3.3 原子位置特征
该类特征着眼于提取局部相邻的两个或是多个原子之间的特征,比如键长、键角、角傅里叶级数[14]、局部化学环境、局部原子性质的差异度、局部泰森多边形参数等。
由于该类特征向量的长度一般和原子个数有关系,也无法使用类似于我们在2.1.3.1章节提到的解决库伦矩阵原子个数不确定的方法,因为引入类似的“虚无原子”会影响一些特征对于晶胞的描述。因此需要保证输入的数据集中的化合物之间原子个数相同。
2.1.3.4 特征提取总结
通过上述我们系统开发的特征提取模块,我们共获取到了767个特征,其中包括170个物质组分特征,273个原子位置特征,324个结构特征,这些特征构成了我们的“特征库”。接下来,给予我们的特征库,我们开发了一系列的特征自动筛选方法。
2.1.4 特征自动筛选
特征筛选的过程共分为3个阶段:(1)去除“无效”特征;(2)去除“相似”特征;(3)保留与目标值关联度高的特征。
首先,是要将自身数值变化过小的特征去除掉。由于这一步是预处理的过程,因此尽量避免误删掉有用的特征。一种比较常规的做法是,如果某个特征内有95%的数据分布在5%的值域内,说明数据的分布集中在一个很小的范围内,又或者说该特征的相对方差很小,这样对于模型学习所能提供的价值就比较有限。我们可以认为这个特征的意义不大,可以删除[23]。
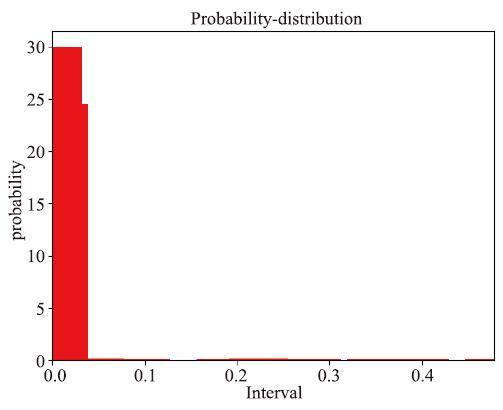
比如,一种极端情况为,该特征上的数据全部都为同一个值,方差为0,这样的特征显然是没有任何意义的。下图展示的是特征抗扭截面系数wt_CN4在本案例的数据上的频率分布直方图,可以看到绝大部分数据都集中在最左侧一个很小的区域内,这样的数据可以认为是没有价值的。
图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4特征变量wt_CN4的频率分布直方图
Fig.4Histogram of frequency distribution of feature variable wt_CN4
需要注意的是,变化小的特征变量未必意味着在物理学意义上一定就不是有价值的特征。由于我们主要还是基于数理统计方法对特征进行筛选,没有考虑特征的物理意义,变化大特征对模型训练的影响大于变化小的特征,因此我们还是对变化小的特征予以筛除,以保持整个方法的一致性。
按设定的阈值我们保留一部分剩余特征(如为300到350个之间)。而在这一阶段被丢弃的特征主要为原子位置信息和化学键的信息等原始单一特征。这进一步说明了原始的晶胞结构信息是无法直接作为模型输入的。
接下来,我们需要将变化趋势“相似”的特征剔除掉。因为变化趋势相似的特征往往意味着不能为模型带来新的信息,或者说该特征可以由其他特征推断出来。
以往的研究者比如Pabitra[23]等人使用了K-NN或者K-Means聚类[24]的方式来判断特征之间的相似性,然而这种评价方式受特征值域的影响很大,并不客观。
因此,我们使用spearman相关系数来刻画两个特征之间的相似度。为此,我们计算了每两个特征之间的spearman相关系数,并以此构建了一个相关系数矩阵,来描述每两个特征之间的关系。
假设该矩阵为M,则
或者说,矩阵中每一个元素表示了对应两个特征之间的spearman相关系数,相关系数越高,说明两个特征越“相似”。因此,我们希望我们筛选出来的特征集合的总体相似度尽量小。假设筛选出来的最优特征集合为
我们没有找到多项式时间的方法来解决该问题(或许不存在这样的方法)。我们在这里使用 “蒙特-卡罗”方法[25]来找到一个近似的最优解。我们随机地生成10000个特征集合(特征集合即上面提到的),并在这些特征集合中,找到总体相关系数最小的一个特征集合,作为此阶段输出的特征集。经过反复的尝试,我们认为设置此阶段剩余的特征数大致在30-50之间,可以在模型准确性和运行效率之间取得一个比较不错的平衡,具体数字由算法自动确定。此阶段剩余的特征基本保留着组分特征、位置特征、结构特征这三类特征中比较常见的特征,比如空间群序号、熔点、以及一些化学键的性质等。
