全文HTML
--> --> -->众所周知, 对于分子结体系, 分子的末端基团不同、电极界面结构不同[55-57], 都会导致分子结与电极间的作用不同. 据此, 利用分子结界面间的作用强弱对界面结构的依赖关系, 通过对作用力的测量可以推断出分子与电极之间的结构及作用类型. 这对于精确识别分子结界面结构, 特别是对于实时控制界面的动态结构实现精准构建分子器件具有重要意义. 为了理解不同末端基团对界面结构的识别功能, 在第一性原理计算基础上, 构建了以二苯乙烯为分子骨架[56], 分别以—S, —SH, —NH2, —NO2为末端基团的分子结. 研究了各分子结在电极拉伸和断裂过程中表现出的动力学差异, 并通过分子-电极结合能、扩展体系分子轨道空间分布以及自然键轨道 (natural bond orbital, NBO)系统分析了不同分子末端与金电极间相互作用的内在物理机理. 在此基础上结合已有实验及理论结果[56-62], 讨论了含S或含N的末端基团在体系拉伸断裂过程中表现出的对金电极界面的识别功能.
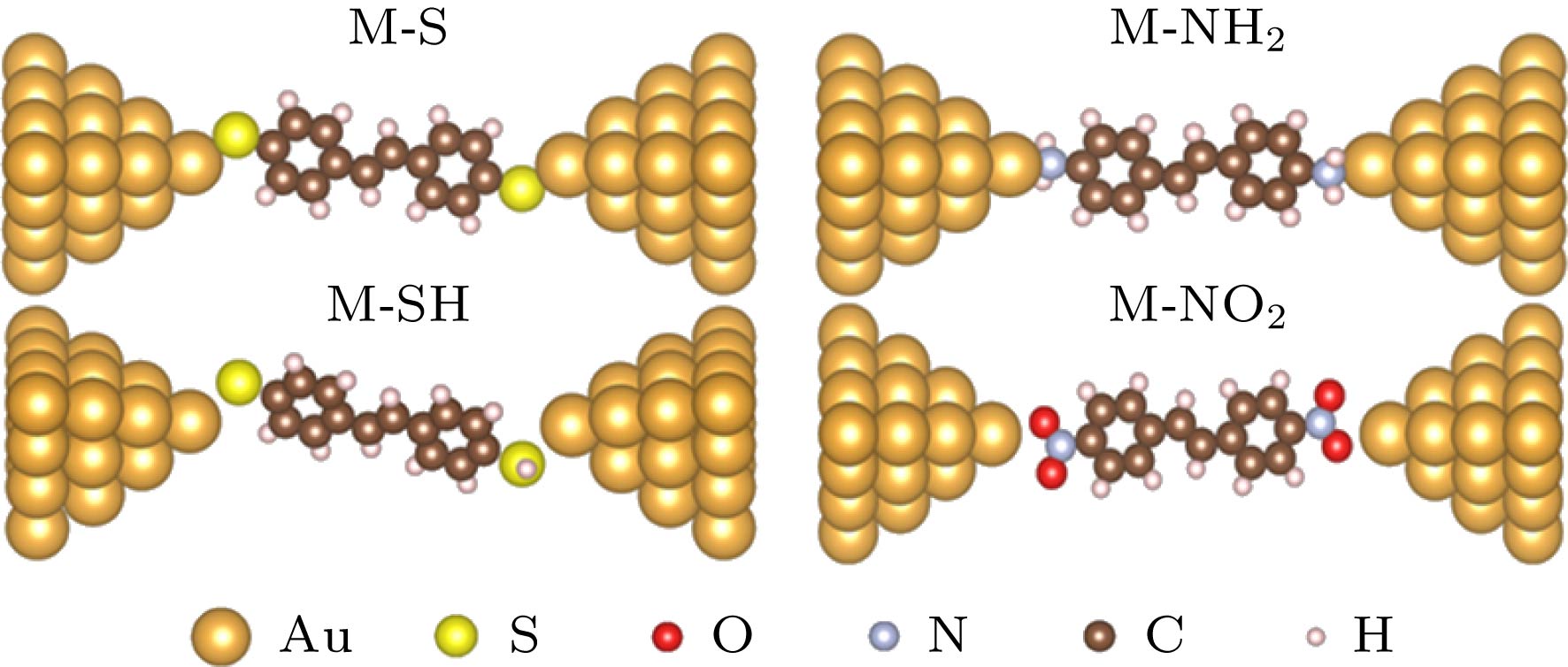 图 1 M-S, M-SH, M-NH2和M-NO2分子结体系的界面构型
图 1 M-S, M-SH, M-NH2和M-NO2分子结体系的界面构型Figure1. Interface configurations for M-S, M-SH, M-NH2 and M-NO2 molecular junctions.
