全文HTML
--> --> -->近年来, 电场诱导极性材料储氢逐渐受到人们的关注[6—11]. 电场极化增强了主体材料与H2的静电作用, 从而提高对H2的吸附强度; 而将外电场移除, 又可实现快速脱氢. 通过对电场诱导极性材料(单层BCN, BC2N, BN, AlN和 C纳米材料等)储氢的研究[6—8,10,11], 表明电场能显著改善其储氢性能. 然而, 所需外加的电场强度约为0.04—0.05 a.u. (1 a.u. = 5.1 × 1010 V/m), 如此强的电场, 目前还无法实现. 因此, 如何降低所需的电场强度是电场储氢需要解决的关键问题.
采用极性更强的材料是降低场强的一种可能方式. MgO是具有较强极性的离子化合物. 虽然H2在MgO晶体表面的吸附很早就受到人们的关注, 而理论与实验研究表明H2通常在MgO晶体表面形成弱的物理吸附[12—16]. 纳米材料如团簇因其极大的比表面积和独特的电子结构而具有比相应体相材料更优越的吸附性能. 虽然团簇储氢面临诸多挑战, 如实验研究比较困难; 如何能在温和条件下操作的储氢以及寻找具有高存储密度和优良循环动力学性能的新储氢体系等[2]. 通过理论研究能在原子、分子水平上探究储氢的微观机制, 从而设计合理的储氢方法, 并通过调控团簇的尺寸及组成, 为寻找适宜的储氢体系及相关的实验研究提供理论指导[3,17]. Kwapien等[18]利用从头计算方法研究了H2在(MgO)n (n = 6, 8)上的吸附及解离. Chen 等[19]也在B3LYP/6-31G水平上研究了H2在 (MgO)9和(MgO)12幻数团簇上的吸附性质. 虽然 (MgO)n团簇的储氢能力较MgO晶体有所改善, 但其吸附能仅为–0.03—–0.08 eV, 而将MgO材料置于外电场中, 可进一步提高其储氢性质. 实验上Sun等制备了多孔MgO材料, 发现外加电场后其储氢能力得到一定程度的提高[9]. 我们之前的理论工作也证实电场能显著改善(MgO)9储氢性质, 且所需电场强度仅为0.025 a.u.[20]. 鉴于此, 我们关心若减小团簇尺寸, 能否进一步降低所需外场强度?具有立方结构的(MgO)4是幻数团簇, 特别稳定, 同时也是MgO岩盐结构的基本组成单元[21—23], 因此本文研究了电场中(MgO)4的储氢性质, 结果发现电场能显著改善其储氢性质, 重要的是所需电场小于大尺寸的(MgO)9团簇, 仅需外加0.010 a.u.的电场就可使其对H2的吸附能提高到–0.225 eV, 表明减小团簇尺寸是降低电场强度的可能方式. 电场中(MgO)4中最多能吸附16个H2, 相应的质量密度为16.7 wt%, 表明(MgO)4是一种可能的电场储氢材料.





3.1.电场中(MgO)4的结构
优化得到无外场时(MgO)4的最稳定结构如图1所示. 该结构为具有Td对称性的立方结构, 因此所有Mg—O键长均为1.96 ?, 与之前文献报道的结果一致[21—23]. NPA电荷布居分析表明(MgO)4中Mg将其3s电子向O 2p轨道转移, 从而形成了Mg阳离子和O阴离子(图1左侧结构图中蓝色代表失电子的原子, 即Mg原子, 而红色小球则代表得电子的O原子). 由于(MgO)4的高对称性, 每个Mg原子转移的电荷均相同, 为1.493 e, 因此Mg与O通过强的静电相互作用结合.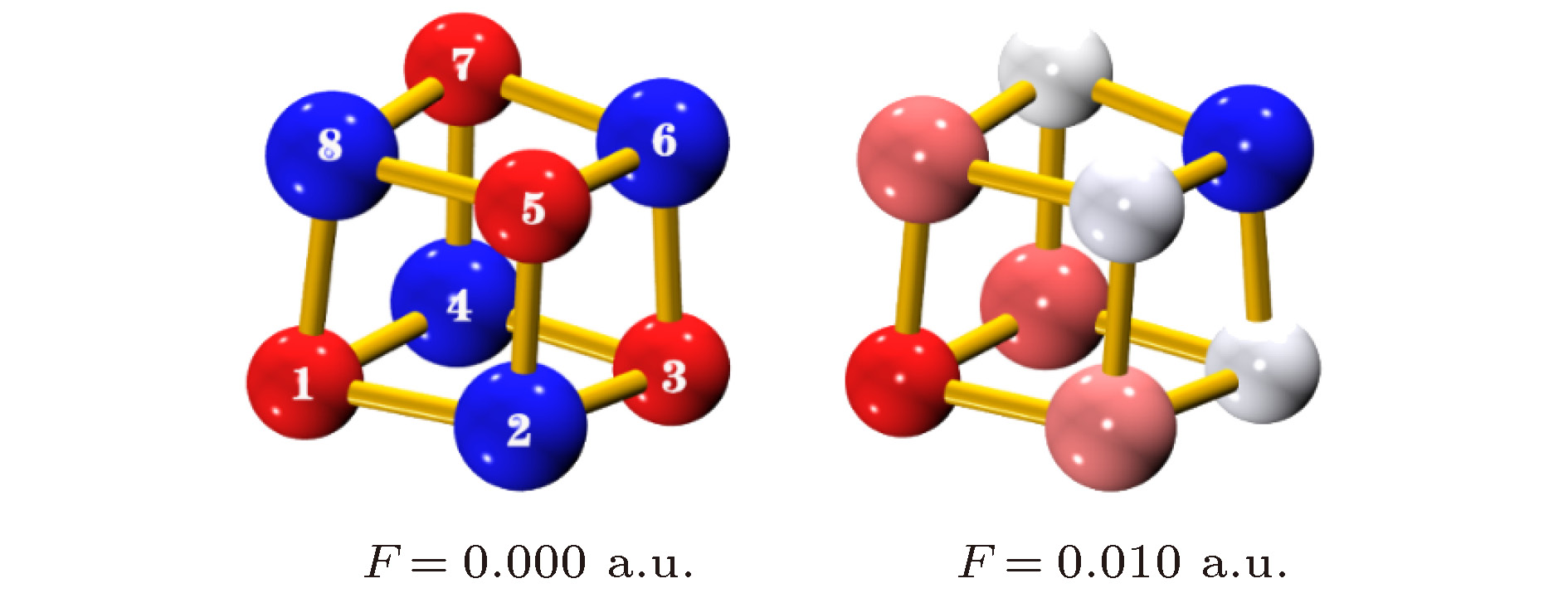 图 1 不同场强下(MgO)4的稳定结构(原子颜色与电荷得失相关, 由蓝色至红色表示失电子越多到得电子越多)
图 1 不同场强下(MgO)4的稳定结构(原子颜色与电荷得失相关, 由蓝色至红色表示失电子越多到得电子越多)Figure1. The stable structures of (MgO)4 under the electric fields with different intensities(the color is correlated with the gaining or losing of electrons, from blue to red, it represents the variation of from the losing to obtaining electrons).