最后一个阶段,我们希望保留对于目标值的预测贡献度最高的特征。在Ouyang[22]等人的文章里提到过使用LASSO的方法筛选特征,我们在这个阶段也采用了这样的方法。将本案例中的40个特征作为LASSO回归的输入,来预测目标值,然后对每一个特征值的LASSO权重进行排序,留下LASSO权重最大的10个特征。之所以在这个阶段留下10个特征,一方面是因为筛选出的10个特征已经足够描述该化合物,因为我们的实验表明前10个特征的LASSO权重在95%以上,也就是说在构建模型时候,如果使用比较“稀疏化”的模型,那么后30个特征对模型的贡献度很小;另一方面是因为接下来的特征重组算法比较耗时,为了减小时间的开销,因此尽量降低此阶段输出特征的数量。
LASSO特征筛选算法大致介绍如下:
首先,使用L1-正则化的线性回归算法,对整个数据集进行拟合,得到如下所示的特征权重向量
其中X为训练样本,y为目标值,
接下来,对输入的40个特征按照其在特征权重向量
同时,对于一些很经典的结构描述符,比如库伦矩阵,我们提高它的优先级,使其可以不参与筛选而直接使用。当然,出于计算量的考量,它不会参与后面的特征重组算法。这种方式也是为了提升我们算法的“下限”,当算法失效,筛选出来的10个特征不足以描述晶胞时,通过像库伦矩阵这样的结构描述符至少还可以保留一定的晶胞信息。
最后,基于上述筛选的特征,我们进行部分特征的重组。
2.1.5 特征重组算法
为了能自动地从最基本的化学式和结构特征中提取出和目标值更为相关的特征,我们提出一个自动化的特征重组算法[22],算法的输入是带有基本特征的数据,如元素相对原子质量、原子半径、晶胞结构等,经过一些算术操作后,输出一系列的特征组合。
特征重组的方法是,对原始特征的一些数学运算的叠加。比如,(1)中的IE表示第一电离能,
换句话讲,特征重组算法并不能创造新的“物理学意义”上的特征,而是增强了原始输入特征的泛化性能。
我们参照了一些比较经典的方法[10, 22],在特征重组算法中,使用了一些基础而简单一元和二元运算符,一元运算符包括:1/xx2,x3,1/x2,1/x3,log(x),exp(x),log(x),exp(x),1/log(x),1/exp(x)这几种;二元运算符使用了加减乘除四种运算符。
通过我们的算法提取出的特征,相比于输入的原始特征而言,会显著地增加和目标值之间的相关性。我们进行了如下的对比试验:在Materials Project的钙钛矿ABX3数据集上,在使用简单结构特征和组分特征作为原始输入特征的情况下,使用线性回归算法得到的结果,与不使用特征重组算法,直接将原始特征作为模型输入时的结果,进行对比,如表2所示。可以看到特征重组算法对于模型性能提升有很大帮助。
Table 2
表2
表2特征重组算法的性能比较
Table 2
| 评价指标 | 对照组(不使用重组算法) | 特征重组 |
|---|---|---|
| RMSE | 1.38 | 0.78 |
| R2-Score | 0.41 | 0.81 |
| Pearson Coefficients | 0.77 | 0.90 |
| Spearman Coefficients | 0.54 | 0.77 |
新窗口打开|下载CSV
2.2 模型训练模块
