 ,1, 王龙,2
,1, 王龙,2Progress in elucidating the origin of eukaryotes
Zhiwei Gao,1, Long Wang,2通讯作者: 王龙,博士,副教授,研究方向:分子遗传与进化。E-mail:wanglong@nju.edu.cn
编委: 刘钢
收稿日期:2020-04-18修回日期:2020-05-6网络出版日期:2020-10-20
| 基金资助: |
Received:2020-04-18Revised:2020-05-6Online:2020-10-20
| Fund supported: |
作者简介 About authors
高志伟,在读本科生,专业方向:生物化学与分子生物学。E-mail:

摘要
作为重大进化谜题,真核生物起源的研究对于解码真核基因组、阐释真核细胞内部结构之间的关系有重要启示作用。在1977年美国微生物学家Carl Woese发现古细菌并提出三域生命之树之后,大量研究显示古细菌与真核生物在进化上存在着密切联系。21世纪以来,系统发育分析方法不断改进,泉古菌门(Crenarchaeota)、广古菌门(Euryarchaeota)之外与真核生物更加相似的新古细菌门类也相继被发现,这些证据更加支持将真核生物与古细菌合并为一域,形成二域生命之树。目前,通过宏基因组技术发现的Asgard古细菌是与真核生物进化距离最近的原核生物。然而,真核生物祖先的身份以及线粒体起源的时间等核心问题仍是学术界争论的焦点。本文结合近年来国内外研究成果,从生命之树的形态变化与真核生物演变的具体机制两个角度梳理了目前对真核生物起源的认知过程、现有水平和研究前景,以期为揭示真核生物起源进程的后续研究提供参考与指引。
关键词:
Abstract
Knowledge of the origin of eukaryotes is key to broadening our understanding of the eukaryotic genome and the relationship among internal structures within a eukaryotic cell. Since the discovery of archaea in 1977 and the proposal of three-domain tree of life by the American microbiologist Carl Woese, the intimate relationship in evolution between eukaryotes and archaea has been demonstrated by considerable experiments and analyses. From the beginning of the 21st century, with the development of phylogenetic methods and the discovery of new archaeal phyla more related to eukaryotes, increasing evidence has shown that Eukarya and Archaea should be merged into one domain, leading to a two-domain tree of life. Nowadays, the Asgard superphylum discovered via metagenomic analysis is regarded as the closest prokaryotes to eukaryotes. Nevertheless, several key questions are still under debate, such as what the ancestors of the eukaryotes were and when mitochondria emerged. Here, we review the current research progress regarding the changes of the tree of life and the detailed eukaryotic evolutionary mechanism. We show that the recent findings have greatly improved our knowledge on the origin of eukaryotes, which will pave the way for future studies.
Keywords:
PDF (1503KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
高志伟, 王龙. 真核生物起源研究进展. 遗传[J], 2020, 42(10): 929-948 doi:10.16288/j.yczz.20-107
Zhiwei Gao.
真核生物起源被称为生物界仅次于生命起源的第2大谜团[1]。加拿大著名微生物学家Roger Stanier曾指出:“原核生物与真核生物细胞之间的差异是当今世界中已发现的最大的单一不连续性”[2]。虽然许多原核生物已经具有带有膜的内部组织结构[3],但真核生物的复杂程度却远高于此。真核生物具有核膜包被的细胞核,将转录与翻译过程分隔开,为基因调控提供更多选择[4];众多细胞器组成的内膜系统使细胞具有精密高效的运输和代谢功能[5]。这些结构增强了真核生物的环境适应能力,使其演化为现存物种中数目占大多数的生命形式。
从原核到真核这一巨大转变通常在教科书中被过度简化为一个迅速的单一事件[6]。然而从时间上看,化石记录显示目前最早的真细菌[7,8]和古细菌化石距今约35亿年,相比之下,真核生物化石距今约18亿年[9],与利用分子钟计算的17~19亿年时间相一致[10],由此推算出的原核生物和真核生物的出现时间相差17亿年之久。阐释这一漫长历史过程中真核生物的演变历程对人们认知真核生物乃至人类自身的细胞和基因组结构至关重要。
真核生物的祖先是什么?真核生物演变的具体机制是什么?这两大核心问题也成为学界长久以来争论的焦点。其中,尤其是真核生物在生命之树上所处的关键节点位置,以及线粒体的起源问题上,依然存在有很大的分歧。近年来,与真核生物进化密切相关的Asgard古细菌超门的发现[11,12]以及其在实验室条件下培养研究的实现[13],为解决这一僵局带来了光明。因此,本文在回顾历史的基础上,重点聚焦近年来在该领域取得的重大研究进展,从生命之树和演化机制两个角度深入阐述真核生物的起源问题。
1 生命之树中的真核生物
1.1 古细菌的发现和三域生命之树的提出
1977年,美国微生物学家Carl Woese通过16S/18S rRNA序列系统发育分析偶然发现了古细菌。他们据此将原核生物分为两大类——真细菌和古细菌,并将真细菌、古细菌和真核生物各分为一个“域”(Domain),作为比“界”(Kingdom)高一级的分类系统,分别命名为细菌域(Bacteria)、古菌域(Archaea)和真核域(Eukarya)[14]。自此,真核生物在分类学中的位置迎来巨变,利用保守基因为所有细胞生命建立分类系统的方法开始流行。20世纪80年代末,基于基因重复(gene duplication)[15]和SSU rRNA[16]的研究发现真核生物虽与古细菌形态差异大,但属于姐妹群(sister groups),具有一个独立于真细菌之外的共同祖先。据此,Woese等[16]提出了著名的三域(three primary domains, 3D)生命之树(tree of life/ universal tree),包含细菌域、古菌域(分为泉古菌门和广古菌门)和真核域(图1)[17],长期以来得到各类教科书和文献的广泛认可[18]。20世纪80年代到21世纪初,许多研究发现古细菌和真核生物在分子层面上高度相关,直接或间接地为Carl Woese的三域生命之树提供证明。在遗传信息复制、转录和翻译水平上,真核生物与古细菌具有许多真细菌不具有的相似性。例如,免疫学实验[19]和进化计算[20]都证明古细菌依赖于DNA的RNA聚合酶与真核生物的结构同源性。真核生物与古细菌的启动子和转录起始因子、30种共有的核糖体蛋白、翻译因子、DNA复制相关的部分酶、组蛋白等都体现出真细菌所缺乏的相似性[21]。此外,真核生物和古细菌具有多种相似的、维持细胞基本生存的酶和代谢通路,如液泡ATPase、分泌通路、辅酶A生物合成的部分酶以及嘧啶和精氨酸生物合成的部分酶等[21]。古细菌细胞分裂系统内的部分蛋白甚至与真核生物的内体分选复合体同源[22,23]。如此多的进化关联性使古细菌成为揭示真核生物起源的重要研究对象[24]。