体系结构优化基于杂化的密度泛函理论, 计算采用B3LYP杂化交换关联泛函, 选用LanL2DZ基组, 在Gaussian 09程序包上进行. 在优化分子结的过程中固定了两端金电极的最外一层金原子, 其他金原子以及中间分子的自由度全部放开进行优化. 在此优化过程中先找到电极之间的平衡距离, 然后再对分子结进行拉伸或压缩. 在拉伸或压缩分子结的过程中逐步改变电极距离并对体系进行几何结构优化, 其中两电极中被固定的金原子层之间的垂直距离定义为电极距离d. 每一次拉伸或压缩均将两电极最外层金原子等步长地向外或者向里移动一小段距离, 其他金原子和分子则以前一步优化好的结构作为初始结构进行优化. 最后利用优化后体系的单点能(E)与电极距离(d)之间的关系进一步计算出电极对分子的作用力

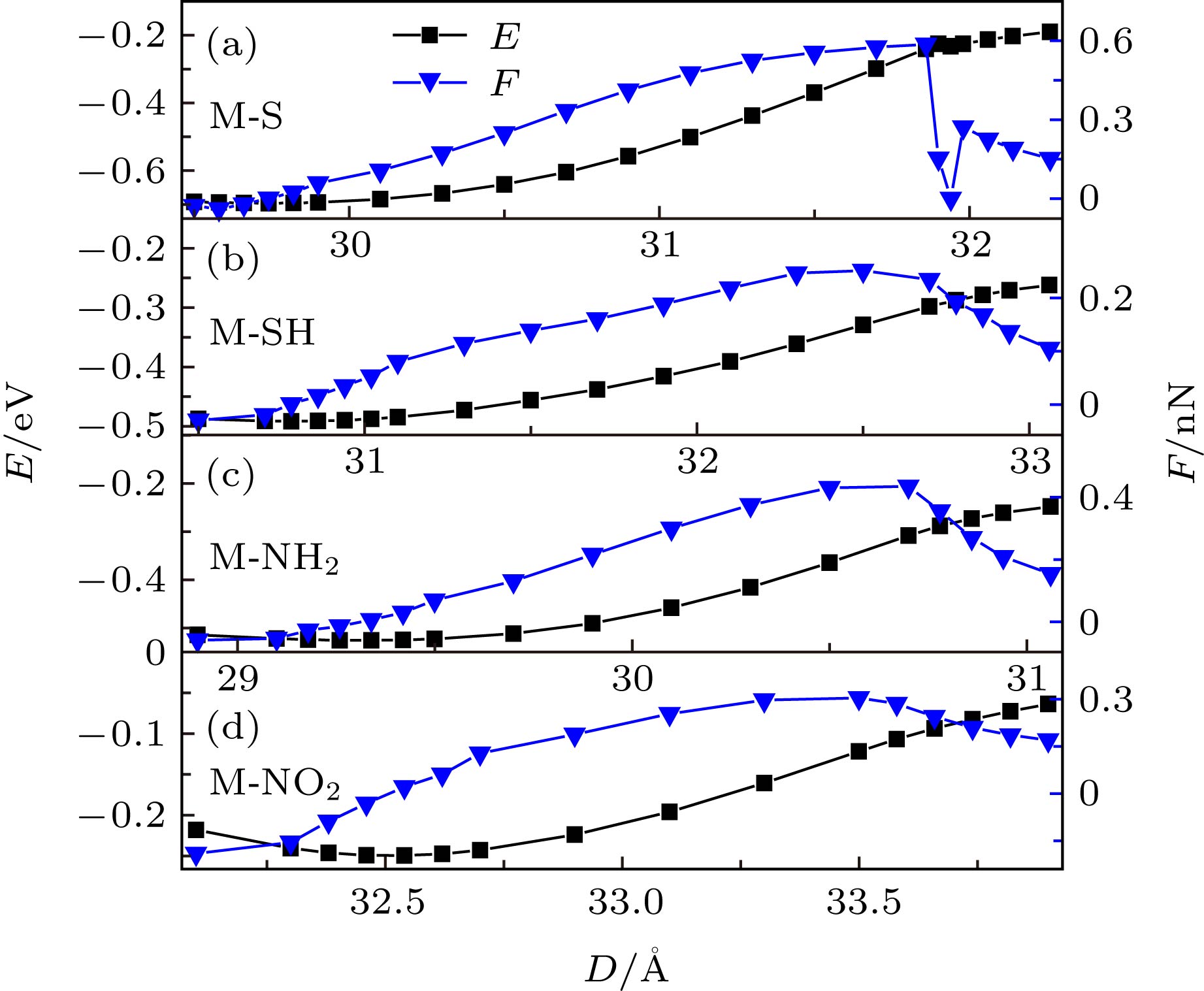 图 2 M-S, M-SH, M-NH2和M-NO2分子结体系的能量及作用力随电极距离的变化曲线
图 2 M-S, M-SH, M-NH2和M-NO2分子结体系的能量及作用力随电极距离的变化曲线Figure2. Energy and force curves as functions of electrode distances for M-S, M-SH, M-NH2 and M-NO2 molecular junctions
 图 3 M-S, M-SH和M-NH2分子结体系的拉伸过程及分子相对于电极的旋转演化过程