我们进一步优化了强度分别为0.005 a.u. 和0.010 a.u. 的不同方向的外电场中(MgO)4的稳定结构, 结果表明当外场方向沿O1-Mg6体对角线方向时, 团簇的能量最低, 其稳定结构如图1所示(场强为0.005 a.u. 时 (MgO)4的结构与场强为0.010 a.u. 时相似, 图中未给出). 一方面电场中(MgO)4仍保持立方结构, 表明团簇能承受外电场, 可用于电场储氢. 另一方面, 由于Mg为阳离子而O为阴离子, 外加电场后Mg顺电场方向移动而O逆电场方向移动, 因此(MgO)4的几何结构有一定畸变. 表1给出了不同强度的电场中(MgO)4的Mg/O原子NPA电荷及Mg—O键长. 由于电子逆着电场方向运动, 导致沿场强方向(即O1-Mg6方向)电荷重新布居. 电场中Mg6, O3, O5和 O7(图1右侧蓝色或篮白色原子)均失电子, 而O1, Mg2, Mg4和Mg8(图1右侧红色或淡粉原子)得电子. 因此与无电场时相比, Mg和O原子均可分为两类: 一类是电荷绝对值增加的Mg6/O1; 另一类是电荷绝对值减少的Mg2, Mg4, Mg8和O3, O5, O7. 第一类Mg6/O1原子的电荷由无电场时的1.493 e/–1.493 e增加为场强为0.010 a.u. 时的1.554 e/–1.512 e, 第二类的Mg/O原子的电荷则减少为1.460 e/–1.474 e. 相应地, Mg—O键也可以分为两类: 第一类以Mg6/O1为端点的Mg—O键长RI由无电场时的1.96 ?缩短为场强0.010 a.u. 时的1.93 ?; 而其他Mg—O键长(即第二类RII)则拉长至1.99 ?. 电场诱导电荷重新分布, 使(MgO)4极化, 随着电场逐渐增强, 其偶极矩由无电场时的0 Debye 分别增加为场强为0.005 a.u. 和0.010 a.u. 时的1.67和3.33 Debye.
| F/a.u. | QMgI/QMgII | QOI/QOII | RI | RII |
| 0 | 1.493/1.493 | –1.493/–1.493 | 1.96 | 1.96 |
| 0.005 | 1.476/1.527 | –1.484/–1.503 | 1.94 | 1.97 |
| 0.010 | 1.460/1.554 | –1.474/–1.512 | 1.93 | 1.99 |
表1不同场强下(MgO)4中两类Mg/O原子的NPA 电荷(e)及Mg—O键长(?) (QMgI/QMgII和QOI/QOII分别是Mg2, Mg4, Mg8/Mg6和O3, O5, O7/O1上的电荷; Mg—O键长RI = R12 = R14 = R18 = R63 = R65 = R67, 而RII = R23 = R25 = R43 = R47 = R85 = R87)
Table1.The NPA charges for the two types of Mg/O atoms and the Mg—O distances (QMgI/QMgII and QOI/QOII are the charges of Mg2, Mg4, Mg8/Mg6 and O3, O5, O7/O1, respectively. The Mg—O distances RI = R12 = R14 = R18 = R63 = R65 = R67, while RII = R23 = R25 = R43= R47 = R85 = R87).