我们将主流的分类和回归算法都集成到一个名为“ModelTraining”的模块内。当用户使用时,首先选择“Classification or Regression”,接下来选择具体的学习器。比如,如果用户选择了Regression,那么接下来可以继续选择“Random Forest Regression”[18]。接下来是算法参数的设置,以回归森林算法举例,图5表示的是用户参数设置的交互式界面。在这个界面里,用户可以设置模型学习的相关参数,比如随机森林算法的Max Depth参数等。参数命名方式和scikit-learn保持一致。
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5用户设置模型参数界面
Fig.5User setting model parameter interface
并且,用户也可以通过勾选的方式选择启用我们的自动超参数调节算法。这样就省去了手动调节超参数的麻烦。
2.2.1 自动化超参数调节
一些复杂的学习器,如随机森林,往往需要用户输入很多的超参数。超参数的选取对于模型的性能非常重要,而很多情况下默认参数的模型表现往往不尽如人意。因此就需要用户对机器学习模型的调参具有一定的经验。
而本文正是致力于帮助计算机和机器学习相关经验不足的材料研究者们,因此本文实现了自动化的超参数优化算法,并在前端交互界面为用户提供了接口(如图5),用户可以通过选择自动化超参数优化的方式,来跳过超参数选择的过程,从而简化了用户训练模型的难度。
使用我们的自动化超参数优化算法后,一般情况下模型可以获得比默认参数下更好的效果。我们进行了如下的对比试验:在Materials Project的钙钛矿ABX3数据集上,在使用章节2.1.3中获取到的特征作为原始输入特征的情况下,分别使用默认参数下的随机森林模型和附加了我们的超参数优化算法后的随机森林模型进行对比,结果如表3所示。
Table 3
表3
表3超参数优化算法的性能比较
Table 3
| 评价指标 | 对照组 (默认参数) | 超参数优化 |
|---|---|---|
| RMSE | 1.38 | 0.82 |
| R2-Score | 0.41 | 0.67 |
| Pearson Coefficients | 0.77 | 0.85 |
| Spearman Coefficients | 0.54 | 0.70 |
新窗口打开|下载CSV
2.3 可靠性评估模块
我们设计并集成了相关的模型评估的工具箱,使用RMSE、MAE、pearson相关系数、spearman相关系数和R2-Score决定系数共五种评价指标。并且,对于随机森林算法,我们引入并实现了了Julia Ling[17]等人提出的关于随机森林的不确定性这一评价指标。不确定性指标通过分析训练集和测试集的差异性,以及训练集自身的分布和噪声的影响,对随机森林中每一个简单学习器(即决策树)的影响,计算其造成的variance和bias的均方根平均数。具体地,不确定性的计算公式为:
其中,S为训练集的大小,
关于可靠性性评估更详细的说明参见第3章应用案例。
3 应用案例
下面使用2个案例讲述用户是如何使用我们的材料数据挖掘与机器学习工具,其中一个是对于材料领域和机器学习相关知识都不是那么了解的用户,我们如何通过一些设置和自动化的方法让其以最简单的步骤构建起属于自己的机器学习流程;另一个是对于相关知识和目的都比较明确,我们的工具是如何保证足够的扩展性,为其提供充足的支持的。3.1 钙钛矿的“结构-带隙”模型构建
案例:构建一个“结构-带隙”构效关系模型,预测ABO3型钙钛矿结构化合物的能隙。拟选取的数据来源Materials Project、Citrination、MDF、ASE几个开放的材料计算数据库。3.1.1 用户输入
首先打开数据导入界面后,选择使用所有候选数据库中的数据,即对界面中所有数据库复选框打勾。