图 3 M-S, M-SH和M-NH2分子结体系的拉伸过程及分子相对于电极的旋转演化过程Figure3. Stretching processes for M-S, M-SH and M-NH2 molecular junctions and rotation-evolution processes of the molecules relative to the electrodes of the molecular junctions.
M-SH分子体系在3.08 nm时达到平衡, 在拉伸过程中, 中间分子初始处于倾斜状态, 随着电极距离的增加, 分子末端的S原子逐渐移动到两尖端金原子的连线上. 当电极距离大约为3.27 nm时(如图2所示), 分子结断裂. 可以看出, M-SH分子体系的平衡距离和断裂距离均大于M-S分子体系, 这是因为H原子的存在一定程度上延长了分子与电极的作用长度. M-SH分子体系断裂时, 中间分子绕着两末端S原子所在的轴发生了大约30°的旋转(如图3(b)所示). 分子结断裂前的最大拉力约为0.25 nN, 这一作用力明显小于—S末端从金电极上断裂下来的作用力. 由此可以预测, 实验中构建分子结所用的功能分子若以—SH为末端, 通过分子结断裂力的测量, 可以判断末端—SH上的H原子是否发生解离. 我们注意到, 在Xu等[59]的实验统计结果中, 0.23 nN处出现了明显的断裂峰, 这表明其实验组装的分子结样品中, H原子确实有一定的概率未从—SH基末端上解离下来就直接连到了金电极上. 综合以上—SH基中H原子解离和未解离的情况可见, 分子结断裂所需拉力大小与电极界面存在明显的对应关系, 利用分子结断裂力的大小不仅可以判断电极尖端处原子排列的构型, 还可以判断末端基团中H原子是否发生解离.