2
3.2.电场中单个H2在(MgO)4上的吸附
为了确定无外场时H2在 (MgO)4上的稳定吸附结构, 我们优化了H2在 (MgO)4上的各种可能吸附方式, 包括H2分别吸附在单个Mg/O原子上方, Mg—O键上方及立方体表面上等. 结果表明H2只能稳定地吸附在(MgO)4的单个Mg/O原子上, 与(MgO)9的结果相似[20]. 由于(MgO)4的高对称性, 其表面所有Mg和O原子分别等价, 因此只有两种稳定吸附结构如图2所示. H2在Mg上为侧位吸附(H2分子的方向与H2分子到 Mg离子的连线垂直), 而在O上为端位吸附(H2分子的方向与H2分子到 O离子的连线一致).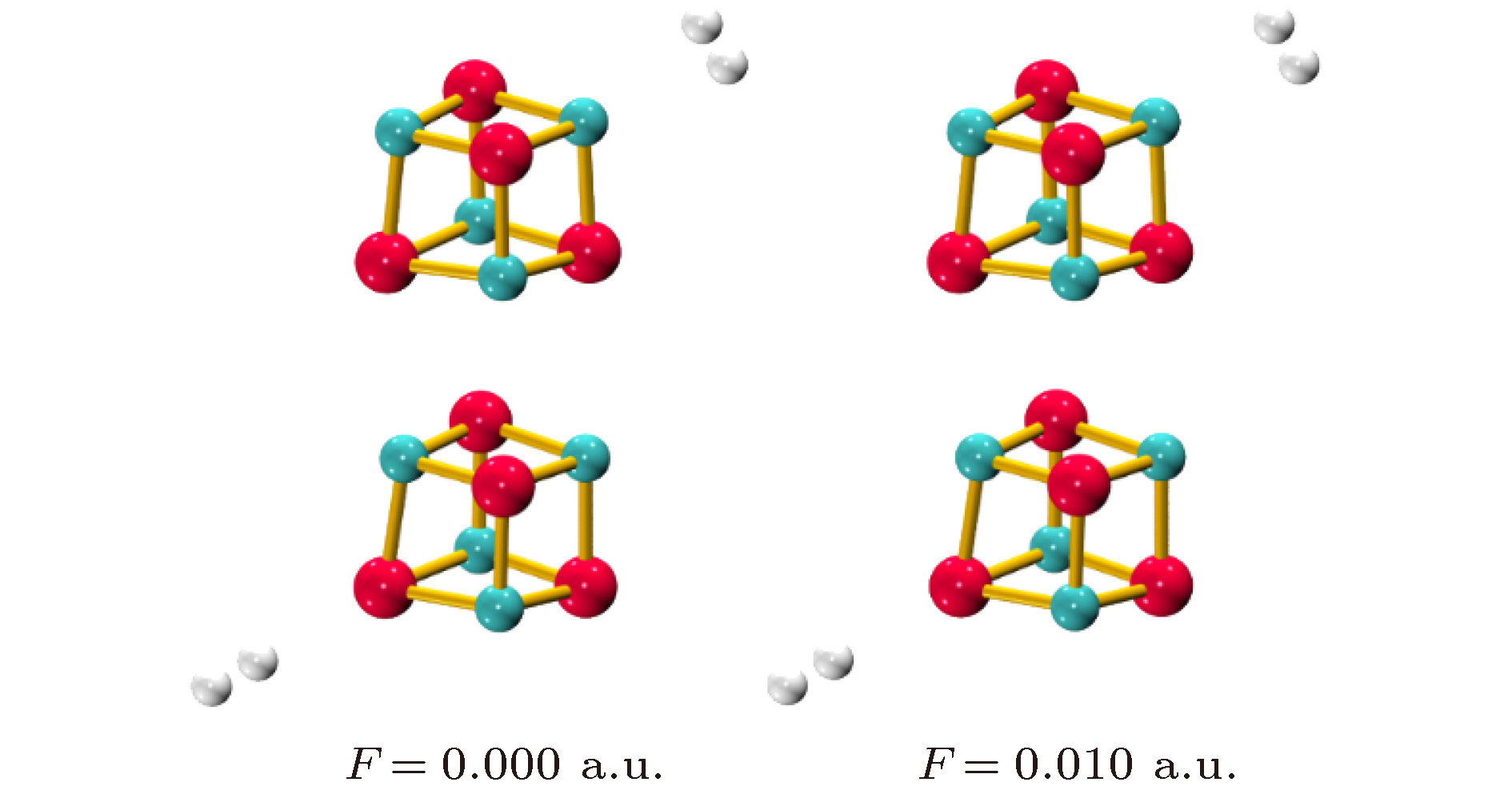 图 2 不同场强条件下H2在(MgO)4上的稳定吸附结构 (绿色球Mg原子; 红色球O原子; 灰色球H原子)
图 2 不同场强条件下H2在(MgO)4上的稳定吸附结构 (绿色球Mg原子; 红色球O原子; 灰色球H原子)Figure2. The stable structures of H2 adsorbed on (MgO)4 under the electric fields with different intensities (the green, red and gray balls are the Mg, O and H atoms, respectively)
电场中 (MgO)4结构的对称性降低, 所有Mg/O原子不再等价. 如前所述, Mg和O原子均分为两类. 一类是电荷绝对值增加的Mg6/O1, 另一类是电荷绝对值减少的Mg2, Mg4, Mg8和O3, O5, O7. 由于外加电场相对较弱, 当H2分子吸附在电荷上述六个绝对值减少的原子上时, 仍能形成吸附, 但其吸附能仅为–0.03—–0.06 eV(0.010 a.u.), 低于无电场时的吸附能; 而在Mg1及O6上时的吸附能高于无电场时的吸附能, 因此这里我们只讨论H2在这两个原子上的吸附, 相应的吸附结构和吸附能、结构参数及NPA电荷数据分别如图2和表2所示. 图2给出了场强为0.010 a.u.时, H2在即Mg6/O1上的稳定吸附结构(由于场强为0.005 a.u. 时的吸附结构与场强为0.010 a.u. 时的稳定结构相似, 图中未给出). 由于外加电场相对较弱, 与无电场时相似, H2在Mg和O上的吸附结构仍为侧位吸附和端位吸附. 分子动力学模拟结果表明中这些吸附结构在300 K条件下仍能稳定存在. 以0.010 a.u.时H2在Mg原子上的吸附结构为例, 其总能量随时间的演化如图3所示, 在整个时间范围内, 总能量以–31458.12 eV为中心波动, 且H2一直稳定吸附在(MgO)4上, 表明电场中的吸附体系是稳定的.
| Site | F/a. u. | Ea/eV | RH—H/? | RH—Mg/O/? | QH | QH2 |
| H2 on Mg | 0 | –0.118 | 0.751 | 2.217 | 0.023/0.023 | 0.046 |