然后,在目标性质文本框中输入band_gap。同时,由于我们的用户对于特征的筛选并不了解,按照我们的默认建议,使用库伦矩阵这一比较经典的结构描述符作为“高优先级特征”,即不参与筛选,必定被使用的特征。同时,启用我们的自动化特征重组方法增强特征的表征能力。
用户输入完成后的界面如图6所示。
图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6用户输入界面-案例1
Fig.6User input interface-case 1
通过根据用户的输入参数进行数据的读取和特征构建,我们获得了原始数据并完成了对数据的特征向量化。本案例中共得到数据923条。
3.1.2 特征筛选
通过特征提取模块,共获取到了767个特征。接下来是特征筛选环节,在本案例中,我们在第一个阶段的筛选留下了350个特征;第二个阶段留下了40个特征;第三个阶段留下了10个特征。
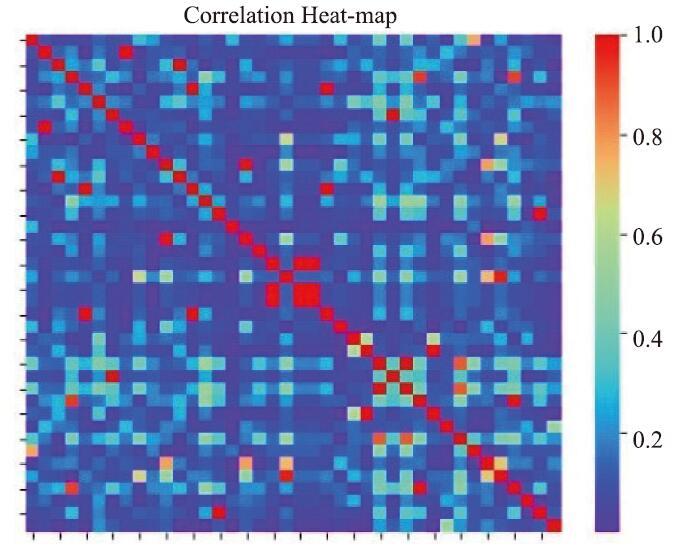
在本案例中,第一阶段筛选出来的特征数为328,作为第二阶段的输入特征。这些特征之间的平均spearman相关系数为0.2630,平均相关系数是通过对输入的328×328的相关系数矩阵中所有两两特征之间的相关系数取平均得到的。而筛选出的40个特征彼此之间的平均相关系数为0.1746。因此说明已经显著降低了特征之间的“相似性”。
通过热度图,图7展示了在本案例中,通过两个阶段的特征筛选后,输出的40个特征中,每两个特征之间的相关性。图中的“温度”越高表示两个特征之间的相关性越高。(因此在对角线上的元素温度是最高的,因为每个特征和它自身的相关性为1。)
图7
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7特征相关性热力图-案例1
Fig.7Feature correlation heatmap-case 1
下表展示了经过了3个阶段筛选出的特征,以及和目标值之间的相关性,可以看到,相关系数最大的特征compound possible和std_dev wt CN_1两个特征的spearman相关系数分别达到了0.46和0.38,这对于单个特征而言,与目标值之间的相关性还是比较大的。
Table 4
表4
表4第3阶段筛选后特征与目标值之间的相关系数-案例1
Table 4
| 特征名称 | 与目标值的相关系数 |
|---|---|
| mean wt CN_2 | -0.1256 |
| max packing efficiency.1 | -0.1197 |
| wt CN_6 | -0.1859 |
| mean row | -0.0046 |
| compound possible | 0.4690 |
| std_dev row | 0.0136 |
| bond #4 | -0.0841 |
| fraction electrons | -0.1233 |
| std_dev wt CN_1 | 0.3843 |
| maximum GSmagmom | 0.0050 |
新窗口打开|下载CSV
获得上述特征后,我们还可调用特征重组算法,算法的输入是上述三个阶段筛选得到的10个特征(不包括作为备用特征的库伦矩阵),输出是获取到的特征的算术组合。本案例中我们只保留2个重组特征,因为在特征重组算法的迭代过程中,其内部也使用到了前面提到过的LASSO算法作为特征的筛选,而当算法运行到最后一个迭代轮次时,特征之间的权重分布的差距已经非常大。图8展示了本案例中在最后一个迭代轮次时特征的权重分布。(在本案例中特征重组算法每一迭代轮次会获取20个重组特征,但是后面的特征的权重过小,以至于柱状图中都无法展示其高度。)
图8
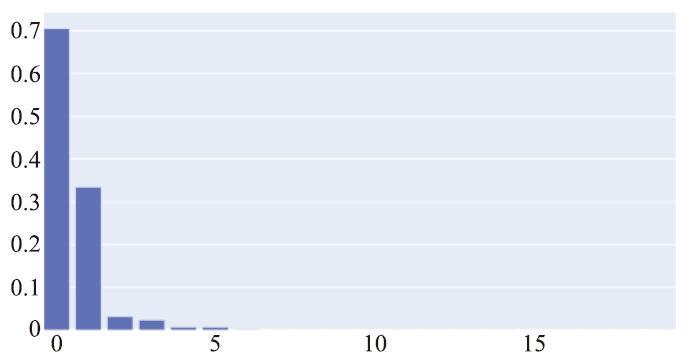 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8特征重组中的特征权重分布