另外, 比较M-S体系与M-SH体系的作用力曲线可以看出, 断裂时, 作用力的变化明显具有不同的特征, M-S体系断裂时, 作用力在最大值处发生了突变式的降低, 而M-SH体系断裂时, 作用力则逐渐地、相对平滑地由最大值减小. 这一差异可以从两种末端基团与电极的不同作用方式上去理解. 一般认为, H原子解离后, S与金电极的作用方式是强的共价键作用, 而H原子未解离的—SH基与金电极之间只能通过弱的配位键作用. 共价键作用在断裂时, 共用电子对分离, 因此作用力在共用电子对分离时产生突变. 而配位键相互作用不存在电子对的分离过程, 因此作用力的变化是一个逐渐变化的过程. 为了理解M-S体系与M-SH体系在外力作用下变化过程的差异, 图4给出了M-S体系与M-SH体系在不同电极距离下的占据前线轨道的电子分布图. 图中显示, M-S分子体系在能量最低点 (d = 2.97 nm)和分子结断裂前(d = 3.20 nm), 最高占据轨道(highest occupied molecular orbital, HOMO)和HOMO-1是离域性非常好的分子轨道, 这表明M-S分子体系在断裂前, S与Au之间一直保持着良好的共价键结合. 当M-S分子体系断裂后, 在断裂的一端, 没有占据分子轨道同时扩展到分子与电极上, 这表明断裂一端的S与Au之间的共价键完全被破坏. 对于M-SH分子体系, 电极处于平衡距离 (d = 3.08 nm) 时, 只有HOMO轨道有一定的扩展性, 但电子扩展到分子中的比重非常小, 其他占据轨道基本为局域轨道. 而体系断裂前, HOMO也变成完全局域的分子轨道. 这说明即使在平衡距离下, 分子与电极的耦合也明显弱于M-S分子体系. 由于M-SH体系的占据轨道或者主要由电极贡献, 或者主要由分子贡献, 因此M-SH体系分子与电极的作用表现出了明显的配位键特征, 所以明显弱于M-S体系的共价键结合. 这一差异从分子与电极的结合能上也可以明显看出. 基于能量曲线, 分子结一端的结合能可以近似表示为



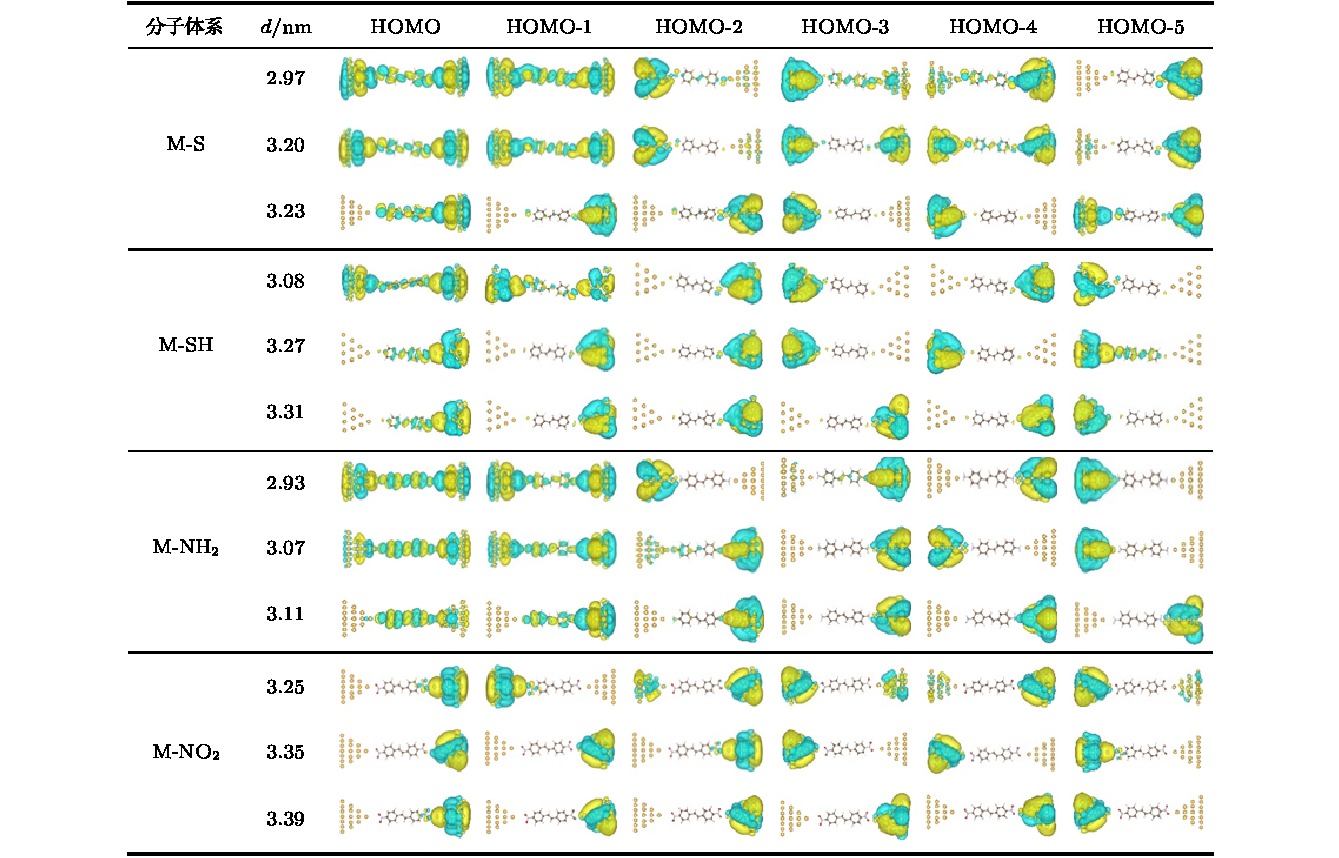 图 4 M-S, M-SH, M-NH2和M-NO2分子结体系在能量最低点以及体系断裂前后的轨道空间分布图
图 4 M-S, M-SH, M-NH2和M-NO2分子结体系在能量最低点以及体系断裂前后的轨道空间分布图Figure4. Spatial distributions of molecular orbitals for M-S, M-SH, M-NH2 and M-NO2 molecular junctions at the lowest ground-state energy points, before and after the breaks of the systems.