| 0.005 | –0.172 | 0.752 | 2.179 | 0.030/0.030 | 0.060 | |
| 0.010 | –0.225 | 0.753 | 2.093 | 0.044/0.044 | 0.088 | |
| H2 on O | 0 | –0.060 | 0.750 | 2.365 | 0.044/–0.068 | –0.024 |
| 0.005 | –0.101 | 0.755 | 2.248 | 0.069/–0.110 | –0.041 | |
| 0.010 | –0.150 | 0.763 | 2.136 | 0.094/–0.157 | –0.063 |
表2H2在(MgO)4上的吸附能、到团簇距离RH-Mg/O, H—H键长RH—H及H和H2的NPA 电荷
Table2.The adsorption energies Ea, H—H bond lengths RH—H, distances between H2 and cluster RH2-Mg/O and NPA charges of H atoms and H2 for (MgO)4H2
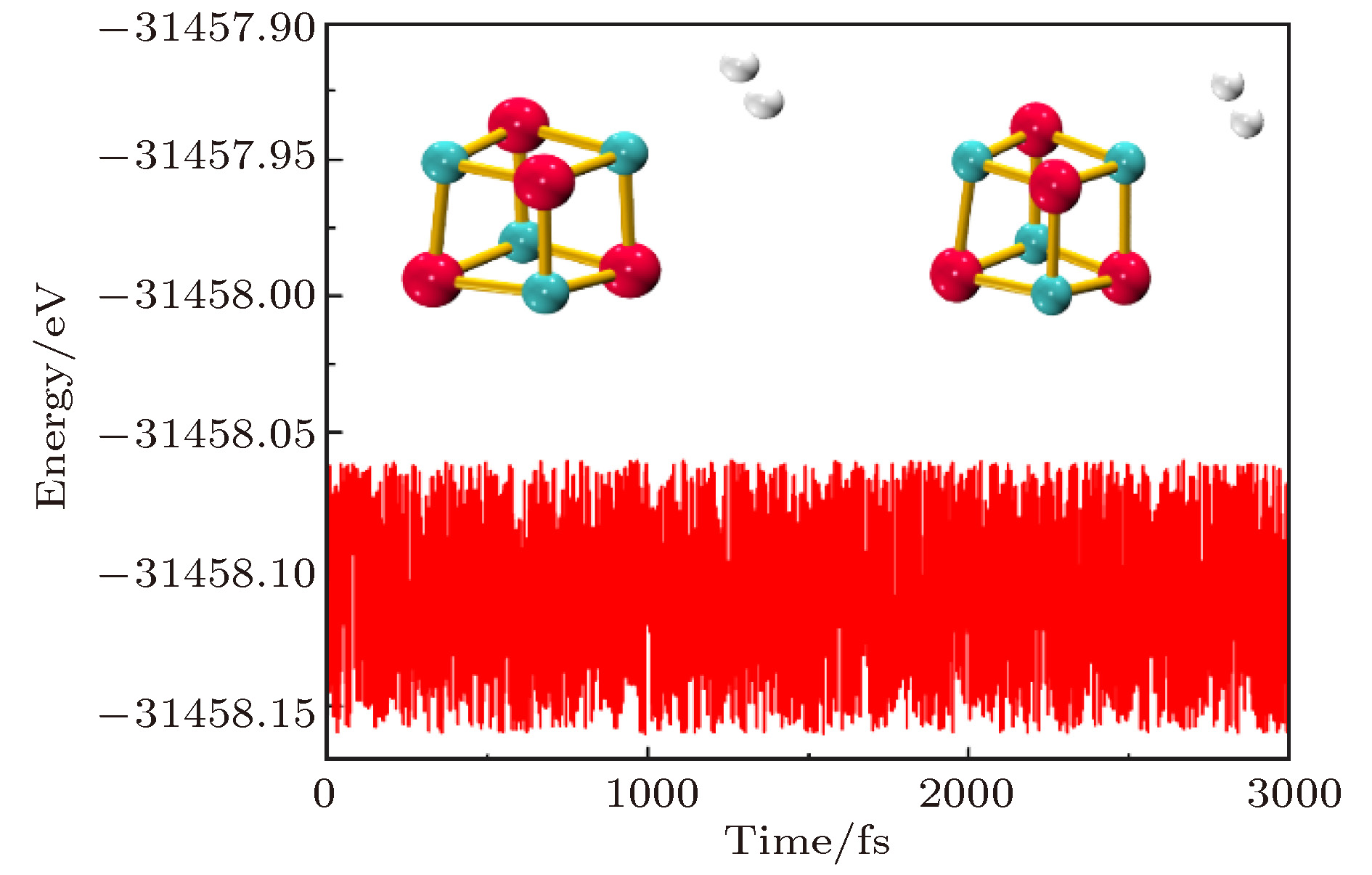 图 3 场强为0.010 a.u. H2吸附在Mg上时总能量随时间的演化(附图中为演化始末的吸附结构)
图 3 场强为0.010 a.u. H2吸附在Mg上时总能量随时间的演化(附图中为演化始末的吸附结构)Figure3. The variation in the energy for the structure of H2 adsorbed on Mg atom under the electric field with the intensity of 0.010 a. u.(the adsorbed structures are also presented in attached map)
结合图1及图2可见, 吸附H2后, (MgO)4团簇的结构未见显著改变, 即吸附过程未显著改变团簇的振动模式, 因此ZPE校正对Ea的影响较小(最大值仅为0.02 eV), 而BSSE的校正则更小(最大值仅为0.01 eV). 表2列出了ZPE及BSSE校正后的平均吸附能Ea, 随着场强增加至0.010 a.u., H2在Mg/O上吸附能均逐渐增大, 由无电场时的–0.118/–0.060 eV 增加到–0.225/–0.150 eV, 达到了理想的储氢吸附强度. 同时H2到Mg/O的距离变短, 由无电场时的2.217/2.365 ? 减少至2.093/2.136 ?. 另一方面, 电场中H—H键长变长, 由无电场时的0.751/0.750 ? 增加至0.753/0.763 ?. 上述结果表明电场使H2与团簇的相互作用增强而H—H键被削弱. 同时, 相同场强条件下, H2在Mg的吸附能总是大于在O上, 表明H2在Mg上吸附更稳定.
2
3.3.电场中(MgO)4与H2的弱相互作用