Fig.8Feature weight distribution in feature recombination
本案例中我们获取到的两个重组特征分别为:
式中的CP为compound possible,Er为Electrone-gativity range,PE为packing efficiency,frac为fraction of valence electrons。
重组特征与目标值band_gap之间的相关性如下表所示。可以看到我们的重组特征和目标值之间相关性非常高,也就对目标值的预测具有很大的贡献。
Table 5
表5
表5重组特征与目标值相关性
Table 5
| 特征名称 | 与目标值的相关系数 |
|---|---|
| Descriptor-1 | 0.79 |
| Descriptor-2 | 0.82 |
新窗口打开|下载CSV
3.1.3 模型训练
在我们的工具中,用户可以选择随机森林,可以选择支持向量机,也可以选择多元线性回归,或是其他学习器。
由于我们的用户对于机器学习算法不熟悉,并不知道该选择什么学习算法,也不知道该如何设置参数。由于我们的工具支持“one-in-multi-out”的计算流程,因此我们可以同时使用多个学习器来对同一份数据进行学习,并且多个学习器之间彼此是相互独立的。在本案例中我们同时使用了较为复杂的随机森林算法和最简单的线性回归算法作为学习器,如图9。
图9
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图9同时使用两个学习器
Fig.9Using two learners at the same time
关于参数设置,用户在此案例中由于对机器算法不了解,因此决定启用我们的自动化超参数优化算法来自动选取超参数。
3.1.4 模型评估
当整个计算过程完成后,我们可以打开学习算法工具箱查看模型的训练结果,即各种量化的评价指标,这些指标是使用测试集进行计算的。
图10展示了该案例的模型评估结果,不仅给出了如前所述的一些评价指标,还绘制了一条“predicted-label”散点图,每一个点对应着一条数据,横轴为模型预测的目标值值,纵轴为实际的目标值,所以用户可以根据散点群距y=x这条直线的距离,直观看出训练结果的好坏。
图10
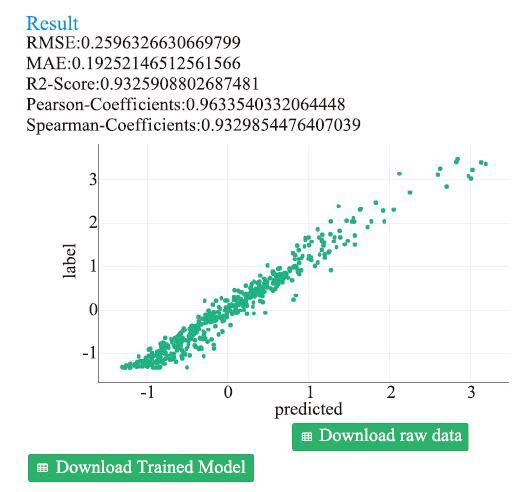 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图10可靠性评估界面-案例1
Fig.10Reliability evaluation interface-case 1
同时由于我们使用的是随机森林算法,我们也可以查看模型的不确定性,如图11所示。图中灰线的部分即为对应样本的不确定性。
图11
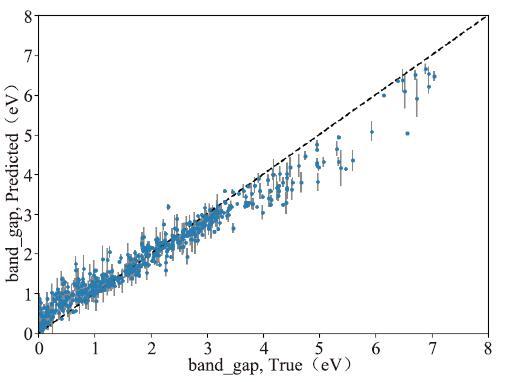 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图11随机森林算法不确定性曲线图
Fig.11Random forest algorithm uncertainty curve
不确定性是随机森林算法特有的一种评价指标,它说明了使用随机森林算法,在指定数据集上,所能达到的准确度的“上限”是多少。该项指标计算了每一个数据,所造成的不确定性(又或者说对模型性能的影响),下图中,每一个蓝色的小圆点代表了本案例中钙钛矿数据集里的一个样本,而其对应的灰线则代表着该样本造成的不确定性。
3.1.5 模型导出
