全文HTML
--> --> -->贵金属作为助催化剂负载到BiOBr表面是常见的改性策略, 当光催化剂中引入贵金属时可将光生电子和空穴的重新分配, 贵金属和半导体之间产生肖特基(Mott-Schottky, M-S)势垒, 确保在M-S界面上电荷载流子高效单向转移, 有效抑制光生电子-空穴对复合[8-10]. 研究发现, 随着贵金属颗粒尺寸的不断减小, 其表面自由能逐渐增大, 催化活性也随之增强. 因此, 研究者把更多工作集中到制备单原子贵金属沉积光催化剂表面领域. 2011年, 张涛课题组[11]将单原子Pt负载于FeOx载体上大大提高了CO氧化活性, 第一次提出了“单原子催化剂”的概念. 自此之后, 单原子催化剂成为炙手可热的前沿课题[12-14]: 第一, 贵金属粒子尺寸减小至原子级, 可充分发挥量子尺寸效应; 第二, 贵金属独特的电子轨道和单原子活性中心不饱和配位环境, 以及金属原子和载体之间可能会发生电荷转移导致金属-载体强相互作用, 从而使单原子催化剂活性得到了大幅度提升[15]; 第三, 考虑到贵金属价格昂贵的事实, 提高贵金属的原子利用率. 然而, 实验过程中常遇到难点是由于载体表面过于活泼, 金属原子容易发生团聚, 活性位点常被掩蔽. 为了避免这一问题的出现, 研究们不仅严格控制金属单原子的负载量, 而且选择能够紧密结合金属单原子的半导体衬底[16], 只是目前关于单原子催化剂的复杂反应机理尚不清晰, 实验上寻找合适基底的方法尚显盲目.
迄今, 国内外研究者借助高速发展的计算机技术和日益完善的密度泛函理论, 通过模拟计算得到理论结果可指导并设计新型高效光催化剂, 为反应机理研究提供新方法新思路[17-19]. 基于此, 本文采用第一性原理方法, 通过构建暴露不同原子终端的BiOBr{001}表面, 确定贵金属Pt在不同暴露原子端晶面上的优先吸附位置, 分析其驰豫结构、电子性质、光学性质和电荷转移趋势, 为实验上制备单原子Pt/BiOBr光催化剂提供有效的理论指导和基础数据.
2.1.计算方法
本工作所有模拟计算都是在Materials Studios软件的CASTEP模块中完成. 计算中采用赝势平面波方法和广义梯度近似(generalized gradient approximation, GGA)下PBE(Perdew–Burke–Ernzerhof)交换关联泛函[20], Broyden-Fletcher-Goldfarb-Shanno (BFGS)方案作为最小化算法来弛豫所有原子[21]. 其中, 选用超软赝势来描述离子实与价电子之间的相互作用[22,23], 处理的基态价电子构型分别为Bi 6s26p3, O 2s22p4, Br 4s24p5和Pt 5d96s1. 所有的结构模型采用平面波截断能(Ecut-off)是500 eV, 选用Monkhorst-Pack方式为布里渊区的积分计算方案, 将k点网格设置为4 × 4 × 1, 结构优化和性质计算中的收敛标准: 能量变化、作用于每个原子应力、最大内应力和原子最大位移分别不大于1 × 10–5 eV/atom, 0.03 eV, 0.05 GPa和0.003 ?.2
2.2.结构模型
BiOBr晶体结构属于四方晶系(tetragonal), 空间族为P4/nmm(No.129), 晶格参数是a = b = 3.923 ?, c = 8.105 ?, c/a = 2.066, α = β = γ = 90°, V0 = 124.73 ?3和Z = 2. 基于BiOBr独特的层状结构, 搭建了三种暴露不同原子终端BiOBr{001}表面模型, 分别是BiOBr{001}-BiO, BiOBr{001}-1Br和BiOBr{001}-2Br (图1(a)—(c)). 为模拟贵金属单原子-半导体载体之间的相互作用, 选用了2 × 2 × 1超晶胞, 真空层为15 ?, 排除c轴方向上周期性原子相互作用的干扰[24]. 为研究单原子Pt吸附于BiOBr{001}-BiO不同位置的电子性质和电荷转移行为, 建立顶位 (top site, T)、桥位 (bridge site, B)和穴位 (hollow site, H)三类吸附位置模型, 如图1(d)—(f)所示. 图 1 不同原子暴露终端BiOBr{001}面及单原子Pt吸附于BiOBr{001}-BiO不同位置的晶体结构模型 (a) -BiO; (b) -1Br; (c) -2Br; (d) TPt; (e) BPt; (f) HPt
图 1 不同原子暴露终端BiOBr{001}面及单原子Pt吸附于BiOBr{001}-BiO不同位置的晶体结构模型 (a) -BiO; (b) -1Br; (c) -2Br; (d) TPt; (e) BPt; (f) HPtFigure1. The crystal structure model of BiOBr{001} surface with different atom exposure terminations and single atom Pt adsorbed on different positions of BiOBr{001}-BiO: (a) -BiO; (b) -1Br; (c) -2Br; (d) TPt; (e) BPt; (f) HPt.