为深入理解电场中 (MgO)4与H2的相互作用, 我们采用QTAIM的拓扑分析法对其电子结构进行了研究. 拓扑分析中, 电子密度梯度为零的位置称为临界点(CP). 根据电子密度Hessian矩阵负本征值的个数, 临界点可分为四种. 若三个本征值均为负值, 称为核临界点(NCP); 若有两个负本征值, 该临界点常位于两个成键原子之间, 称为键临界点(BCP); 若只有一个负本征值, 该临界点常处于环的中间, 称为环临界点(RCP); 若没有负的本征值, 称为笼临界点(CCP). 拓扑分析得到电场中H2在Mg/O上吸附结构的临界点如图4所示.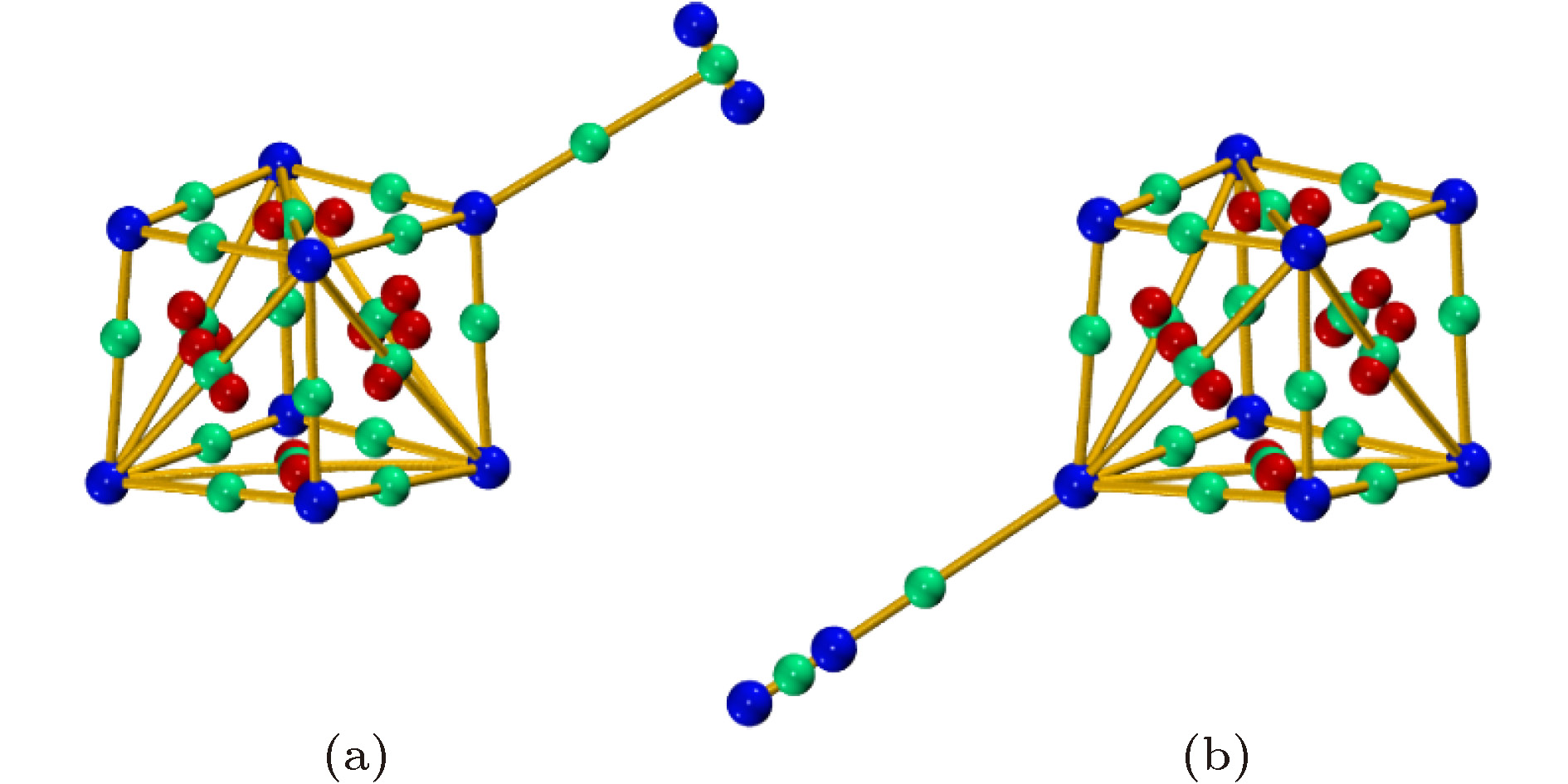 图 4 F = 0.010 a.u. 电场中H2在Mg/O上稳定吸附结构的临界点 (蓝色球为核临界点, 绿色球为键临界点; 红色球为环临界点)
图 4 F = 0.010 a.u. 电场中H2在Mg/O上稳定吸附结构的临界点 (蓝色球为核临界点, 绿色球为键临界点; 红色球为环临界点)Figure4. The critical points for the stable adsorption structures of H2 on Mg/O under the electric field with F = 0.010 a.u.(the blue, green and red balls represent the nuclear, bond and ring critical points, respectively)
由图4可见, 除了Mg/O原子的NCP和各表面的RCP外, 还有对应于Mg—O键、H—H键的BCP, 特别是出现了Mg—H键和O—H键的BCP, 表明吸附的H2与吸附位的Mg/O原子间存在相互作用. 表3列出了相应BCP的拓扑参数, 分析这些参数可以了解相应BCP的成键性质. 负的拉普拉斯电荷密度(








| F/a. u. | H2 on Mg | H2 on O | |||||||||
| BCP | ρ | ${\nabla ^2}\rho $ | H(r) | ELF | BCP | ρ | ${\nabla ^2}\rho $ | H(r) | ELF | ||
| 0.000 | Mg—H | 0.013 | 0.059 | 0.002 | 0.029 | O—H | 0.011 | 0.038 | 0.002 | 0.045 | |
| H—H | 0.266 | –1.167 | –0.292 | 1.000 | H—H | 0.265 | –1.167 | –0.292 | 1.000 | ||
| Mg—O | 0.056 | 0.403 | 0.005 | 0.057 | Mg—O | 0.056 | 0.403 | 0.005 | 0.057 | ||
| 0.005 | Mg—H | 0.014 | 0.063 | 0.002 | 0.031 | O—H | 0.015 | 0.049 | 0.002 | 0.060 | |
| H—H | 0.260 | –1.165 | –0.292 | 1.000 | H—H | 0.261 | –1.142 | –0.287 | 1.000 | ||
| Mg—O | 0.054—0.059 | 0.382—0.426 | 0.004—0.005 | 0.056—0.059 | Mg—O | 0.055—0.059 | 0.390—0.427 | 0.004—0.005 | 0.057—0.059 | ||