当然,古细菌和真核生物之间也存在许多差别。最为显著的是膜脂,古细菌采用醚键相连,而真核生物采用酯键相连;古细菌的甘油磷酸骨架使用甘油-1-磷酸,而真核生物和真细菌使用甘油-3-磷酸,两者由不相关的酶合成。古细菌还拥有独特的产甲烷代谢途径、DNA拓扑异构酶和DNA聚合酶等[21]。此外,真核生物与真细菌也有代谢[25,26]和膜化学性质[27]的部分相似之处。
图1
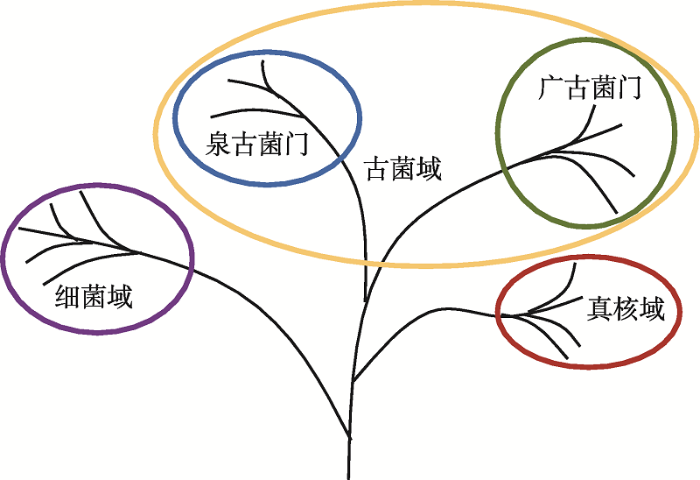 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1Carl Woese的生命之树
Carl Woese依据16S/18S rRNA系统发育分析发现古细菌后,将自然界中的生物分为真核域、细菌域和古菌域三域。根据文献[17]修改绘制。
Fig. 1Tree of life proposed by Carl Woese
1.2 二域与三域的争端
虽然Carl Woese提出的三域系统(图2A)受到广泛认可,但同时期,Lake等[28]依据核糖体结构证据、Rivera和Lake[29]依据延伸因子EF-1α同系物保守区有共同氨基酸插入的证据,提出真核生物是泉古菌门的一个姐妹群,即真核生物是从古细菌域之内产生的,是二级域,而一级域仅为真细菌和古细菌,形成二域(two primary domains, 2D)生命之树(图2B)。人们对应地提出了许多演化模型支持二域系统,实质均为古细菌与细菌之间的相互作用[30],如支持泉古菌门(Crenarchaeota)与细菌相互作用的泉古菌假说(eocyte hypothesis)[28,29],广古菌门(Euryarchaeota)与细菌相互作用的氢假说(hydrogen hypothesis)[31]和互养共栖假说(syntrophic hypothesis)[32]等。图2
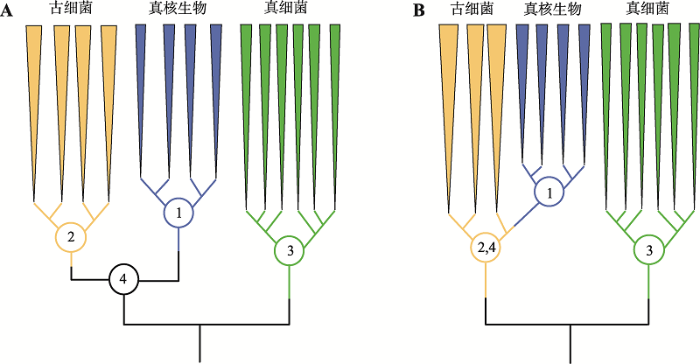 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2三域与二域生命之树的对比
A:三域系统;B:二域系统。AB图中的1、2、3分别对应真核域、古菌域和细菌域的最近共同祖先;真核域与古菌域是两个距离遥远的姐妹群,具有一个最近共同祖先4。B图中真核域的最近共同祖先1直接产生自古菌域内,所以古菌域的最近共同祖先2同时也是古菌域和真核域的最近共同祖先,这与A图的情况不同。根据文献[21]修改绘制。
Fig. 2Comparison between the two-domain and three-domain tree of life
21世纪的第一个10年中,生物信息学蓬勃发展。从系统发育重建的角度看,三域与二域的争端依然激烈。在2003~2009年发表的7篇文献中,通过真核生物、真细菌、古细菌物种取样,测定基因组中的系统发生信号(phylogenetic signal)标记,进行大规模系统发育学分析,3篇文献得出三域系统结果[33,34,35],4篇文献得出二域系统结果[36,37,38,39]。7篇文献采用的数据和方法比较如表1所示。
Table 1
表1
表12003~2009年间发表的7篇阐释生命之树中古细菌与真核生物关系的文章比较
Table 1
| 文献 | 三域中使用的标记数目 | 取样种类(数目) | 氨基酸位置数 | 方法 | 结果 | 优势 | 缺陷 |
|---|---|---|---|---|---|---|---|
| Harris等[33] | 50 | 细菌(25); 泉古菌门(1); 广古菌门(7); 真核生物(3) | 与所分析基因相关 | 单基因分析; 最大似然法; 最大简约法; 距离法 | 3D | 选取数据范围从此前的SSU rRNA扩展到其 他基因 | 当时的基因数据有限,单基因分析可靠性不强 |
| Ciccarelli 等[34] | 31 | 细菌(150); 泉古菌门(4); 广古菌门(14); 真核生物(23) | 8090 | 分散基因串联 | 3D | 取样物种广泛, 增加了客观的 HGT过滤方法 | 先在域内对齐序列,跨域整合时可能将非同源基因对齐 |
| Yutin等[35] | 136 | 与所用基因 相关 | 与所分析基因相关 | 单基因分析; 最大似然法 | 3D | 数据集丰富,分析的基因数目多 | 单个基因提供的系统发育信号弱 |
| Rivera和Lake[36] | Archaeoglobus fulgidus全基因组 | 细菌(2); 泉古菌门(1); 广古菌门(2); 真核生物(2) | 不适用 | 基因组条件 重建 | 2D; 真核生物与泉古菌门为姐妹群 | 使用全基因组 分析 | 取样物种少,受HGT和基因丢失影响大 |
| Pisani等[37] | 未提供数据 | 细菌(97); 泉古菌门(4); 广古菌门(17); 真核生物(17) | 与所分析基因相关 | 超树法 (Supertree) | 2D; 真核生物与广古菌门为姐妹群 | 综合众多数据集信息,在单基因 树基础上建立二级树 | 过滤步骤后剩余的有效数据少,且拥有单基因分析的局限性 |
| Cox等[38] | 45 | 细菌(10); 泉古菌门(3); 广古菌门(11); 真核生物(16) | 5521 | 分散基因串联; 贝叶斯方法; 最大似然法; 最大简约法 | 2D; 真核生物与泉古菌门为姐妹群 | 除简单模型外使用了更多更复杂的模型,首例使用新模型证明了2D系统结果 | 复杂模型的参数估计的精确性难以保证 |
| Foster等[39] | 41 | 细菌(8); 泉古菌门(8); 奇古菌门(2); 广古菌门(6); 真核生物(11) | 5222 | 分散基因串联; 贝叶斯方法; 最大似然法; 最大简约法 | 2D; 真核生物是一个由泉古菌门和奇古菌门构成的群的姐妹群 | 同上,作者相同,选取的数据更多,使用了新发现的古细菌门类数据 | 复杂模型的参数估计的精确性难以保证 |
新窗口打开|下载CSV
值得注意的是,上述7篇文献使用数据在很大程度上有重叠,大多基于蛋白相邻类的聚簇(Cluster of Orthologous Groups of proteins, COG)数据库,最终却得到了截然不同的结论,集中体现了系统发育学方法在解决真核生物起源时面临的典型难题:
(1)长期的碱基替换导致基因组中遗留的系统发育信号很微弱,若使用单基因位点或较短的16S rRNA、18S rRNA分析跨域的物种,背景噪声大,可以得到的有效信息很少,如前述Harris等[33]与Yutin等[35]两文的方法正如此。同时,单个基因可能受基因特异性事件影响(如基因重复、丢失和转移),不能反映真实的物种进化情况[40,41]。
(2)为解决单基因信号微弱的问题,许多研究采用了分散基因串联(concatenation)的方法[34,38,39],即将多个不连续的基因直接串联组合为“普遍基因”(universal gene),序列对齐后再统一分析。但此举的局限性在于真核生物起源分析的范围广,真核生物、古细菌和真细菌3个类群跨域分析时很可能将非同源蛋白强制对齐,在组合基因时人为引入偏差[21]。此外,可供大范围跨域分析的“普遍基因”本身数量也十分有限。
(3)同样为解决单基因信号微弱的问题,有研究采用全基因组比对,但局限性在于可用的全基因组数据比单基因数据少,且计算更加复杂。选取物种数少时,结果受抽样本身影响大,例如Rivera和Lake[36]仅仅使用了7个全基因组,偶然误差较大。但该方法随着未来基因组数据的快速增加和组学分析方法的不断进步,有望提供更多可靠的信息。
(4)不同物种间的水平基因转移(horizontal gene transfer, HGT)和基因丢失都可能抹去进化印记。HGT和基因丢失可能发生在真核生物进化的各个阶段(图3),机制包括转化、转导和接合等[42,43,44]。在Cox等[38]的研究中,检测到选取的80个基因有30个受到HGT影响,数据舍弃不用。Akanni等[45]利用超树方法分析了392个真细菌和51个古细菌基因组,在古细菌进化树基部发现了两个来自真细菌的大规模HGT,基因来源涉及δ-变形杆菌、梭菌和放线菌。对此,Cotton等[46]和Cohen等[47]认为,与转录、翻译相关的核心基因的负选择压大,受HGT的影响应比一般代谢通路的基因小,是更加合适的分析对象。
图3
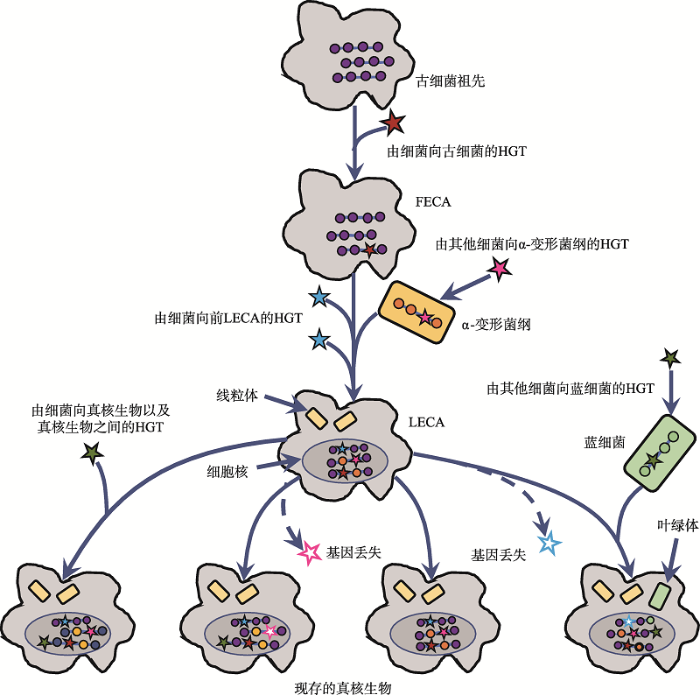 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3真核生物进化过程中的水平基因转移(HGT)与基因丢失
HGT可能发生在古细菌祖先→FECA→LECA→现存真核生物演化进程中的各个阶段。线粒体和叶绿体的内共生过程也为HGT的发生提供条件。HGT和基因丢失均能削弱现存生物基因组中遗留的系统发育信号。根据文献[17]修改绘制。
Fig. 3HGT and gene loss during the evolution of eukaryotes