3.1.体相BiOBr
基于第一性原理方法对BiOBr原胞进行结构优化, 弛豫后的晶格参数是a = b = 3.914 ?, c = 8.821 ?, c/a = 2.253和α = β = γ = 90°, 均在误差允许范围内且与已报道数据基本一致[25]. 如图2(a)所示, 体相BiOBr能带结构投影在布里渊区, 将能量为零设为费米能级(EF). 纯BiOBr晶体带隙(Eg), 即价带势能最高处(valence band maximum, VBM)和导带势能最低处(conduction band minimum, CBM)的距离是2.332 eV, 比实验值2.6—2.9 eV略低[26,27], 与GGA泛函本身的缺陷有关[25,28]. 由于载流子的有效质量和迁移性与能带弯曲度直接相关, 图2(a)可知BiOBr的CBM能带是高度疏散的, 表明光生电子的有效质量低和迁移率高, 光生电子在寿命结束之前跃迁到CBM的概率更大. 此外, VBM位于G和F点之间, 而CBM位于G点, 处于布里渊区的不同高对称点, 证实了BiOBr具有间接带隙的特征. 图2(b)所示体相BiOBr的总态密度(total density of states, TDOS)和原子态密度图(projected density of states, PDOS). 结果表明, BiOBr价带是由Br 4p 和O 2p轨道主导, 两者轨道能量接近且发生杂化, 表明Bi和O原子之间形成共价键; 而导带主要由Bi 6p 轨道贡献, 由此可以推断, 光生电子的跃迁路径类型为p-p型, 即从O 2p 和Br 4p跃迁到Bi 6p. 电子结构分析结果与先前DFT方法计算结果完全一致[6,28,29], 进一步证明我们采用的计算方法和结果是合理且值得信赖的.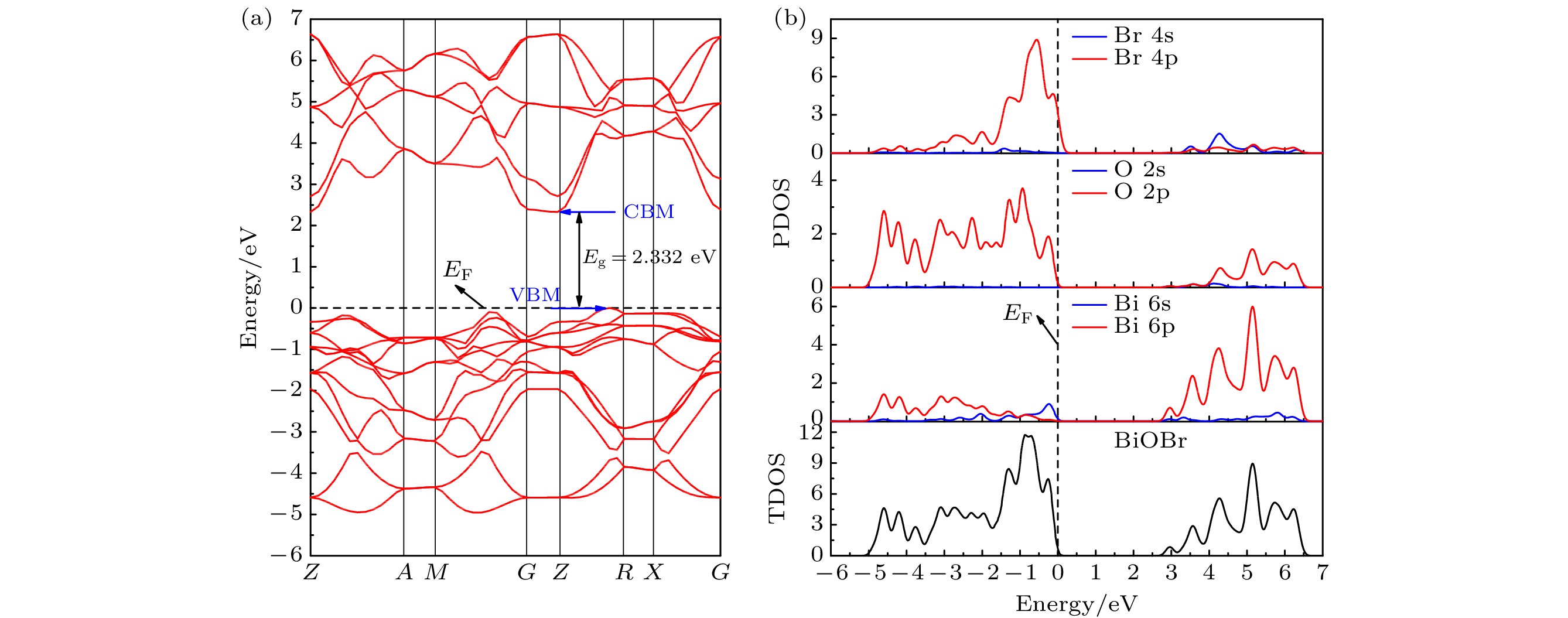 图 2 体相BiOBr的(a)能带结构以及(b)总态密度图和原子态密度图
图 2 体相BiOBr的(a)能带结构以及(b)总态密度图和原子态密度图Figure2. (a) Band structure and (b) total density of states and projected density of states of bulk BiOBr.
2
3.2.不同原子暴露终端的BiOBr{001}表面