| 0.010 | Mg—H | 0.016 | 0.069 | 0.002 | 0.034 | O—H | 0.019 | 0.063 | 0.002 | 0.080 | |
| H—H | 0.255 | –1.162 | –0.291 | 0.999 | H—H | 0.255 | –1.102 | –0.278 | 0.999 | ||
| Mg—O | 0.051—0.061 | 0.341—0.533 | 0.004—0.005 | 0.055—0.060 | Mg—O | 0.025—0.061 | 0.358—0.444 | 0.004—0.005 | 0.054—0.060 | ||
表3H2在(MgO)4上吸附结构的拓扑参数
Table3.The topological parameters for the adsorption structures of H2 on (MgO)4
约化密度梯度(RDG(r))是研究弱相互作用的有力工具, 其定义为
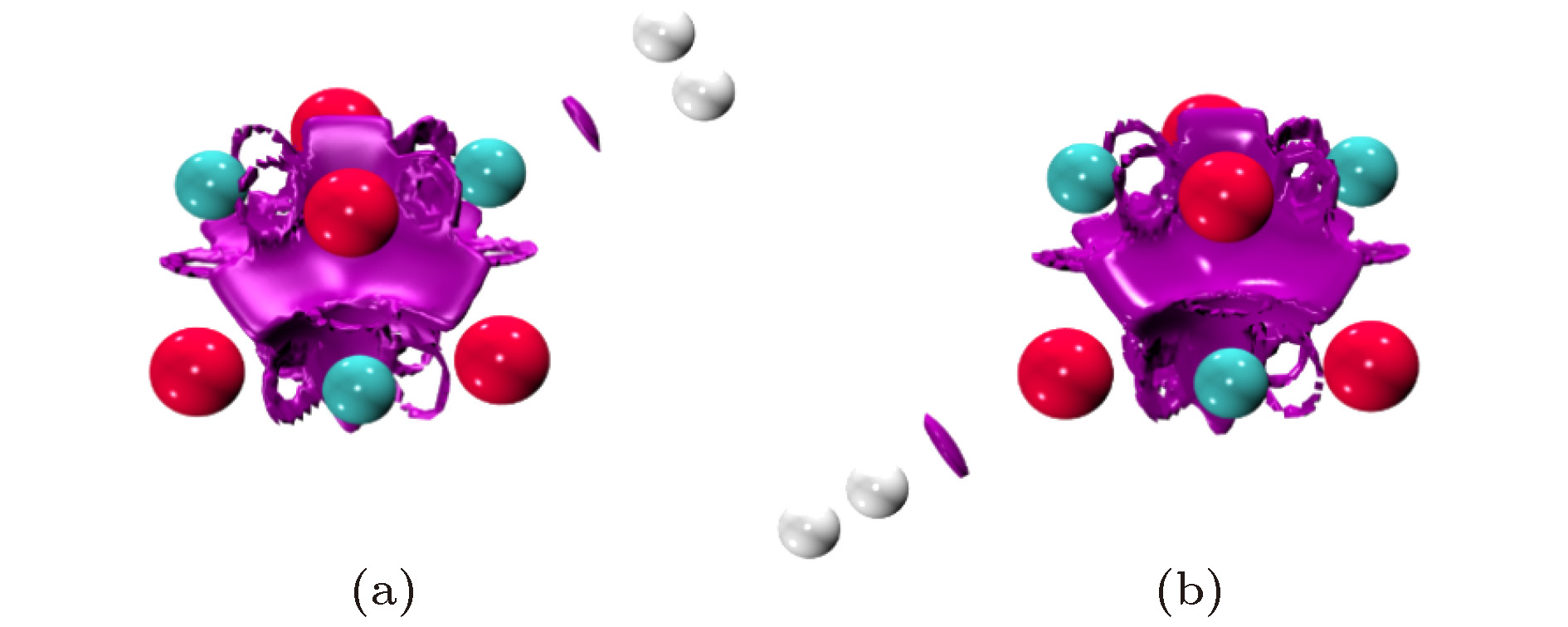 图 5 F = 0.010 a. u. 电场中H2在Mg/O离子上吸附结构的RDG等能面
图 5 F = 0.010 a. u. 电场中H2在Mg/O离子上吸附结构的RDG等能面Figure5. The RDG isosurface for the adsorption structures of H2 on Mg/O under the electric field with F = 0.010 a. u.
如何知道这两个弱相互作用的类型?根据QTAIM理论, RCP电子密度Hessian矩阵的第二个本征值(λ2)为正值, 而BCP则为负值. 同时弱相互作用强度与ρ(r)相关, 如范德瓦耳斯作用具有很小的ρ(r), 而位阻效应等互斥作用则具有相对较大的ρ(r). 因此λ2的正负号(记为[λ2])与ρ(r)的乘积(记为[λ2]ρ(r))可作为确定弱相互作用类型的指标函数. 利用[λ2]ρ(r)结合RDG(r), 不仅可以定位体系中弱相互作用的位置, 同时也能确定弱相互作用的类型. 利用[λ2]ρ(r)值对RDG(r)等能面进行着色, 形成填色图, 只需考察弱相互作用区域的颜色, 即可确定其类型. 蓝色表示氢键、卤键等具有较强吸引的相互作用; 绿色表示范德瓦耳斯吸引作用; 而红色则表示位阻效应对应的互斥作用. 电场中(MgO)4吸附H2体系的RDG(r)等能面填色图如图6所示. 由图可见, 位于(MgO)4表面的区域为红色, 表明其间是互斥作用, 而H2与Mg/O原子间则是绿色的弱相互作用区域, 表明其间是范德瓦耳斯吸引作用.
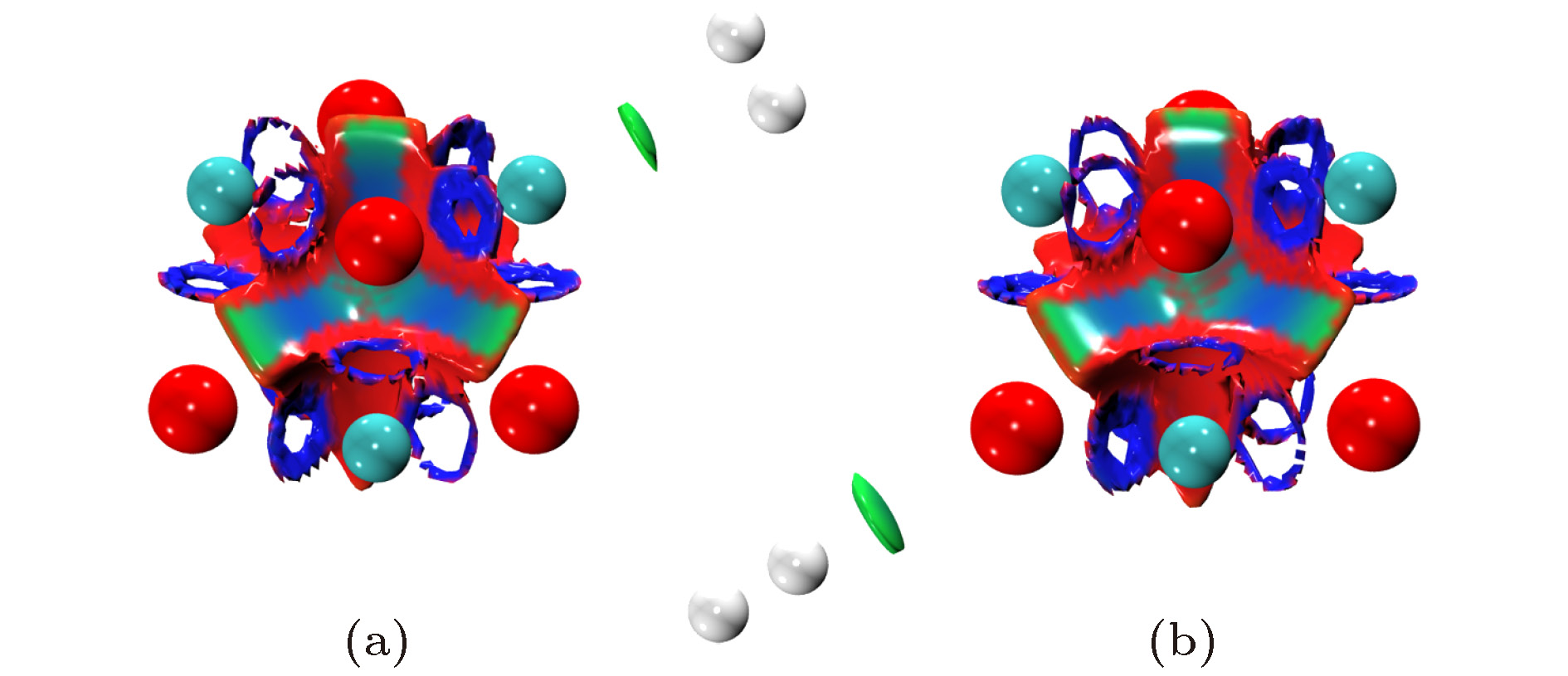 图 6 F = 0.010 a. u. 电场中H2在Mg/O离子上吸附结构的填色RDG等能面
图 6 F = 0.010 a. u. 电场中H2在Mg/O离子上吸附结构的填色RDG等能面Figure6. The color-filled map of RDG isosurface for the adsorption structures of H2 on Mg/O under the electric field with F = 0.010 a. u.