| 体系 | M-S | M-SH | M-NH2 | M-NO2 |
| 结合能E/eV | 0.505 | 0.229 | 0.277 | 0.186 |
| 成键轨道数 | 1 | 0 | 0 | 0 |
| 孤对电子数 | 2 | 2 | 1 | 3 |
| NBO净电荷 | S (–0.056) | S (0.020) | N (–0.910) | O (–0.412) |
表1M-S, M-SH, M-NH2和M-NO2体系分子与电极间的结合能、末端原子与电极间的成键轨道数、末端原子的孤对电子数以及末端原子的NBO净电荷数
Table1.Binding energies between the molecules and the electrodes, the numbers of bonding orbitals between the terminal atoms and the electrodes, the numbers of lone electrons on the terminal atoms and the NBO net charges on the terminal atoms for M-S, M-SH, M-NH2 and M-NO2 molecular junctions.
M-NH2分子体系电极的平衡距离约为2.93 nm, 断裂距离约为3.07 nm, 电极的平衡距离和断裂距离都小于M-S和M-SH体系的相应的数值, 这是由于N原子为第二周期元素, 其半径小于第三周期S原子的半径. 断裂前的最大拉力约为0.45 nN(如图2所示), 这一数值比饱和碳链末端氨基与金电极之间的断裂力(0.6—0.7 nN)明显小[61]. 这是因为, 受苯环中π电子的影响, 与苯环相连的氨基上的轨道变为sp2杂化; 而饱和碳链末端的氨基上的轨道受饱和碳链的影响具有明显的sp3杂化特点. 当然, 与苯环相连的氨基, 在与尖端的金原子相连的时候, 金原子对其轨道产生了影响, 使其轨道由原来纯的sp2杂化变成sp2与sp3混合杂化的特点. 从而导致了—NH2中的孤对电子不能像纯的sp3杂化的—NH2中的孤对电子一样与尖端金原子充分接触发生作用, 因此进一步导致M-NH2分子体系断裂力明显小于饱和碳链末端氨基从金电极上断裂下来所需的作用力[61]. 断裂后, 分子和电极由于弛豫作用而发生回缩现象, 同时分子绕两末端基团中N原子的连线发生了一定角度的旋转(如图3(c)所示). 另外, 在我们以前的研究中发现, 苯环上的—NH2与平面金电极相连时不出现sp3杂化现象[6], 因此从平面金电极上断裂下来所需的作用力只有不到0.2 nN[6], 因此—NH2末端对金电极界面构型也具有识别功能.