构建了三种暴露不同原子终端的BiOBr{001}面模型(BiOBr{001}-BiO, BiOBr{001}-1Br和BiOBr{001}-2Br), 系统研究其几何构型、电子结构、光学性质和电荷转移行为. 优化后的表面结构模型俯视图如图3所示. 结果发现, 由于表面原子重构, BiOBr{001}-BiO表面Bi原子向内偏移, 而次层O原子向外偏移, 导致表面Bi—O键和中央Bi—O键的键长差异很大, 且表面Bi—O—Bi的键角由114.6°缩小为78.3°. 值得注意的是, BiOBr{001}-1Br和BiOBr{001}-2Br表面Br原子之间的距离稍微变大, 但表面Bi—O(Bi—Br)键和中央Bi—O(Bi—Br)键的键长变化很小, 这可能是由于暴露BiO端产生了悬挂键, 而暴露Br端并未产生悬挂键, 其表面重构低所致. 图 3 (a) BiOBr{001}-BiO, (b) BiOBr{001}-1Br和 (c) BiOBr{001}-2Br结构优化后的俯视图
图 3 (a) BiOBr{001}-BiO, (b) BiOBr{001}-1Br和 (c) BiOBr{001}-2Br结构优化后的俯视图Figure3. The optimized top view of (a) BiOBr{001}-BiO, (b) BiOBr{001}-1Br, and (c) BiOBr{001}-2Br.
图4给出了三种暴露不同原子端BiOBr{001}表面体系的能带结构图和态密度图, 对比体相BiOBr电子结构, BiOBr{001}-BiO表面体系的价带和导带位置均向低能方向移动, 价带顶下移到–2.020 eV, 导带底下移到费米能级附近(图4(a)), 且在导带下方出现表面态能级(surface states energy level, SEL), 结合图4(b)可知, 该表面态能级是由表面Bi 6p轨道贡献, 使BiOBr{001}-BiO体系呈p型半导体的n型表面性质[18]. 此外, BiOBr{001}-BiO的VBM和CBM属于不同布里渊区, 仍保持间接带隙特点(图4(a)), 有助于抑制光生电子-空穴对的复合. BiOBr{001}-1Br表面能带结构和态密度的特征与体相结构几乎一致(图4(c)—(d)), 与暴露1Br原子端仅仅断开双Br原子层之间微弱的范德瓦耳斯力有关. 图4(e)— (f)显示BiOBr{001}-2Br的能带结构图和态密度图, 价带上方出现了表面态能级, 由表面原子Br 4p电子态贡献, 使得BiOBr{001}-2Br体系展现出p型半导体的p型表面性质. 此外, BiOBr{001}- 2Br表面体系仍然是间接带隙.