范德瓦耳斯作用本质是静电作用, 因此我们对(MgO)4吸附H2体系的NPA电荷进行了计算并列于表2. H2吸附在Mg上, 两个H原子均带正电荷, H2上的净电荷为正, 表明电荷由H2分子向团簇转移. 当H2吸附在O上时, 近O离子端的H原子带正电荷, 另一端H原子带负电荷, 表明H2被极化. 同时H2上的净电荷为负, 表明电荷由团簇向H2分子转移. 随着电场的增强, H2吸附在Mg/O上时的电荷绝对值均单调增加, 表明电场促进了H2与团簇间的电荷转移. 另一方面, 我们还计算了电场中(F = 0.010 a.u.)与无电场时H2吸附在Mg/O上的电荷密度差分, 如图7所示. 图中的暖色/冷色和实线/虚线分别表示电子增加和减小的区域. 当H2吸附在Mg上时, Mg与H2间电子密度增加, 而相反的另一侧电子密度减小, 表明电场中H2被极化; 当H2吸附在O上时, 极化效应更加明显, O与H2间电子密度减少, 而相反的另一侧电子密度增加. 这表明电场中氢分子被有效极化. 综合上述分析, 一方面电场促进了H2与(MgO)4间的电荷转移, 另一方面电场使团簇及H2被有效极化, 增强了其间的静电作用, 从而提高了(MgO)4对H2的吸附. 另外, 在相同的场强条件下, 当H2吸附在Mg上时, 转移的电荷量总是大于吸附在O上, 因此H2吸附在Mg上时更稳定.
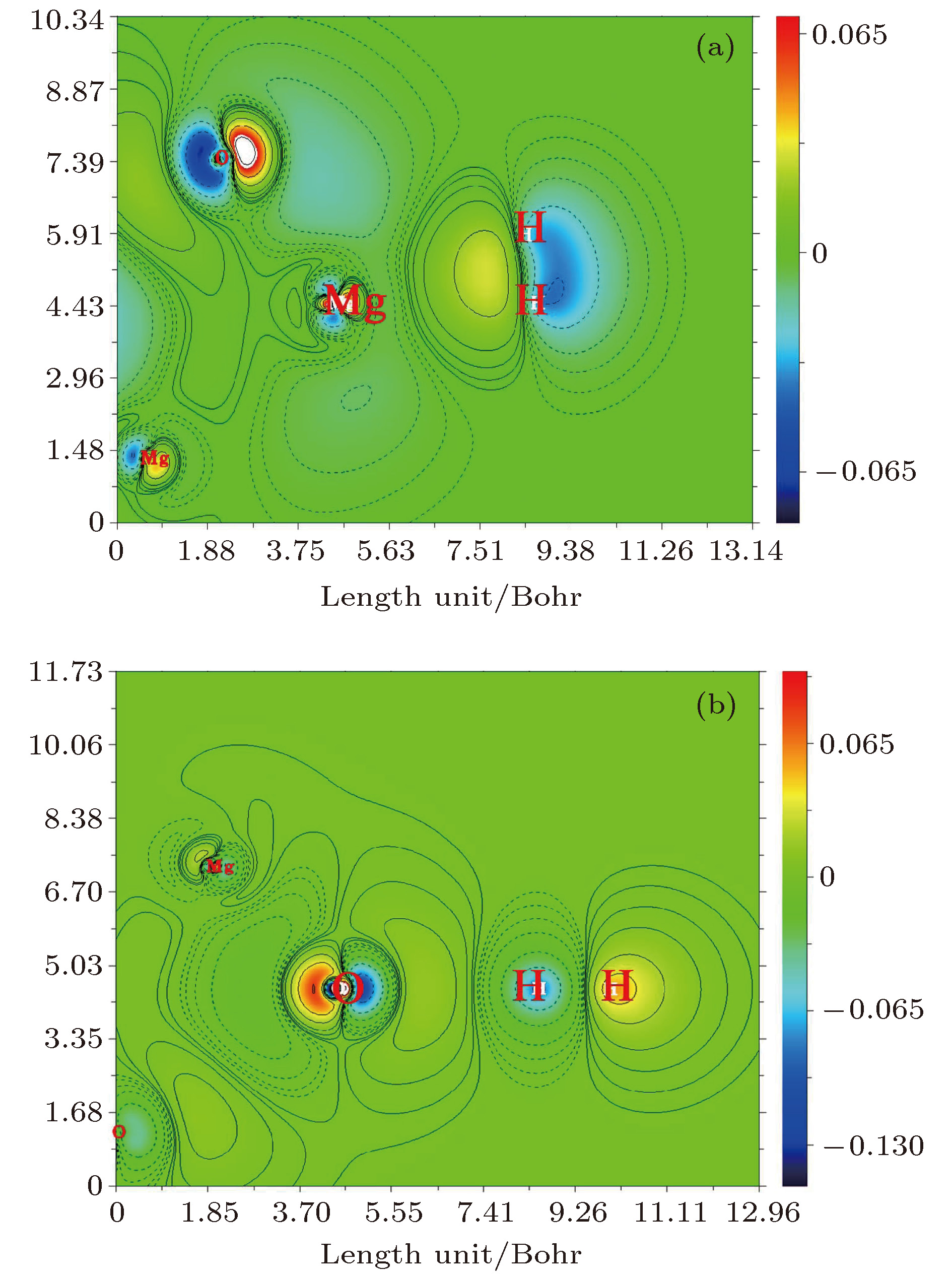 图 7 H2在Mg (a)和O (b)原子上吸附结构的电荷密度差分
图 7 H2在Mg (a)和O (b)原子上吸附结构的电荷密度差分Figure7. The color-filled contours of charge density difference for adsorption structures of H2 on Mg (a) and O (b), respectively
2
3.4.电场中多个H2在(MgO)4上的吸附