(5)长支吸引(long branch attraction, LBA),由于不同域间的基因差别可能很大,在同源性高的物种之外可能形成进化距离遥远但仍然相互靠近的长支,造成偏差[48,49]。早在1999年,Tourasse等[50]便认为三域树结果是受LBA影响产生的。
(6)建立的模型本身设计过于简单,模拟能力差[48]。
近10年来,随着模型的复杂化和基因数据的积累,人们尽量规避上述6点对结果的影响。2011~ 2013年发表的3篇文献结果都支持二域系统[48,51,52]。2014年,Rochette等[53]的系统发育分析显示,真核生物与古细菌间的联系比真细菌紧密得多,但与泉古菌门和广古菌门没有密切联系,真核生物分支接近古细菌并很可能从中发出。与此同时,泉古菌和广古菌以外古细菌门类的发现为二域系统提供了更多的支持,相关内容将在下一部分阐述。
1.3 新古细菌类群的发现对生命之树的影响
2004年,Venter等[54]首次采用不经实验室培养直接测定环境中微小生物基因组的“全基因组鸟枪测序”方法,即通常所说的宏基因组(metagenome)技术,研究海水中的微生物样本,发现了148个新细菌种类和120万个此前未知的基因。此后,人们不断利用宏基因组技术发现多个泉古菌门、广古菌门之外的古细菌新门,为生命之树带来巨大变化。2008年后,奇古菌门(Thaumarchaeota)[55]、初古菌门(Korarchaeota)[56]、曙古菌门(Aigarchaeota)[57]相继被发现。其中奇古菌门是被发现的第一个新古细菌门类,而曙古菌门是第一个完全由宏基因组学数据重建的基因组,并且发现了与真核生物同源的泛素系统。依据16S rRNA测序结果,这3个新门与泉古菌门一起归为一个单系群(monophyletic group),称为“TACK”超门(TACK superphylum)或变形古菌(Proteoarchaeota)[58,59]。通过改进的最大似然估计或贝叶斯网络模型,人们发现真核生物从TACK超门内分支出来或是TACK超门的一个姐妹群(图4A)[17,60]。这表明真核生物分支是从古细菌域内发出的,支持上一部分所叙述的二域系统,否定三域系统。新发现的TACK超家族成员含有大量一般只在真核生物中表达或与其相关的真核生物标志蛋白(eukaryotic signature proteins, ESPs),如肌动蛋白、微管蛋白的古细菌同源物、转录翻译因子类似的蛋白等[11]。TACK超门成为追溯真核细胞起源的重要研究材料。
2014年以来,通过改进的宏基因组技术——解码宏基因组(genome-resolved metagenomics),人们又在TACK超门中发现新的古细菌门:深古菌门(Bathyarchaeota)、地古菌门(Geoarchaeota)和韦斯特古菌门(Verstraetearchaeota) (图4B)[17]。广古菌门外还存在着一个深枝DPANN超门(DPANN superphylum),包含丙盐古菌门(Diapherotrites)、小古菌门(Parvarchaeota)、谜古菌门(Aenigmarchaeota)、纳古菌门(Nanoarchaeota)和纳盐古菌门(Nanohaloarchaeota) (图4B)[17]。这些发现极大地丰富了生命之树中古细菌的分支。
近5年来的一个重要突破是发现了与真核生物遗传关系更近的Asgard古细菌。Asgard古细菌大多生活在海底,分类上原先属于海洋底栖类群B (Marine Benthic Group B, MBG-B)、古代古细菌群(Ancient Archaeal Group, AAG)、深海古细菌群(Deep-sea Archaeal Group, DSAG)和海洋热液喷口群(Marine Hydrothermal Vent Group)[61]。2015年,瑞典乌普萨拉大学Ettema课题组在北冰洋Loki’s Castle附近的海底沉积物中利用宏基因组技术重建出一种新的古细菌基因组,命名为洛基古菌门(Lokiarchaea),蛋白质组中的重要部分(175个蛋白质,占比3.3%)都与真核蛋白相似,为ESPs,参与膜的变形和细胞形状的形成过程[11]。此后,又发现了与Lokiarchaea门相近的3个新门:奥丁古菌门(Odinarchaeota)、索尔古菌门(Thorarchaeota)和海姆达尔古菌门(Heimdallarchaeota)[12,62]。这4个新门被一并命名为Asgard超门(Asgard superphylum)[11,12] (图4B),是真核生物的姐妹群。其中,海姆达尔古菌门与真核生物距离最近[63]。
图4
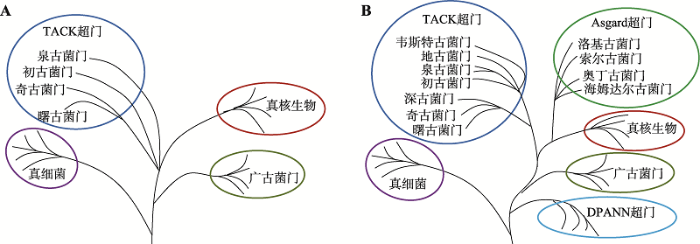 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4发现新门类古细菌后的生命之树
A:新发现TACK超门古细菌。真核生物是TACK超门的一个姐妹群,或者直接从TACK超门中产生。B:新发现DPANN超门和Asgard超门古细菌。DPANN超门形成古细菌的一个深枝,Asgard超门与真核生物进化距离最近,真核生物是Asgard超门的一个姐妹群,或者直接从Asgard超门中产生。根据文献[17]修改绘制。
Fig. 4Tree of life after the discovery of new archaeal phyla
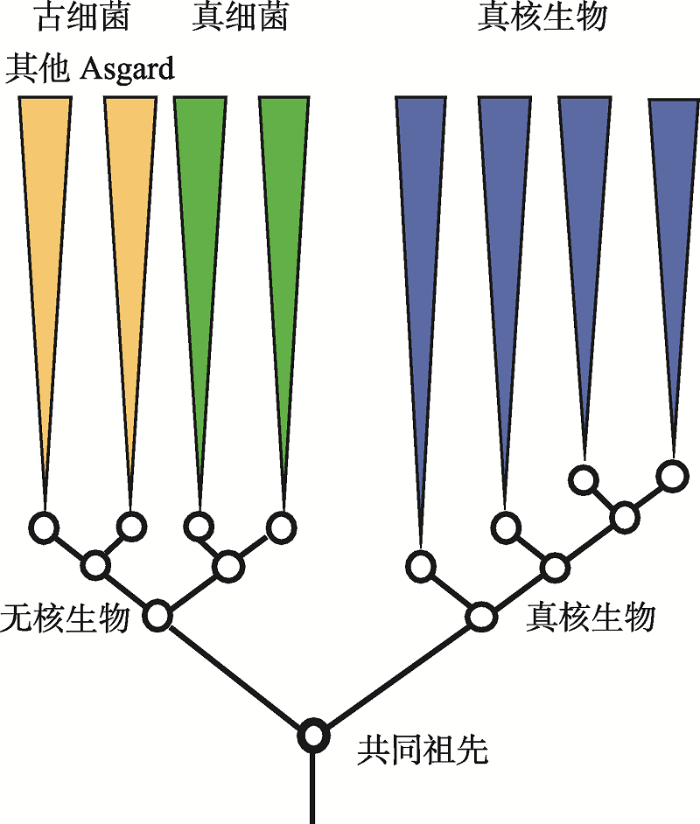
不过,新的发现也引发了学术界的争议。一方面,Da Cunha等[64]指出,Ettema课题组的取样存在污染,最终结果也因为取到演化速度快的序列而造成LBA效应,并且去除数据集中的蛋白延伸因子EF2便足够破坏真核生物与Lokiarchaea的密切关系。他们认为Lokiarchaea和Asgard超门是广古菌门的姐妹群,而非真核生物的姐妹群,且真核生物为单系群,根源通往TACK超门中Thaumarchaeota的分支,最终结果是三域系统。Ettema课题组的Spang等对此进行了反驳,认为没有证据表明取样污染,去除EF2依然能够证明Asgard与真核生物属于一个单系群,而Da Cunha等使用了不合适的方法导致三域系统结果[65]。此后双方仍有争论,但未有结果[66,67]。最近,Williams等[49]针对Da Cunha等[64]的结果,使用相同软件的不同最大似然方法包分析相同数据,得到与原文不同的二域系统结果;他们进一步使用多个新模型分析35个基因的对齐结果,真核生物均与Asgard古细菌形成姐妹群;基于“超树”和基因合并对齐方法分析3000多个古细菌和真核生物基因的结果显示真核生物是从古细菌域内产生的,因此认为“生命之树”应当为二域系统。另一方面,Harish等[68,69,70]以蛋白质折叠作为分析对象,将前人证明三域系统的模型[71,72]扩展为非平稳模型(non- stationary model),推算出的生命之树树根将细胞生命分为无核生物(Akaryote,即古细菌与真细菌)和真核生物二域,形成了一个与此前二域、三域生命之树均不同的二域系统(图5)。分析结果还指出,Asgard古细菌与所有其他的古细菌是姐妹群,难以作为一个整体处理。这一结论说明细胞生命内最深层次的分裂位于原核与真核之间。相比常用的蛋白质氨基酸序列[49],Harish等[73]认为蛋白质结构域在生化角度上非冗余和更保守,更适合作为进化标记进行系统发育学分析。然而,这个新的二域生命之树是选取不同进化分子标记的结果,违背了此前众多研究所证明的真核生物与古细菌间的密切关系,因此争论依然存在。
对于Asgard古细菌与真核生物的密切关系也存在不同解读。Fournier和Poole[74]便试图以现代哺乳动物的祖先合弓纲(Synapsida)与现代爬行动物的进化关系作为平行案例,说明仅从真核生物与Asgard古细菌的谱系关系无法解决二域/三域争论,此外还应确定古细菌和真细菌之间的根源。
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5有争议的新二域生命之树
依据蛋白质折叠分析结果构建的生命之树将生物分为真核生物和无核生物两域。根据文献[73]修改绘制。
Fig. 5The controversial new two-domain tree of life
虽有上述争论,但发现新的、进化上更接近于真核生物的Asgard种类依然是重要的研究方向。Seitz等[75]发现了新的Asgard古细菌海拉古菌门(Helarchaeota)。深圳大学李猛课题组利用开源数据库中的现有数据分析Asgard古细菌16S rRNA序列,发现了5个此前未知的Asgard亚组(subgroup),并且为Asgard古细菌的转录活性混合营养型生活方式提供了证据[76]。他们利用几个沿海沉积物样本重建出15个基因组,其中包括一个新的Asgard门类“Gerdarchaeota”[77]。最近,他们设计的新SSU rRNA引物完成了Asgard中一种不能与通用引物匹配的新类群的扩增工作[78]。Ettema课题组未发表的结果也显示发现了5个新Asgard类群,其中Idunnarchaeota是目前与真核生物最接近的类群[79]。