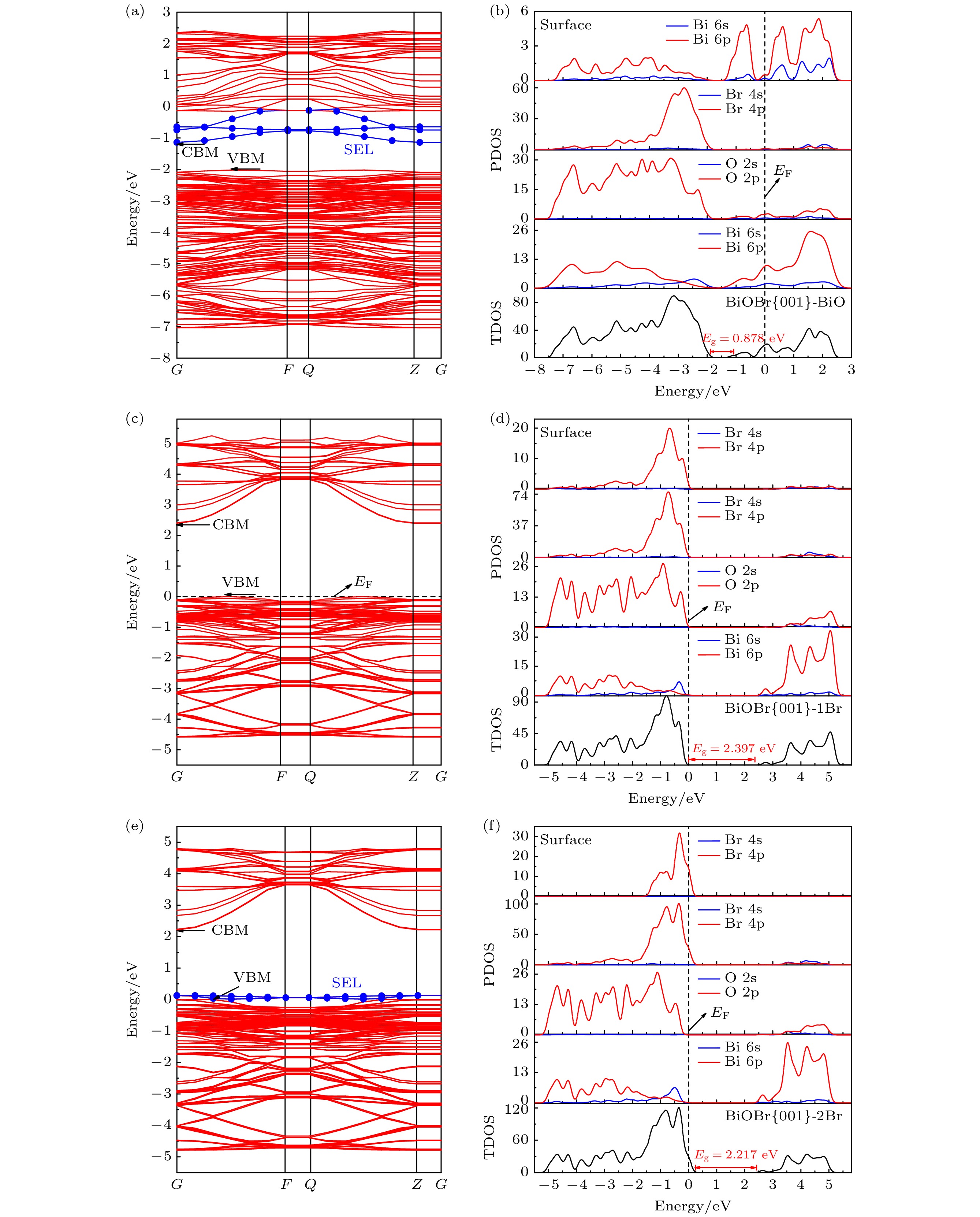 图 4 不同原子暴露终端BiOBr{001}面的能带结构和态密度图 (a), (b) -BiO; (c), (d) -1Br; (e), (f) -2Br
图 4 不同原子暴露终端BiOBr{001}面的能带结构和态密度图 (a), (b) -BiO; (c), (d) -1Br; (e), (f) -2BrFigure4. The band structures and density of states of BiOBr{001} surface with different atom exposure terminations: (a), (b) -BiO; (c), (d) -1Br; (e), (f) -2Br.