为研究电场中(MgO)4的储氢质量密度, 我们优化了场强为0.010 a.u.时多个H2在(MgO)4上的吸附结构. 由于H2只能吸附在单个Mg/O原子上方, 因此首先优化了多个H2吸附在(MgO)4中单个Mg/O上的结构, 发现每个Mg/O原子上最多能吸附2个H2分子. 因此我们又优化了16个H2吸附在(MgO)4上的结构(如图8所示), 当继续增加H2分子数目时, 则不能形成稳定的吸附结构, 表明电场中(MgO)4最多能吸附16个H2, 相应的质量密度为16.7 wt%, 达到了美国能源部制定的质量密度需达到6 wt%的要求, 表明(MgO)4是一种优良的电场储氢材料. 吸附的16个H2的键长为0.747—0.752 ?, 到吸附的Mg/O原子间的距离为2.316—2.958 ?, 由于先吸附的H2对后吸附H2的位阻效应, 相应每个H2分子的平均吸附能也减小为–0.09 eV.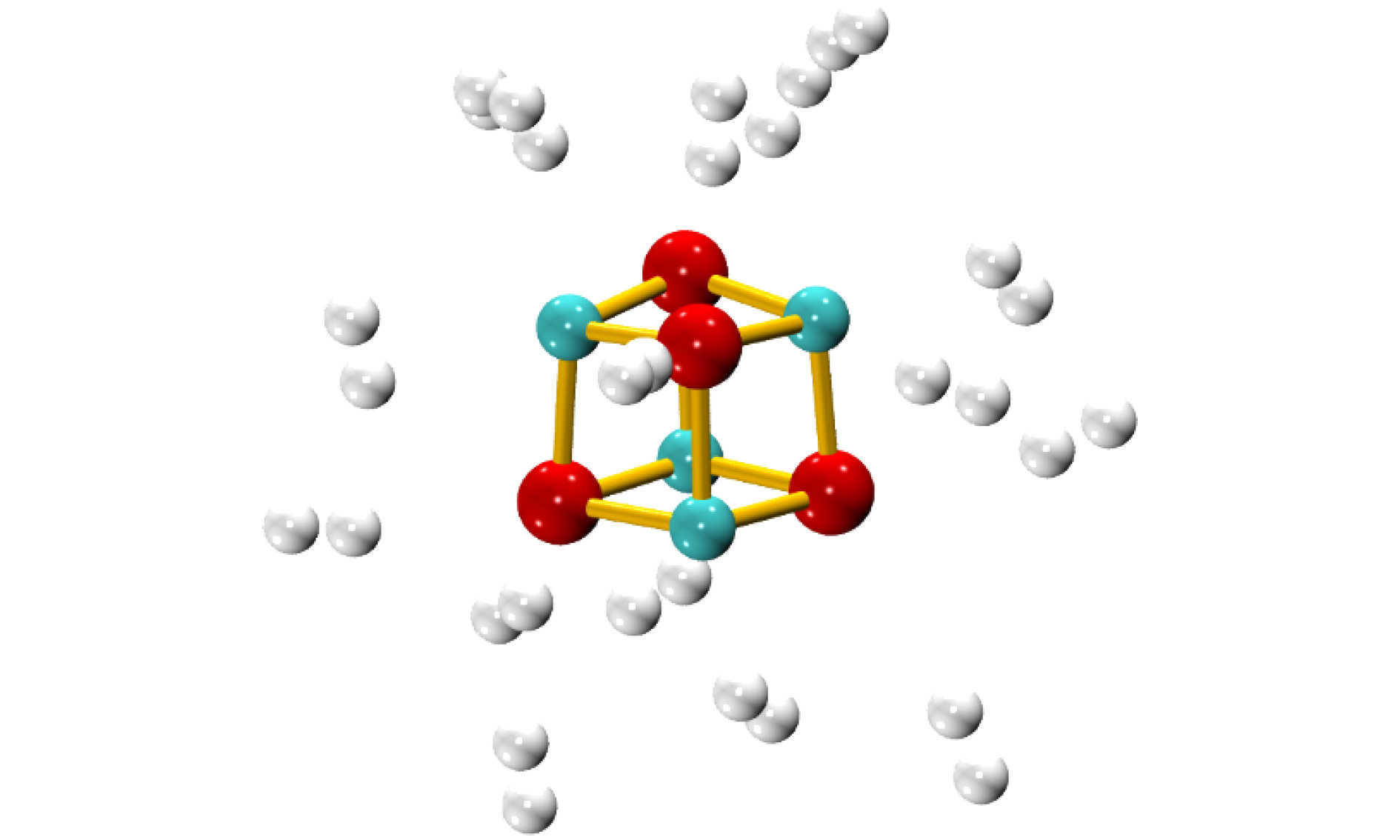 图 8 F = 0.010 a.u. (MgO)4吸附16个H2的稳定结构(绿色球为Mg原子, 红色球为O原子; 灰色球为H原子)
图 8 F = 0.010 a.u. (MgO)4吸附16个H2的稳定结构(绿色球为Mg原子, 红色球为O原子; 灰色球为H原子)Figure8. The adsorption structures of 16 H2 on (MgO)4 under the electrical field with the intensity of 0.010 a. u. (the green, red and grey balls are Mg, O and H atoms, respectively).
将小尺寸 (MgO)n幻数团簇担载于轻质基底材料上形成复合体系, 可能进一步提升其储氢性能, 并具有更复杂、更丰富的物理过程. 另外一些稳定的幻数团簇具有原子特性, 称为超原子. 以这些超原子为单元组装形成的新型复合材料, 仍能保持超原子单元本身优良的物化性能[30]. 因此以小尺寸 (MgO)n幻数团簇为单元形成的复合物或组装材料, 仍可能具有较好的电场储氢性能, 而其中涉及的尺寸和电场效应、存储密度及储氢机理等也是值得进一步研究的课题.
感谢国家超级计算机中心在广州和深圳提供的计算资源.