总之,随着模型的复杂化、基因序列和蛋白质序列数据的积累以及新门类古细菌的发现,学术界对于生命之树的争论已逐渐由三域系统向二域系统学说倾斜。主流观点认为真核生物产生自古细菌域内,Asgard超门是目前与其进化距离最接近的古细菌。在未来,生命之树的形态以及真核生物的定位仍可能不断改变,但Carl Woese的三域生命之树仍将长期存在于教科书与人们的脑海中。
2 真核生物的起源机制
2.1 真核生物起源机制的研究现状
关于真核生物的起源,首先需要明确的两个概念是FECA和LECA。FECA (first eukaryotic common ancestor)指所有真核生物的最早共同祖先;而LECA (last eukaryotic common ancestor)指所有现存真核生物距今最近的共同祖先[17]。FECA与LECA间的阶段常被称为进化树上的茎期(stem phase)[80]。大量原生生物和多细胞生物的基因组学和细胞生物学研究已证明,LECA拥有现代真核生物的大部分遗传和细胞结构特征,如细胞核、线粒体、细胞骨架、囊泡和运输系统、细胞周期等[6]。保守估计LECA基因组有至少4000~5000个基因[80,81]。从理论上分析,原核生物向真核生物的转变大致涉及以下方面:由厌氧向兼性厌氧、好氧代谢方式的转变,共生与内共生,吞噬作用的产生,基因组嵌合体的变化,细胞核的出现,信号通路的复杂化,被膜细胞器的出现,细胞周期的出现,细胞体积的增大等等[1]。研究真核生物起源机制的关键在于以FECA和LECA为坐标,理清茎期中这些进化事件的先后关系和具体过程。
结合实验数据与理论分析,一些局部的问题已经得到充分阐释,例如GTPase、longins、COPs和ESCRT-III复合物等真核生物标志性蛋白的进化历程,内膜系统和核孔复合物等真核细胞结构的组建过程已在大量综述中有详尽描述[6,82,83]。
然而,从整体上解决真核生物的起源机制始终绕不开线粒体的起源问题(图6)[6,84]。作为细胞的能量工厂,线粒体为真核生物在“大氧化事件”后在有氧环境中的辐射进化提供了必要条件;真核生物内膜系统与细胞核的产生也与线粒体的内共生密切相关。
2.2 线粒体与真核生物起源
线粒体普遍存在于现代真核生物体内,且随不同物种展现出丰富的多样性[85]。许多真核生物(多营寄生生活)为适应厌氧环境,将线粒体简化为线粒体相关细胞器(mitochondrion-related organelles, MROs)[86,87]。极端的例子如Monocercomonoides sp. 的线粒体已完全丧失[88]。但学界普遍认为线粒体的简化丧失是发生在LECA之后比较新的进化行为,LECA拥有线粒体结构[30,81,89]。图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6线粒体内共生是引发真核生物起源的重要事件
现有的主流观点认为一种类似于现存Asgard超门的古细菌宿主内共生一种α-变形菌后形成胞内的线粒体,原核细胞逐渐演化为真核细胞。根据文献[6]修改绘制。
Fig. 6The endosymbiosis of mitochondrion was an important event in eukaryogenesis
真核生物祖先最初通过内共生途径获得线粒体。1905年,俄国植物学家Konstantin Mereschkowsky便提出叶绿体由内共生形成的构想;20世纪20年代,Wallin将此推广至线粒体[90]。40多年后,美国生物学家Lynn Sagan (后来的Lynn Margulis)重拾内共生理论,在著名的On the Origin of Mitosing Cells一文中提出真核生物的细胞器包括线粒体和叶绿体在内都是由内共生细菌演化而来的[91],引发巨大争议。不过,随后对于线粒体、叶绿体的基因和蛋白的分析显示它们都拥有与真核谱系相距甚远的原核特征,分别对应α-变形菌和蓝细菌,为内共生学说提供了佐证[92,93,94]。后来在Symbiosis in Cell Evolution一书中,Margulis正式提出线粒体是原始真核生物吞噬革兰氏阴性好氧细菌后,通过长期内共生作用形成的[90],逐渐得到学术界的广泛认可。自此,线粒体的内共生起源成为真核生物起源研究的核心点。
2.2.1 线粒体内共生的早期与晚期之争
长期以来,学界在线粒体内共生发生的时间问题上存在争论。主要观点分为两派:一派认为宿主吞噬好氧的α-变形菌前,已经先通过不同途径形成细胞核,成为具有真核细胞特征(含细胞核、动态细胞骨架、内膜系统)的过渡体,具有原始吞噬功能等,被称为晚期线粒体模型(“Mito-late” models);另一派认为,宿主首先与好氧的α-变形菌内共生形成带有线粒体的原核细胞,随后促进原核细胞演化出细胞核、内膜系统等真核生物特征,被称为早期线粒体模型(“Mito-early” models)。
20世纪80年代,研究者在现存真核生物中发现了不含线粒体的物种,如贾第虫属(Giardia)、毛滴虫属(Trichomonas)等,大多是寄生虫。英国演化生物学家Cavalier-Smith将它们归类为源真核生物(archezoans)[95],祖先是细胞内部已具有复杂结构而尚未内共生线粒体的初级真核生物,属于真核生物树中的早期分支谱系[96]。传统的晚期线粒体假说认为,细胞内部的复杂结构以及吞噬营养能力是线粒体起源的前提,细胞核的出现是为了避免细胞骨架牵拉对DNA的损伤[97,98]。基于此的真核生物起源机制有很多,几个影响深远的模型如(图7,A~D)所示[32,97,99~101]。内共生学说提出者Margulis本人认为吞噬宿主是一种类似现代Thermoplasma spp. (属于广古菌门)的嗜酸无壁古细菌,细胞核在嵌合细胞形成后自发进化形成(图7A)[100,102];著名的互养共栖假说(图7B)认为真核生物来自产甲烷菌与δ-变形杆菌的共生作用[32];Cavalier-Smith本人则认为真核生物与古细菌演化自一个共同祖先,真核域与古菌域归属于新壁总域(Neomura) (图7D)[98]。
但是,针对Cavalier-Smith提出的源真核生物概念,21世纪以来的研究表明,此前发现的所有不含线粒体的真核生物均含有产氢体(hydrogenosome)或mitosome等细胞器,与线粒体同源,是上文所说的MROs[1,30,105]。早期支持源真核生物概念的研究也被认为受到分析偏差影响,导致源真核生物概念不再流行[30]。
此后,另一派早期线粒体假说获得了许多拥护者。几个经典的Mito-early模型如(图7,E~G)所示[31,103,104],相似之处在于都是一对古细菌和真细菌首先形成共生关系,真细菌是古细菌代谢产物的受体,随后内共生,形成细胞核等其他结构,其中包含著名的氢假说(图7E)。
早期线粒体假说最有力的论据在于现存生物中尚未发现不含线粒体或其同源物的真核细胞过渡形态,这似乎暗示线粒体的内共生应发生在一个较早的时期,很可能早于细胞核。Von Dohlen等[106]在非吞噬作用的原核细胞体内发现存在其他原核细胞的现象也表明,内共生或许不需要真核生物祖先拥有复杂的吞噬营养系统。
从线粒体与真核细胞其他结构的关系角度也支持早期线粒体假说。Martin等[4,107~110]提出了对线粒体、细胞核和真核细胞分裂形式出现顺序的推测。在线粒体始祖进入宿主胞质形成内共生体后,两者间的基因重组和启动子的相互关联导致内共生体(原始线粒体)基因在宿主细胞质内表达[107]。这种嵌合的原核基因组模糊了宿主与内共生体间的区别,彼此出现基因片段交换,如从线粒体基因组向宿主基因组转移II型内含子等。然而,原核生物的共转录翻译机制无法解决内含子剪接速度比蛋白翻译速度慢从而产生缺陷肽链的矛盾,细胞核的出现则为转录和翻译提供了物理屏障[4]。随后染色体被局限在细胞核内,失去与细胞膜的接触,在细胞分裂时无法自动分离,使细胞体积较原核生物显著增加[108]。同时,胞质内由于富含线粒体产生的ATP,蛋白表达水平显著升高。为解决染色体分离问题,可能出现了依赖于微管的染色体分离方式,最终形成减数分裂、真核细胞周期和有丝分裂过程[109,110]。此外,线粒体早期内共生释放外膜囊泡(outer membrane vesicles, OMVs)的过程也有利于解释真核生物内膜系统的快速形成[111],详细机制可参阅McBride[112]的综述。
图7
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7有关线粒体起源的模型