为衡量不同原子暴露端BiOBr{001}体系的稳定性, 依据下面公式[30]计算其表面能(Esurf):
| Surface | Esurf/(J·m–2) | Erel/(J·m–2) | Esurf /(J·m–2) [16] | W/eV | VBM | CBM | Eg/eV | SEL |
| {001}-BiO | 2.244 | –0.137 | 2.2—2.4 | 2.576 | G –2.020 | G-F –1.142 | 0.878 | –1.142—0.126 |
| {001}-1Br | 0.005 | –0.002 | –0.2—0.3 | 7.203 | G-F 0 | G 2.397 | 2.397 | –3.825—0 |
| {001}-2Br | 2.142 | –0.025 | 2.2—2.5 | 7.566 | G-F 0 | G 2.217 | 2.217 | 0.011—0.128 |
表1不同原子暴露终端BiOBr{001}面的表面能和电子性质计算结果
Table1.The calculation results of the surface energy and electronic properties of the BiOBr{001} surface with different atom exposure terminations.
从表1数据可以看出, BiOBr{001}-BiO表面能为2.244 J·m–2, 是三种体系中最高值, 表明以BiO为终端的{001}面最不稳定, 这与切割表面后产生的悬挂键有关, BiOBr{001}-1Br体系最低表面能说明该表面体系稳定性最好, 这与上述该表面体系类似BiOBr体相结构的结论一致. 我们还得知在BiOX (X = F, Cl, Br)晶体的三种原子暴露终端结构中, {001}-1X面的结构稳定性都是最好的[31,32]. 此外, 从表1数据可知, BiOBr{001}-BiO表面的价带顶下移到–2.020 eV, 具有良好的氧化能力, 三者带隙大小排序分别是{001}-1Br >{001}-2Br > {001}-BiO, 与文献报道完全吻合[33].
为研究三种暴露不同原子端表面体系的光学性质和电荷转移行为, 图5和图6分别给出其光学吸收谱图和差分电荷密度图(采用0.5 eV剪刀算符修正光学性质, 使其更接近实验值). 与体相BiOBr相比, BiOBr{001}-BiO表面体系在200 nm处光吸收强度大幅度减弱, 但吸收带边出现明显的红移, 在300—600 nm的波长范围内有较强的光吸收(图5), 可能归因于导带下方出现的SEL作为电子跃迁的“跳板”, 降低了光生电子跃迁到导带的能量阈值. 而BiOBr{001}-1Br和BiOBr{001}-2Br表面体系的光学性质与体相BiOBr无明显差别, 这与1Br表面体系类似体相结构有关, 而BiOBr {001}-2Br表面体系尽管在价带上方出现SEL, 由于位置太浅且表现p型特征, Kong等[32]采用DFT方法计算不同原子暴露终端BiOI{001}表面的光学性质, 得到的趋势与我们的计算结果一致.