用户可以在训练完成后的学习算法工具箱中,点击Download Trained Model按钮,下载训练好的pickle格式的模型。pickle是基于python的一个开源代码库[5],模型文件下载到用户本地后用户可以使用库函数很方便地读入存储于文件中的模型,并且与scikit-learn完全兼容,都进来的模型可以直接使用scikit-learn的预测函数进行预测。
3.2 A2BCO6的“结构-体弹性模量”模型构建
案例:构建一个“结构-体弹模量”构效关系模型,使用了Materials Project和Ctrination作为数据来源,并筛选出化学式满足“A2BCO6”格式的化合物作为数据集,预测的目标值为体弹性模量Bulk Modulus。讲述一个对机器学习和物理学方面都比较了解的用户将如何使用我们的工具。3.2.1 用户输入
首先同样地,该用户需要添加一个数据导入模块。
由于该用户希望构建的是体弹性模量(Bulk Modulus)的模型,所以,为了区别其它弹性模量(比如剪切模量和杨氏弹性模量),该用户决定只使用Materials Project和Ctrination数据库。因为在Materials Project数据库和Ctrination数据库有较丰富的体弹性模量数据。
接下来在选择化合物体系的界面,因为该用户的体系在候选当中没有,因此选择Customize并在输入框中输入“A2BC{O}6”,之所以氧原子要用中括号是因为,在该化学式体系中,区别于可以使用任何原子替代的“A”、“B”、“C”原子,氧原子是不可以被替代的。比如说,在该用户的化合物体系中可以包含Si2LiAlO6(标准化学式为LiAl(SiO3)2)和C2NaMgO6两种化合物,但不可以包含Si2LiAlS6。
我们这种划分数据集的方式一定程度上借鉴了ASE[7]数据库中对于数据的划分方式。ASE数据库中包括了ABX3、A2BCX4、ABX2等数据集,并按照此种方式进行了划分。
接下来,该用户在目标性质文本框中输入elasticity.K_VRH。但是需要注意的的一点是,体弹性模量在Materilas Project和Ctrination当中的命名方式是不同的(在Materilas Project中命名为elasticity.K_VRH,在Ctrination中命名为Bulk modulus),因此需要在我们定义的语义文本中加入以下内容:
Table 6
表6
表6语义文本片段-案例2
Table 6
| 语义文本中关于体弹性模量的片段 |
|---|
| {‘Property_Name’: { ‘elasticity.K_VRH’: { ‘MaterialsProject’:’elasticity.K_VRH’, ‘Ctrination’: ‘Bulk modulus’ } }, ‘Units’: { ‘elasticity.K_VRH’: { ‘MaterialsProject’: ‘GPa’, ‘MDF’: ‘ GPa ‘, ‘OurStandard’: ‘ GPa ‘ } } } |
新窗口打开|下载CSV
最后同样地,用户勾选库伦矩阵作为结构特征。
用户输入完成后的界面如图12所示。本案例共获取到201条数据。
图12
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图12用户输入界面-案例2
Fig.12User input interface-case 2
3.2.2 特征获取
特征获取阶段和案例1大致相同,共获取到了767个特征,其中包括170个组分特征,273个原子位置特征,324个结构特征,这些特征构成了“特征库”。
接下来是特征筛选环节,我们的算法在第一个阶段的筛选留下了350个特征;第二个阶段留下了40个特征;第三个阶段留下了10个特征。