A~D为晚期线粒体模型,E~G为早期线粒体模型。A:Margulis认为古细菌宿主热原体菌(Thermoplasma)首先与螺旋菌(Spirochaete)内共生,逐渐演化出细胞核,后形成线粒体[100]。B:互养共栖假说,产甲烷古细菌与δ-变形菌在代谢上存在氢的交换。一个产甲烷菌被多个δ-变形菌包围后,δ-变形菌间的相互作用促进胞质融合,形成内质网、细胞核等结构,δ-变形菌基因组随着基因的转移逐渐消失,线粒体的内共生最后发生[32]。C:内生核假说,泉古菌与G-真细菌互相作用,泉古菌形成细胞核,真细菌形成细胞质,再与α-变形菌内共生形成线粒体[101]。D:Cavalier-Smith认为真核生物与古细菌演化自一个共同祖先,同属新壁总域[97,99]。E:氢假说,宿主是严格依赖氢且严格自养的厌氧古细菌,共生体能进行有氧呼吸,但通过厌氧异养代谢为宿主提供氢气[31]。F:古细菌宿主在形成真核细胞的其他复杂结构前首先与耗氧的α-变形菌内共生形成线粒体[103]。G:基于硫元素的代谢循环,宿主是产H2S的古细菌,线粒体祖先是耗H2S的α-变形菌[104]。根据文献[30]修改绘制。
Fig. 7Models of the origin of mitochondrion
近10年来,有关Mito-late和Mito-early模型的问题仍有争论。延续Mito-early假说风潮,主流观念认为线粒体祖先先于宿主吞噬作用出现,直接侵入古细菌细胞内发生内共生,导致真核细胞的产生[53,113]。然而,发现Asgard古细菌Lokiarchaeota后,基因组重建出大量细胞骨架相关的ESPs,开启了具有吞噬作用的原核生物谱系从古细菌域内部进化的可能性,支持Mito-late模型[11,114]。但后续基于宏基因组重建的研究显示,Asgard古细菌不具备吞噬营养功能,吞噬作用的形成涉及来自古细菌、真细菌以及真核生物新基因的组合[115],宿主是否具有吞噬功能依然未知。不过,限于主流观念大多是依据对当今真核生物组织和功能方面的观察所作的进化推断,始终缺乏确切的实验分析证据,Hampl等[116]便认为没有充分的理由肯定线粒体产生于细胞吞噬作用之前。
总的来说,目前Mito-early假说更为流行,但也面临着其他一些无法解释的现象:真核生物的嵌合性基因组中,大多数来自细菌的基因无法回溯到线粒体的祖先α-变形菌。2016年,Pittis和Gabaldón[2,117]首次通过比较真核生物与距离最近的原核生物蛋白间的亲缘距离,发现α-变形菌与真细菌来源蛋白之间的进化距离短于真细菌与古细菌来源蛋白之间的进化距离,推测线粒体内共生前,真核细胞祖先可能已与其他真细菌内共生形成嵌合基因组,胞内有内质网和高尔基体,但还不是进化成熟的真核细胞。Ettema[118]将其称为介于Mito-early和Mito-late间的中期线粒体(Mito-intermediate)假说。但此结果引发许多质疑。William和Nelson-Sathi[119]认为作者采用了错误的统计和分析方法;Esposti也认为Pittis和Gabaldón的方法早已被证明失准[53],低估了α-变形菌祖先对真核生物谱系的影响[120]。此外,目前认为与真核生物祖先关系最密切的Asgard古细菌本身也存在大量其他真细菌基因[17]。又鉴于位于现存α-变形菌纲深枝的磁球菌(Magnetococcus)具有一个α-变形菌成分仅占1/3的嵌合基因组,Esposti[120]推测久远的线粒体祖先本身很可能就是一个嵌合基因组,而非仅有α-变形菌的血统[121],所以Pittis与Gabaldón的最终结论显得有些草率。
至今,线粒体内共生的早期与晚期之争依旧无法定论,不过可以确定的是,线粒体的内共生发生在TACK和Asgard古细菌基因组中所编码的ESPs基因出现之后。若真如Esposti所说,线粒体祖先是一个嵌合基因组的话,这段长达30余年的争论便可以画上句号。
2.2.2 线粒体祖先的确定
探索线粒体始祖细菌的性质有助于人们了解具体的内共生过程。长久以来,线粒体祖先与α-变形菌纲相关的结论为学术界广泛接受[93],但与其中哪一谱系更加接近的问题始终没有解决,给重建线粒体与现存α-变形菌谱系的最近祖先(last common ancestor)带来困难。
大量通过单基因或全基因组基因串联建树的结果常常将线粒体与立克次氏体目(Rickettsiales)相关联[122,123]。而当引入Pelagibacterales目(SAR11)基因组数据作系统发育分析时,则发现Pelagibacterales目是线粒体-立克次氏体目一枝的姐妹群[124],或直接是线粒体的姐妹群[125]。后来的分析表明,这些结果是受到线粒体和Pelagibacterales目之间趋同相似性影响带来的偏差[126,127]。随后,依赖于核基因编辑和线粒体基因编辑的线粒体蛋白数据库的分析结果支持线粒体发源自立克次氏体目内[128,129]。
但是,上述将线粒体与立克次氏体目关联的研究也存在缺陷。第一,线粒体、立克次氏体目以及上文提到的Pelagibacterales目(SAR11)与其他α-变形菌纲的物种相比进化更加迅速且已经失去了许多基因,在基因组层面A+T占比颇高,很容易招致LBA偏差[89]。对此,一些研究的应对措施是选用较早由线粒体基因组转移到核基因组的基因进行分析[128],但不可忽视的一点是前文所提到的核基因组中存在的碱基替换、基因丢失和HGT问题。第二,推测的立克次氏体目在地球上的存在时间不够长,目内处于主干地位的Wolbachia属仅有大约5亿年历史[120]。第三,立克次氏体目的代谢方式也与线粒体差别很大,普遍不存在现代真核生物的厌氧性状,不编码调节线粒体嵴的MICOS蛋白和功能等同于线粒体ATP合酶抑制亚基的相关结构[120]。
另一种通过单基因数据集和多基因串联数据集推断网络的方法将线粒体与其他α-变形菌纲分支相连,如红螺菌目(Rhodospirillales)的红螺菌属(Rhodospirillum)和根瘤菌目(Rhizobiales)的苍白杆菌属(Ochrobactrum)[89]。从代谢相似性来看,有分析认为线粒体可能更加接近于红螺菌属[120]。还有研究认为线粒体来自α-变形菌纲内的一个独立深枝[127]。
2018年,Martijn等[130]通过宏基因组重建得到12个α-变形菌纲谱系(包含几个新发现的谱系),将线粒体放在了这些谱系的进化分支之外,是所有测序的α-变形菌纲序列的姐妹群,与此前的众多结果存在矛盾。然而,2019年Fan等[131]的结果则又显著支持线粒体来源于α-变形菌纲内的传统观点,并指出Martijn等在规避异源基因的处理时可能存在模型过度拟合的缺陷。
形成这一僵局的原因来自多方面。首先,许多现存的α-变形菌纲生物受到与其他细菌之间的双向水平基因转移(HGT)影响[132],线粒体始祖本身甚至可能就有一个嵌合体基因组[120];其次,线粒体基因在进化上比一般自由生活的细菌更加迅速[128],且线粒体基因组向核基因的转移与时间呈现非线性关系[133],为模型的建立带来困难;最后,α-变形菌纲作为一个成员变异性极大的纲,现存成员间的进化关系仍在调整,例如Mu?oz-Gómez等[134]的研究已将Holosporales目从“广义的立克次氏体目”(Rickettsiales sensu lato)中分离出来。环境微生物基因组的测定也在不断发现新的α-变形菌纲物种。
综上,线粒体始祖的判定仍是探索真核生物起源道路上亟需解决的问题。
2.2.3 古细菌宿主的确定
长期以来的系统发育基因组学研究已经基本确定线粒体内共生的宿主与古细菌相关[84]。宏基因组技术出现之前,古细菌分为广古菌门和泉古菌门两门。宿主身份有颇多说法。泉古菌假说支持宿主属于泉古菌门[28,29];氢假说(图7E)[31]与互养共栖假说(图7B)[32]支持宿主属于广古菌门中的产甲烷菌(methanogens);Searcy等[135]与Margulis等[100]支持宿主属于广古菌门中的热原体菌(Thermoplasma) (图7A)。这些模型虽彼此对立,但共同点是线粒体内共生仅涉及一个古细菌宿主和一个真细菌内共生体。然而,与此不同的其他一些模型则认为宿主是两种原核生物的嵌合体,这些观点被统一称作三元假说(ternary hypothesis)。其中较为流行的是内生核假说(endokaryotic hypothesis) (图7C),推测宿主的细胞核起源于一种古细菌,而细胞质起源于一种真细菌,以此解释真核生物基因组中无法追溯到α-变形菌纲的细菌来源基因[101,136]。然而,后期的系统发育检验并没有发现这样一个细菌谱系,这表明内生核假说缺乏证据支持[53]。面对众多模型长期不可调和的僵局,2012年,Godd[137]提出了另一种三元假说。在融合纳古菌属(Nanoarchaeum) (当时未独立为门,属于广古菌门)和燃球菌属(Ignicoccus) (属于泉古菌门)两种古细菌的基因组后,Godd发现可以解释当时的大部分数据,提出宿主可能是两种古细菌的嵌合体(chimera)。这一设想具有很强的创新性,但仍然需要大量的系统发育分析和细胞学研究检验才能令人信服。