 图 5 不同原子暴露端的BiOBr{001}表面的光学吸收谱图
图 5 不同原子暴露端的BiOBr{001}表面的光学吸收谱图Figure5. The optical absorption spectrum of the BiOBr{001} surface with different atom exposure terminals.
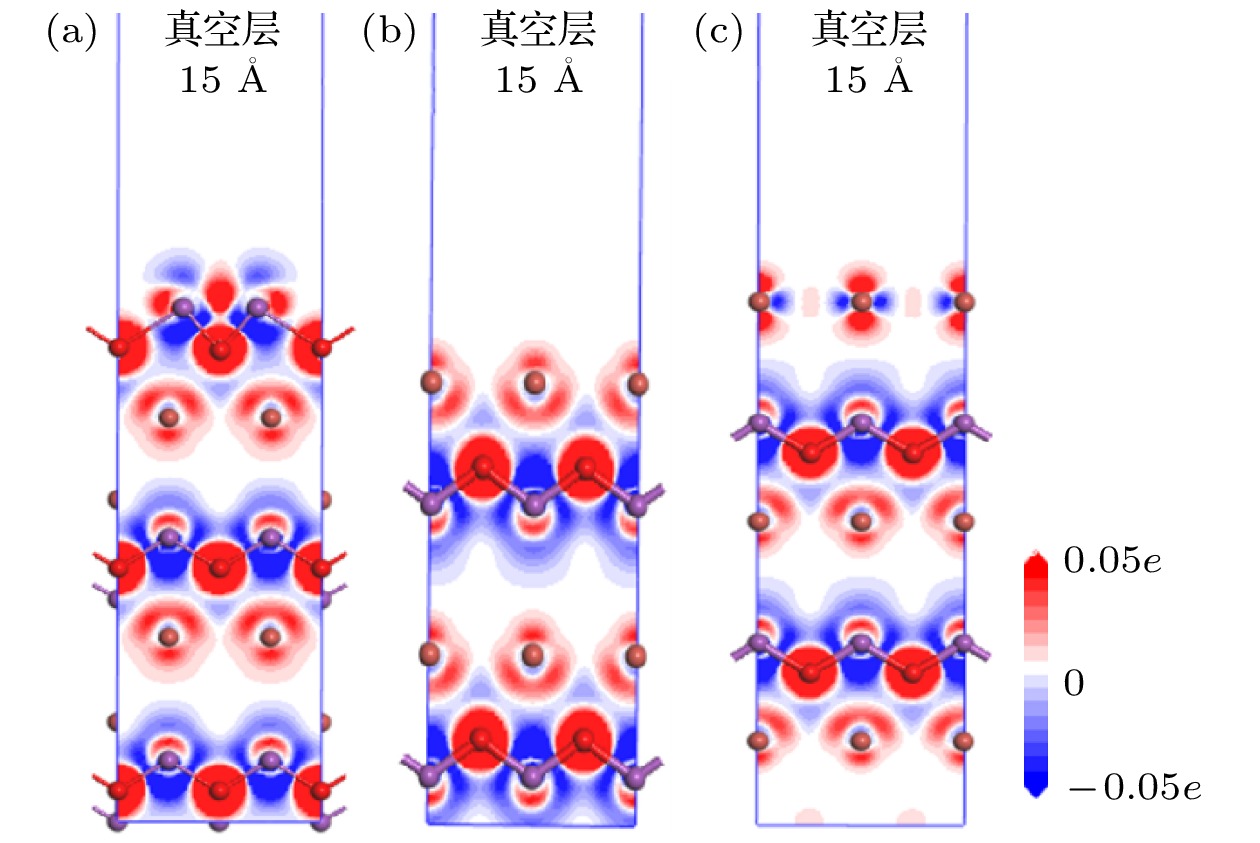 图 6 不同原子暴露端BiOBr{001}表面的差分电荷密度图 (a) -BiO; (b) -1Br; (c) -2Br
图 6 不同原子暴露端BiOBr{001}表面的差分电荷密度图 (a) -BiO; (b) -1Br; (c) -2BrFigure6. The difference charge density of the BiOBr{001} surface with different atom exposure terminals: (a) -BiO; (b) -1Br; (c) -2Br.
由图6可知, BiOBr{001}-BiO体系的表面Bi原子周围富电子区域和贫电子区域共存且交织, Bi原子上下为贫电子区域, 左右为富电子区域, 这与Bi成键的O原子电负性较大, 吸引电子的能力比Bi强有关, 而BiOBr{001}-1Br体系几乎无明显变化, 电子几乎不发生转移, 这也与上述关于电子结构的分析结果很好地一致. 不难发现, BiOBr{001}-2Br体系2Br原子周围的富电子区域和贫电子区域也是共存且交织, 但Br原子上下为富电子区域, 左右为贫电子区域, 与{001}-BiO体系的趋势是相反的.
2
3.3.单原子Pt负载于BiOBr{001}-BiO表面的不同吸附位置
为澄清单原子Pt吸附于BiOBr{001}-BiO体系不同位置的电子性质和吸附行为, 选择了光响应能力最好且功函数最低的BiOBr{001}-BiO体系作为基底, 将单原子Pt吸附于其顶位(T), 桥位(B)和穴位(H) (TPt/BiOBr{001}-BiO, BPt/BiOBr{001}-BiO, HPt/BiOBr{001}-BiO).图7(a),(b)为TPt/BiOBr{001}-BiO体系的能带结构和态密度图. 由图7(a)可发现价带上方出现杂质能级(impurity energy levels, IELs), 是由顶位Pt 5d轨道贡献, 占据–2.3 — –1.5 eV能量范围(图7(b)). 对比BiOBr{001}-BiO能带结构发现(图4(a)), 顶位Pt的引入, 促使其VBM向低能方向偏移, 且保持间接带隙的特征. 然而, 引入桥位Pt, 其价带和导带稍向低能方向偏移并在禁带中出现非常明显的杂质能级, 由桥位Pt 5d态主导(图7(d)). 图7(e),(f)显示, HPt/BiOBr{001}-BiO体系的能带结构与TPt/BiOBr{001}-BiO体系的特征相似, 均在价带上方产生Pt 5d态, 但穴位Pt诱导的杂质能级局域化较显著, 此外, 穴位Pt引入, 使其价带和导带向高能方向移动. 总之, 单原子Pt引入主要对VBM和IELs产生贡献.