图13为第2个阶段的特征筛选后获取到的特征之间的相关性热度图。本案例中,筛选之前特征之间的平均相关性为0.2744,而筛选后的平均相关性为0.1724。
图13
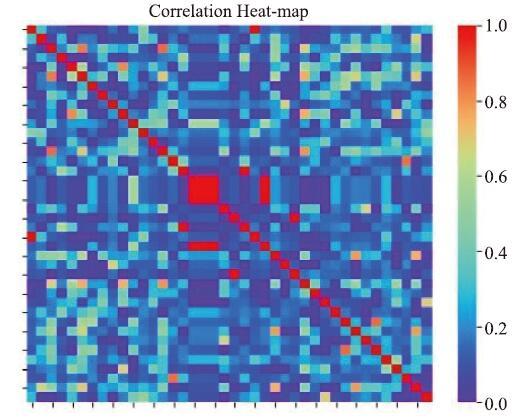 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图13特征相关性热力图-案例2
Fig.13Feature correlation heatmap-case 2
表7展示了第3个阶段的特征筛选后,剩下的10个特征和目标之间的相关性。可以看到range NUnfilled和bond # 4这两个特征与目标值得相关性也都分别达到了0.45和0.32,作为单一特征具有和目标值较强的相关性。
Table 7
表7
表7第3阶段筛选后特征与目标值之间的相关系数-案例2
Table 7
| 特征名称 | 与目标值的相关系数 |
|---|---|
| range NUnfilled | 0.4514 |
| minimum local difference | -0.1821 |
| mean Column | -0.0546 |
| bond #0 | 0.0412 |
| bond #3 | 0.2903 |
| bond #4 | 0.3283 |
| local difference in MendeleevNumber | -0.0359 |
| std_dev melting_point | 0.1888 |
| mean local difference in MeltingT | 0.2480 |
| bond #2 | 0.2513 |
新窗口打开|下载CSV
3.2.3 模型训练
当用户对机器学习模型较为熟悉时,可以直接选择合适的模型。比如该用户认为支持向量回归(SVR)用来学习体弹性模量比较合适,可以使用SVR模块并手动设置参数,如图14。在支持向量回归中,用户可以设置kernel和epsilon两个参数。kernel参数为SVR的核函数,为字符串格式,默认值是“rbf”;epsilon是经验风险和正则项的平衡参数,默认值为0.1。
图14
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图14支持向量回归参数设置界面-案例2
Fig.14Support vector regression parameter setting interface-case 2
4 结论与展望
本文针对材料科学工作者开展机器学习工作门槛较高这一现状,在对前沿相关工作[30,31,32]充分调研的基础上,介绍了基于MatCloud框架开发的一个材料数据挖掘与机器学习模块。具体介绍了我们的数据导入模块,是如何根据我们所制定的规范,完成数据的汇总,以及材料数据挖掘与机器学习工具用户界面的设计与实现、特征提取和模型训练模块的工作原理等细节。最后,通过两个案例,详细阐述了我们的模块是如何简化材料科学研发者上手数据挖掘门槛的。
本文中提到的材料数据挖掘与机器学习工具有些功能还不够完善,因此下一步将继续完善该工具的其他模块。另外,本文中只着眼于使用传统的机器学习方法,在下一步中会尝试使用深度学习方法[11, 19, 21, 27]。
致谢
本文的想法和思路来自一些公司、高校和科研院所一线的建议和反馈,作者对他们表示感谢,同时也感谢匿名审稿人对文章提出的修改意见。利益冲突声明
所有作者声明不存在利益冲突关系。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[J].
DOI:10.1063/1.4812323URL [本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[J].
[本文引用: 1]
[J].