在结合宏基因组和单细胞基因组分析技术发现TACK超门和Asgard超门以后,内共生宿主的身份逐渐明朗,也基本消除了第三个原始真核生物谱系存在的可能性[11,17]。Asgard古细菌中大量ESPs的出现同时否定了其他一些有关病毒影响真核生物起源的理论。这些理论认为真核生物的大量基因,甚至细胞核本身都来自核质大DNA病毒(nucleocytoplasmic large DNA viruses, NCLDVs)[138,139],其基于的证据主要是在NCLDVs中发现了许多与真核生物相关的基因。而如今Asgard古细菌中大量ESPs的发现以及随后纠正的系统发育分析则表明,NCLDVs中的真核样基因实际来自于真核生物病毒的HGT。病毒的高进化速率为此前的系统发育重建带来偏差,导致了NCLDVs是真核生物基因甚至是细胞核源头的判断。事实上,NCLDVs可能是Asgard古细菌或相关谱系的古老成员[90]。
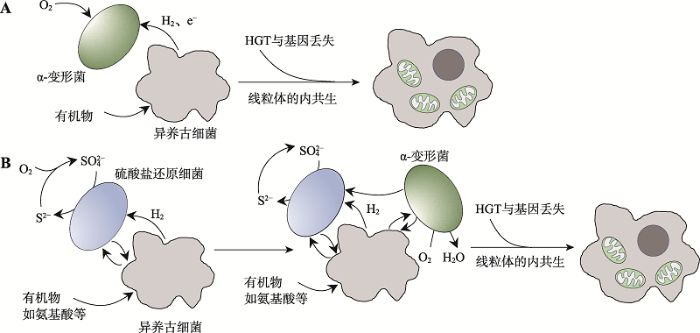
如今,对Asgard古细菌的分析研究已经成为了解真核生物祖先的重要手段。Asgard超门呈现出丰富的代谢方式。宏基因组重建分析推测Asgard超门普遍存在氮和硫代谢循环[61],不能进行吞噬作用[115],Lokiarchaeota存在依赖氢的代谢途径[140],Thorarchaeota存在依赖无机碳和有机碳的混合代谢以及产乙酸途径[62,141],Lokiarchaeota和Thorarchaeota存在卤代有机物代谢途径[142],Heimdallarchaeota存在光能营养途径[143,144],Helarchaeota存在厌氧碳氢化合物代谢途径[75],部分Lokiarchaeota编码镍铁氢化酶[142]。基于这些代谢研究提出的真核生物起源新模型也不断涌现,如电子或氢从有机异养古细菌宿主流向细菌共生体的“逆流模型”(reverse flow model) (图8A)[145]。此外,对水底生物垫内Asgard古细菌与周围其他细菌的基因组综合研究也有助于阐释线粒体祖先与宿主内共生前的共生关系,例如Sagha?等[146]便利用16S/18S rRNA宏条形码和多元统计分析技术,在盐沼池水中发现了Lokiarchaeota与此前知之甚少的细菌门类TA06间的强正相关关系,可能存在共生作用。
图8
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8真核生物起源的逆流模型与E3模型的对比
A:逆流模型;B:E3模型。两个模型的共同点是异养古细菌宿主与α-变形菌内共生形成线粒体;不同点在于E3模型比逆流模型多引入了一个共生菌——硫酸盐还原细菌。
Fig. 8Comparison between reverse flow model and E3 model
由于大量Asgard古细菌的研究结论都只是基于宏基因组分析的结果,细胞生物学层面的研究验证便显得尤为重要。然而,Asgard古细菌的生长环境大多处于海底沉积物中,且可能依赖其他共生细菌生存,这为Asgard古细菌的分离培养和蛋白的提纯带来巨大困难,相关工作开展缓慢。Ak?l和Robinson[147]取得了标志性成果。他们利用质粒载体表达Asgard古细菌的前纤维蛋白编码基因,发现表达产物能够成功与哺乳动物的肌动蛋白结合,即使相距2、30亿年的进化距离依旧与哺乳动物的相关蛋白呈现结构和功能的相似性,这为Asgard超门与真核生物的相似性提供了一个细胞学证据。
近年来,对Asgard古细菌的研究终于迎来里程碑式的成果。日本科学家Imachi等[13]花费12年时间利用连续流动生物反应器,成功从深海沉积物中分离,并首次在实验室环境下提纯培养出一个Asgard古细菌菌株MK-D1,属于Lokiarchaeota。该研究的原始目的是寻找甲烷降解菌[148]。分离到的Lokiarchaeota门古细菌生长缓慢,滞后期长达3~6个月,二倍扩增需14~25天。进一步观察显示,这是一种小型厌氧球菌,细胞可以形成凸起结构、气泡和胞外小泡,没有明显的真核样细胞器,具备厌氧代谢氨基酸的能力,依赖共生菌一起生存。Imachi等据此提出了新的真核生物起源模型——纠缠-吞噬-内生(entangle-engulf-endogenize, E3)模型:厌氧古细菌宿主与一种硫酸盐还原细菌共生,共生细菌是宿主氨基酸代谢产生的脂肪酸和氢气的受体,随后宿主与另一好氧细菌内共生形成线粒体,共涉及3种原核生物。这与此前仅涉及两个原核生物的“逆流模型”[145]存在不同(图8)。逆流模型认为古细菌代谢产生的氢气直接被线粒体祖先消耗,无需引入假设的另一个共生伴侣(图8A),而E3模型中的线粒体起源是处于共生关系下的厌氧宿主古细菌对环境中氧气浓度增加的适应行为(图8B)。
在未来,有关古细菌宿主的研究无疑将借鉴Imachi等的经验,从环境中发现并在实验室条件下培养研究与真核生物进化距离更近的微生物。
3 结语与展望
近几十年来对真核生物起源的研究积累了大量数据、模型和观点,这是一个涉及进化生物学、微生物学、生物化学、分子生物学、细胞生物学和古生物学等多领域的宏大问题。新技术的发明(如16S/18S rRNA测序、基因组测序、宏基因组测序和单细胞全基因组测序)、新物种的发现(如古细菌域、TACK超门和Asgard超门)以及分析方法的改进不断推动我们对于真核生物起源的认知。然而争论依然存在,主流观点将生命之树上的真核生物分支放在古细菌域内,从Asgard超门内发出或是Asgard超门的姐妹群,对应二域系统生命之树;真核生物的始祖是一个类似Asgard超门的古细菌,在与一种α-变形菌纲细菌内共生形成线粒体后促进其他细胞内部结构的进化。
未来的研究重点,一方面在于改进系统发育分析方法,并延续环境微生物的宏基因组重建分析工作。在确定古细菌宿主的角度上,发现新的Asgard古细菌,甚至是其他与真核生物更接近的古细菌新门类。在确定线粒体祖先的角度上,亟需理清α-变形菌纲的谱系关系。目前可获取到基因组数据的α-变形菌纲物种大多与医学与农业相关,或者是部分可在实验室条件下分离培养的菌种[120,130],可能存在一定的取样偏差,不一定能完全代表现存α-变形菌的多样性。因此,在海水、地下水和土壤等环境中发现未培养微生物,扩展α-变形菌纲物种数的工作可能仍是重中之重。此外,发展基因树-物种树(gene tree-species tree)综合分析[149]等更加复杂的计算方法也有望为线粒体的祖先提供更加精确的定位。另一方面,实验室中的细胞生物学研究亟需跟进。在确定古细菌宿主的角度上,借鉴Imachi等[13]的工作,实验室条件下直接培养Asgard古细菌进行基因组测序可以避免宏基因组重建时的误差,直接观察细胞形态和代谢过程。但这面临两个问题,一是需要改进生物反应器系统,缩短Asgard古细菌的分离提纯时间;二是需要研究自然条件下Asgard古细菌的生活环境和共生细菌等,摸索其在实验室中的最适培养条件,加快繁殖速度以供实验需求。在确定线粒体祖先的角度上,Hu[150]通过冷冻电镜发现HeLa细胞线粒体存在与许多现存α-变形菌纲生物相似的出芽增殖方式,不同于传统主流观点所描述的二分裂增殖方式,而这一现象难以在常用的荧光显微镜下观察到。此结果说明或许可以将新一代显微成像技术应用于线粒体祖先的研究中,从形态学层面比较线粒体与立克次氏体等现存α-变形菌的异同,为系统发育学分析提供必要补充。此外,实验室内对真核细胞进化某些步骤的直接模拟实验也有利于我们还原真核生物始祖的内共生效应和细胞器的进化历程。相关研究工作已在开展之中,例如2018年Mehta等[151]报道的在酵母胞质内培养大肠杆菌多达120天的工作。
最后,值得提出的一点是,现在采用的Asgard古细菌等研究对象并不古老,也同真核生物一样走过了十几亿年的辐射进化历程。现存代谢方式丰富多样的Asgard古细菌不可能完全代表原始的真核生物。未来的目标将是通过分析大量现存与即将涌现的数据,提出一个适用于所有真核生物特征的真核生物起源模型。
致谢
本文在撰稿及写作过程中得到南京大学生命科学学院杨四海教授的悉心指导,特此表示由衷的感谢。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 3]
DOI:10.1002/iub.1950URLPMID:30358047 [本文引用: 2]