 图 7 单原子Pt在BiOBr{001}-BiO面不同吸附位置的能带结构和态密度图 (a), (b) TPt; (c), (d) BPt; (e), (f) HPt
图 7 单原子Pt在BiOBr{001}-BiO面不同吸附位置的能带结构和态密度图 (a), (b) TPt; (c), (d) BPt; (e), (f) HPtFigure7. The band structure and density of states of single-atom Pt at different adsorption positions on BiOBr{001}- BiO surface: (a), (b) TPt; (c), (d) BPt; (e), (f) HPt.
为了衡量吸附体系的稳定性及金属-半导体之间相互作用, 根据下面公式[18,30]计算其吸附能:
| Sites | Eads /(J·m–2) | Pt-Bi length/? | VBM | CBM | Eg/eV | Pt 5d states | SEL |
| T | –5.189 | 2.612 | G-F –2.379 | G –0.745 | 1.634 | –2.307— –1.497 | –0.745—0.063 |
| B | –5.427 | 2.568 | F –2.045 | G –0.741 | 1.634 | –1.688 — –1.609 | –0.741—0.517 |
| H | –6.087 | 2.831 | G F –2.101 | G –0.746 | 1.356 | –2.051 — –1.777 | –0.746—-0.013 |
表2单原子Pt在BiOBr{001}-BiO面不同吸附位置的吸附能、功函数和电子性质的计算结果
Table2.The calculation results of the adsorption energy, work function, and electronic properties of single-atom Pt at different adsorption positions on BiOBr{001}-BiO surface.
图8给出了单原子Pt吸附于BiOBr{001}-BiO不同位置的光学吸收谱图. 与BiOBr{001}-BiO体系比较发现, 单原子Pt吸附体系的光学性质均发生变化, 顶位Pt和桥位Pt的引入, 虽然其光学吸收带边无明显向长波方向移动, 但在450—700 nm之间出现明显的吸收拖尾, 光响应能力依然增强. 更令人惊喜地是, HPt/BiOBr{001}-BiO体系的吸收带边红移, 大幅度拓宽光响应范围, 增强光催化活性[9,35,36].
 图 8 单原子Pt在BiOBr{001}-BiO面不同吸附位置的光学吸收谱图
图 8 单原子Pt在BiOBr{001}-BiO面不同吸附位置的光学吸收谱图Figure8. The optical absorption spectrum of single-atom Pt at different adsorption positions on BiOBr{001}-BiO surface
为进一步理解不同吸附位置Pt原子与BiOBr{001}-BiO之间的相互作用和电荷转移情况, 图9分析了三种吸附体系的差分电荷密度, 图中红色表示得电子, 蓝色表示失电子. 结果发现, Pt吸附在BiOBr{001}-BiO表面的顶位、桥位和穴位, Pt原子失去电子而带正电, 说明Pt是良好的电子受体, 特别是顶位Pt和桥位Pt原子处产生的贫电子区域是开放性的, 可预测该体系可有效的活化吸附于其表面小分子(如CO2, CO, O2, N2等)[34,37]. 此外, Mulliken布居分析(见表3)数据进一步证实单原子Pt是良好的电子受体, 且穴位Pt接受电子能力最强(–0.920e).
 图 9 单原子Pt在BiOBr{001}-BiO面不同吸附位置的差分电荷密度图 (a) TPt; (b) BPt; (c) HPt
图 9 单原子Pt在BiOBr{001}-BiO面不同吸附位置的差分电荷密度图 (a) TPt; (b) BPt; (c) HPtFigure9. The differential charge density of single-atom Pt at different adsorption positions on BiOBr{001}-BiO surface: (a) TPt; (b) BPt; (c) HPt.
| Systems | Pt | BiOBr{001}-BiO | TPt/BiOBr{001}-BiO | BPt/BiOBr{001}-BiO | HPt/BiOBr{001}-BiO |
| Work function /eV | 5.650 | 2.576 | 3.300 | 3.254 | 3.001 |
| Pt Mulliken charge ?Q /e | — | — | –0.810 | –0.860 | –0.920 |
表3Pt/BiOBr{001}-BiO体系的功函数W和Mulliken电荷变化值 ?Q
Table3.Work function W and Mulliken charge change value ?Q of Pt/BiOBr{001}-BiO.
基于以上计算结果, 分析探究了Pt/BiOBr{001}-BiO光催化体系电子转移行为, 如图10所示绘制了Pt/BiOBr{001}-BiO体系的光生电子转移机理示意图, Pt(5.650 eV)功函比BiOBr{001}-BiO(2.576 eV)高, 在M-S接触界面, 光生电子从BiOBr{001}-BiO表面定向流动到原子Pt, 直到两者的费米能级达到一致, 由于电子转移过程发生能带弯曲, 接触表面出现损耗层, M-S之间产生肖特基势垒, 确保在M-S界面上电荷载流子发生高效单向转移, 从而有效地抑制了光生电子-空穴的复合.