DOI:10.1088/1361-648X/aa680eURLPMID:28323250 [本文引用: 1]

The atomic simulation environment (ASE) is a software package written in the Python programming language with the aim of setting up, steering, and analyzing atomistic simulations. In ASE, tasks are fully scripted in Python. The powerful syntax of Python combined with the NumPy array library make it possible to perform very complex simulation tasks. For example, a sequence of calculations may be performed with the use of a simple 'for-loop' construction. Calculations of energy, forces, stresses and other quantities are performed through interfaces to many external electronic structure codes or force fields using a uniform interface. On top of this calculator interface, ASE provides modules for performing many standard simulation tasks such as structure optimization, molecular dynamics, handling of constraints and performing nudged elastic band calculations.
[本文引用: 2]
[本文引用: 1]
[J].
[本文引用: 2]
[J].
DOI:10.1364/OE.25.016235URLPMID:28789131 [本文引用: 1]
We proposed two noise-robust iterative methods for phase retrieval and diffractive imaging based on the Pauta criterion and the smoothness constraint. The work is to address the noise issue plaguing the application of iterative phase retrieval algorithms in coherent diffraction imaging. It is demonstrated by numerical analysis and experimental results that our proposed algorithms have higher retrieval accuracy and faster convergence speed at a high shot noise level. Moreover, they are proved to hold the superiority to cope with other kinds of noises. Due to the inconvenience of conventional iteration indicators in experiments, a more reliable retrieval metric is put forward and verified its effectiveness. It should be noted that the proposed methods focus on exploiting the longitudinal diversity. It is anticipated that our work can further expand the application of iterative multi-image phase retrieval methods.
[J].
DOI:10.1103/PhysRevLett.77.2581URLPMID:10061990 [本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
DOI:10.1002/jcc.24764URLPMID:28272810 [本文引用: 1]
The rise and fall of artificial neural networks is well documented in the scientific literature of both computer science and computational chemistry. Yet almost two decades later, we are now seeing a resurgence of interest in deep learning, a machine learning algorithm based on multilayer neural networks. Within the last few years, we have seen the transformative impact of deep learning in many domains, particularly in speech recognition and computer vision, to the extent that the majority of expert practitioners in those field are now regularly eschewing prior established models in favor of deep learning models. In this review, we provide an introductory overview into the theory of deep neural networks and their unique properties that distinguish them from traditional machine learning algorithms used in cheminformatics. By providing an overview of the variety of emerging applications of deep neural networks, we highlight its ubiquity and broad applicability to a wide range of challenges in the field, including quantitative structure activity relationship, virtual screening, protein structure prediction, quantum chemistry, materials design, and property prediction. In reviewing the performance of deep neural networks, we observed a consistent outperformance against non-neural networks state-of-the-art models across disparate research topics, and deep neural network-based models often exceeded the
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[本文引用: 1]
[J].
[本文引用: 1]
[J].
DOI:10.1016/j.commatsci.2012.10.028URL [本文引用: 1]
[J].
DOI:10.1107/S0365110X57000675URL [本文引用: 1]
[J].
DOI:10.1103/PhysRevMaterials.2.083802URL [本文引用: 3]
[J].
[本文引用: 2]
[J].
DOI:10.2307/2346830URL [本文引用: 1]
[J].
DOI:10.2307/2297968URL [本文引用: 1]
[J].
DOI:10.1038/npjcompumats.2015.10URL [本文引用: 1]
[J].
DOI:10.1038/nature16961URLPMID:26819042 [本文引用: 1]
The game of Go has long been viewed as the most challenging of classic games for artificial intelligence owing to its enormous search space and the difficulty of evaluating board positions and moves. Here we introduce a new approach to computer Go that uses 'value networks' to evaluate board positions and 'policy networks' to select moves. These deep neural networks are trained by a novel combination of supervised learning from human expert games, and reinforcement learning from games of self-play. Without any lookahead search, the neural networks play Go at the level of state-of-the-art Monte Carlo tree search programs that simulate thousands of random games of self-play. We also introduce a new search algorithm that combines Monte Carlo simulation with value and policy networks. Using this search algorithm, our program AlphaGo achieved a 99.8% winning rate against other Go programs, and defeated the human European Go champion by 5 games to 0. This is the first time that a computer program has defeated a human professional player in the full-sized game of Go, a feat previously thought to be at least a decade away.
[J].
DOI:10.1016/j.commatsci.2018.01.039URL [本文引用: 1]
[J].
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}