The origin of eukaryotes stands as a major open question in biology. Central to this question is the nature and timing of the origin of the mitochondrion, an ubiquitous eukaryotic organelle originated by the endosymbiosis of an alphaproteobacterial ancestor. Different hypotheses disagree, among other aspects, on whether mitochondria were acquired early or late during eukaryogenesis. Similarly, the nature and complexity of the receiving host is debated, with models ranging from a simple prokaryotic host to an already complex proto-eukaryote. Here, I will discuss recent findings from phylogenomics analyses of extant genomes that are shedding light into the evolutionary origins of the eukaryotic ancestor, and which suggest a later acquisition of alpha-proteobacterial derived proteins as compared to those with different bacterial ancestries. I argue that simple eukaryogenesis models that assume a binary symbiosis between an archaeon host and an alpha-proteobacterial proto-mitochondrion cannot explain the complex chimeric nature that is inferred for the eukaryotic ancestor. To reconcile existing hypotheses with the new data, I propose the
DOI:10.1146/annurev.micro.59.030804.121258URLPMID:15910279 [本文引用: 1]
The phylum Planctomycetes of the domain Bacteria consists of budding, peptidoglycan-less organisms important for understanding the origins of complex cell organization. Their significance for cell biology lies in their possession of intracellular membrane compartmentation. All planctomycetes share a unique cell plan, in which the cell cytoplasm is divided into compartments by one or more membranes, including a major cell compartment containing the nucleoid. Of special significance is Gemmata obscuriglobus, in which the nucleoid is enveloped in two membranes to form a nuclear body that is analogous to the structure of a eukaryotic nucleus. Planctomycete compartmentation may have functional physiological roles, as in the case of anaerobic ammonium-oxidizing anammox planctomycetes, in which the anammoxosome harbors specialized enzymes and is wrapped in an envelope possessing unique ladderane lipids. Organisms in phyla other than the phylum Planctomycetes may possess compartmentation similar to that of some planctomycetes, as in the case of members of the phylum Poribacteria from marine sponges.
DOI:10.1038/nature04531URLPMID:16511485 [本文引用: 3]
The origin of the eukaryotic nucleus marked a seminal evolutionary transition. We propose that the nuclear envelope's incipient function was to allow mRNA splicing, which is slow, to go to completion so that translation, which is fast, would occur only on mRNA with intact reading frames. The rapid, fortuitous spread of introns following the origin of mitochondria is adduced as the selective pressure that forged nucleus-cytosol compartmentalization.
[本文引用: 1]
[本文引用: 1]
DOI:10.1242/jcs.178566URLPMID:27672020 [本文引用: 5]
Eukaryogenesis - the emergence of eukaryotic cells - represents a pivotal evolutionary event. With a fundamentally more complex cellular plan compared to prokaryotes, eukaryotes are major contributors to most aspects of life on Earth. For decades, we have understood that eukaryotic origins lie within both the Archaea domain and alpha-Proteobacteria. However, it is much less clear when, and from which precise ancestors, eukaryotes originated, or the order of emergence of distinctive eukaryotic cellular features. Many competing models for eukaryogenesis have been proposed, but until recently, the absence of discriminatory data meant that a consensus was elusive. Recent advances in paleogeology, phylogenetics, cell biology and microbial diversity, particularly the discovery of the 'Candidatus Lokiarcheaota' phylum, are now providing new insights into these aspects of eukaryogenesis. The new data have allowed the time frame during which eukaryogenesis occurred to be finessed, a more precise identification of the contributing lineages and the biological features of the contributors to be clarified. Considerable advances have now been used to pinpoint the prokaryotic origins of key eukaryotic cellular processes, such as intracellular compartmentalisation, with major implications for models of eukaryogenesis.