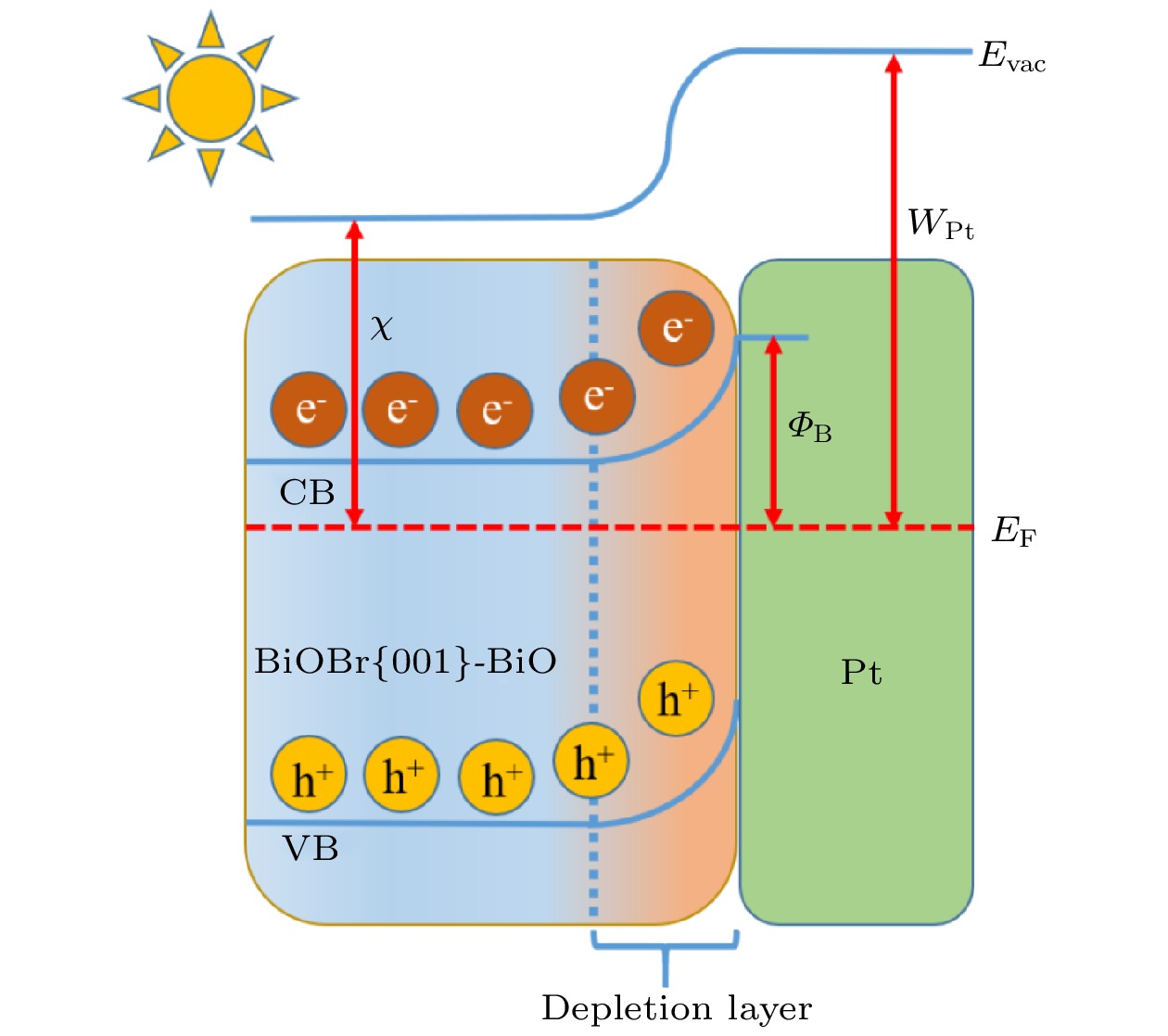 图 10 Pt/BiOBr{001}-BiO光催化剂体系的电子转移机理
图 10 Pt/BiOBr{001}-BiO光催化剂体系的电子转移机理Figure10. Possible electron transfer mechanism of Pt/BiOBr{001}-BiO photocatalyst system.