不同于M-NH2分子与金电极的连接发生在N原子与尖端金原子之间, M-NO2分子与金电极的连接发生在其中一个O原子与尖端金原子之间. 因此, M-NO2分子体系平衡时的电极距离(d = 3.25 nm)和断裂时的电极距离(d = 3.35 nm)明显比M-NH2分子体系的大. 断裂前的最大拉力约为0.33 nN. 在电极的拉伸过程中, 分子末端的—NO2与中间的二苯乙烯分子骨架部分始终保持着平面构型, 即末端的轨道不会因为电极尖端金原子的作用而发生类似于—NH2的sp3杂化. 这是因为, 在—NO2基中, 由于O原子的作用, N原子的成键已达到饱和, 所以不存在多余的孤对电子, 因此M-NO2分子与电极的连接发生在末端的一个O原子与尖端金原子之间. 虽然—NO2中的O原子也存在孤对电子, 但由于O原子的电负性明显强于N原子, 所以不利于O原子与尖端金原子之间产生电子态的耦合, 因此—NO2末端与尖端金原子的作用明显弱于—NH2末端与尖端金原子的作用. 实际上, 整个—NO2是电负性非常强的基团, 会阻隔分子骨架的π电子与金电极发生耦合. 从分子轨道的分布上也可以明显看出M-NH2分子与金电极通过HOMO和HOMO-1发生了明显的耦合作用, 而M-NO2分子中只有离尖端金原子最近的O原子与电极之间发生了一定程度的轨道耦合(图4). 这也导致了M-NH2分子体系的结合能(0.28 eV)明显大于M-NO2分子体系0.19 eV的结合能.
利用自然键轨道 (NBO)分析可以更直观地理解不同分子末端与电极尖端原子的相互作用. 计算显示, M-S体系的末端S原子与电极尖端的Au原子之间存在一个成键轨道和一个反键轨道, 另外S原子还拥有两对孤对电子. 在成键轨道中S的贡献为70.63%, Au的贡献为29.37%, 而反键轨道中S的贡献为29.37%, Au的贡献为70.63%. M-SH, M-NH2和M-NO2体系末端与电极尖端的Au原子之间均未形成成键轨道, 只能通过分子末端的孤对电子与电极相互作用, 其中M-SH体系中末端S原子有两对孤对电子, M-NH2体系中末端N原子只有一对孤对电子, 而M-NO2体系中与电极直接相连的O原子中有三对孤对电子. 由于—NH2中的N原子只有一对孤对电子, 与Au原子的作用更具定域性, 因此相对于M-SH和M-NO2体系, M-NH2体系中分子与电极形成了较强的配位键. M-SH和M-NO2体系由于末端孤对电子较多, 因相互排斥导致相互作用的定域性较差, 因此与电极间作用较弱, 特别是S原子相对于O原子半径较大, 所以M-SH体系的末端与电极间作用更弱.
根据NBO分析, 将分子末端原子在各轨道中的电子占据数求和可以得到末端原子的NBO总电子占据数, 再根据NBO总电子占据数和原子核(实)电荷数进一步计算得到末端原子的NBO净电荷数如表1所列. 表中显示, 对于未成键的M-SH, M-NH2和M-NO2体系, 末端原子的NBO净电荷数与体系的断裂力之间表现出了极大的相关性. 其中M-NH2体系中N原子的NBO净电荷数高达约–0.9 e, 明显高于M-NO2体系中末端O原子约–0.4 e的NBO净电荷数和M-SH体系中末端S原子上约0.02 e的NBO净电荷数. 这一结果显示出, 若分子末端原子与电极间不能形成成键轨道, 末端原子上带有更多的NBO净电荷数, 可使分子与电极间的结合更稳定.