DOI:10.1038/35065071URLPMID:11242044 [本文引用: 1]
Sulphate-reducing microbes affect the modern sulphur cycle, and may be quite ancient, though when they evolved is uncertain. These organisms produce sulphide while oxidizing organic matter or hydrogen with sulphate. At sulphate concentrations greater than 1 mM, the sulphides are isotopically fractionated (depleted in 34S) by 10-40/1000 compared to the sulphate, with fractionations decreasing to near 0/1000 at lower concentrations. The isotope record of sedimentary sulphides shows large fractionations relative to seawater sulphate by 2.7 Gyr ago, indicating microbial sulphate reduction. In older rocks, however, much smaller fractionations are of equivocal origin, possibly biogenic but also possibly volcanogenic. Here we report microscopic sulphides in approximately 3.47-Gyr-old barites from North Pole, Australia, with maximum fractionations of 21.1/1000, about a mean of 11.6/1000, clearly indicating microbial sulphate reduction. Our results extend the geological record of microbial sulphate reduction back more than 750 million years, and represent direct evidence of an early specific metabolic pathway--allowing time calibration of a deep node on the tree of life.
DOI:10.1016/j.gca.2008.08.026URL [本文引用: 1]
[本文引用: 1]
DOI:10.1073/pnas.1110633108URLPMID:21810989 [本文引用: 1]
Although macroscopic plants, animals, and fungi are the most familiar eukaryotes, the bulk of eukaryotic diversity is microbial. Elucidating the timing of diversification among the more than 70 lineages is key to understanding the evolution of eukaryotes. Here, we use taxon-rich multigene data combined with diverse fossils and a relaxed molecular clock framework to estimate the timing of the last common ancestor of extant eukaryotes and the divergence of major clades. Overall, these analyses suggest that the last common ancestor lived between 1866 and 1679 Ma, consistent with the earliest microfossils interpreted with confidence as eukaryotic. During this interval, the Earth's surface differed markedly from today; for example, the oceans were incompletely ventilated, with ferruginous and, after about 1800 Ma, sulfidic water masses commonly lying beneath moderately oxygenated surface waters. Our time estimates also indicate that the major clades of eukaryotes diverged before 1000 Ma, with most or all probably diverging before 1200 Ma. Fossils, however, suggest that diversity within major extant clades expanded later, beginning about 800 Ma, when the oceans began their transition to a more modern chemical state. In combination, paleontological and molecular approaches indicate that long stems preceded diversification in the major eukaryotic lineages.
URLPMID:25945739 [本文引用: 6]
DOI:10.1038/nature21031URLPMID:28077874 [本文引用: 3]
The origin and cellular complexity of eukaryotes represent a major enigma in biology. Current data support scenarios in which an archaeal host cell and an alphaproteobacterial (mitochondrial) endosymbiont merged together, resulting in the first eukaryotic cell. The host cell is related to Lokiarchaeota, an archaeal phylum with many eukaryotic features. The emergence of the structural complexity that characterizes eukaryotic cells remains unclear. Here we describe the 'Asgard' superphylum, a group of uncultivated archaea that, as well as Lokiarchaeota, includes Thor-, Odin- and Heimdallarchaeota. Asgard archaea affiliate with eukaryotes in phylogenomic analyses, and their genomes are enriched for proteins formerly considered specific to eukaryotes. Notably, thorarchaeal genomes encode several homologues of eukaryotic membrane-trafficking machinery components, including Sec23/24 and TRAPP domains. Furthermore, we identify thorarchaeal proteins with similar features to eukaryotic coat proteins involved in vesicle biogenesis. Our results expand the known repertoire of 'eukaryote-specific' proteins in Archaea, indicating that the archaeal host cell already contained many key components that govern eukaryotic cellular complexity.
DOI:10.1038/s41586-019-1916-6URLPMID:31942073 [本文引用: 3]
The origin of eukaryotes remains unclear(1-4). Current data suggest that eukaryotes may have emerged from an archaeal lineage known as 'Asgard' archaea(5,6). Despite the eukaryote-like genomic features that are found in these archaea, the evolutionary transition from archaea to eukaryotes remains unclear, owing to the lack of cultured representatives and corresponding physiological insights. Here we report the decade-long isolation of an Asgard archaeon related to Lokiarchaeota from deep marine sediment. The archaeon-'Candidatus Prometheoarchaeum syntrophicum' strain MK-D1-is an anaerobic, extremely slow-growing, small coccus (around 550 nm in diameter) that degrades amino acids through syntrophy. Although eukaryote-like intracellular complexes have been proposed for Asgard archaea(6), the isolate has no visible organelle-like structure. Instead, Ca. P. syntrophicum is morphologically complex and has unique protrusions that are long and often branching. On the basis of the available data obtained from cultivation and genomics, and reasoned interpretations of the existing literature, we propose a hypothetical model for eukaryogenesis, termed the entangle-engulf-endogenize (also known as E(3)) model.
URLPMID:270744 [本文引用: 1]
DOI:10.1073/pnas.86.23.9355URL [本文引用: 1]
DOI:10.1073/pnas.87.12.4576URLPMID:2112744 [本文引用: 2]
Molecular structures and sequences are generally more revealing of evolutionary relationships than are classical phenotypes (particularly so among microorganisms). Consequently, the basis for the definition of taxa has progressively shifted from the organismal to the cellular to the molecular level. Molecular comparisons show that life on this planet divides into three primary groupings, commonly known as the eubacteria, the archaebacteria, and the eukaryotes. The three are very dissimilar, the differences that separate them being of a more profound nature than the differences that separate typical kingdoms, such as animals and plants. Unfortunately, neither of the conventionally accepted views of the natural relationships among living systems--i.e., the five-kingdom taxonomy or the eukaryote-prokaryote dichotomy--reflects this primary tripartite division of the living world. To remedy this situation we propose that a formal system of organisms be established in which above the level of kingdom there exists a new taxon called a
DOI:10.1038/nrmicro.2017.133URLPMID:29123225 [本文引用: 10]
Woese and Fox's 1977 paper on the discovery of the Archaea triggered a revolution in the field of evolutionary biology by showing that life was divided into not only prokaryotes and eukaryotes. Rather, they revealed that prokaryotes comprise two distinct types of organisms, the Bacteria and the Archaea. In subsequent years, molecular phylogenetic analyses indicated that eukaryotes and the Archaea represent sister groups in the tree of life. During the genomic era, it became evident that eukaryotic cells possess a mixture of archaeal and bacterial features in addition to eukaryotic-specific features. Although it has been generally accepted for some time that mitochondria descend from endosymbiotic alphaproteobacteria, the precise evolutionary relationship between eukaryotes and archaea has continued to be a subject of debate. In this Review, we outline a brief history of the changing shape of the tree of life and examine how the recent discovery of a myriad of diverse archaeal lineages has changed our understanding of the evolutionary relationships between the three domains of life and the origin of eukaryotes. Furthermore, we revisit central questions regarding the process of eukaryogenesis and discuss what can currently be inferred about the evolutionary transition from the first to the last eukaryotic common ancestor.
DOI:10.1038/441289aURLPMID:16710401 [本文引用: 1]
URLPMID:10872322 [本文引用: 1]
DNA-dependent RNA polymerases of archaebacteria not only resemble the nuclear RNA polymerases of eukaryotes rather than the eubacterial enzymes in their complex component patterns but also show striking immunochemical, i.e., structural, homology with the eukaryotic polymerases at the level of single components. Thus, eukaryotic and archaebacterial RNA polymerases are indeed of the same type, distinct from the eubacterial enzymes, which, however, are also derived from a common ancestral structure.
DOI:10.1139/m89-011URLPMID:2541879 [本文引用: 1]
Unrooted phylogenetic dendrograms were calculated by two independent methods, parsimony and distance matrix analysis, from an alignment of the derived amino acid sequences of the A and C subunits of the DNA-dependent RNA polymerases of the archaebacteria Sulfolobus acidocaldarius and Halobacterium halobium with 12 corresponding sequences including a further set of archaebacterial A+C subunits, eukaryotic nuclear RNA polymerases, pol I, pol II, and pol III, eubacterial beta' and chloroplast beta' and beta
URLPMID:20844558 [本文引用: 6]
DOI:10.1073/pnas.0809467105URLPMID:18987308 [本文引用: 1]
In contrast to the cell division machineries of bacteria, euryarchaea, and eukaryotes, no division components have been identified in the second main archaeal phylum, Crenarchaeota. Here, we demonstrate that a three-gene operon, cdv, in the crenarchaeon Sulfolobus acidocaldarius, forms part of a unique cell division machinery. The operon is induced at the onset of genome segregation and division, and the Cdv proteins then polymerize between segregating nucleoids and persist throughout cell division, forming a successively smaller structure during constriction. The cdv operon is dramatically down-regulated after UV irradiation, indicating division inhibition in response to DNA damage, reminiscent of eukaryotic checkpoint systems. The cdv genes exhibit a complementary phylogenetic range relative to FtsZ-based archaeal division systems such that, in most archaeal lineages, either one or the other system is present. Two of the Cdv proteins, CdvB and CdvC, display homology to components of the eukaryotic ESCRT-III sorting complex involved in budding of luminal vesicles and HIV-1 virion release, suggesting mechanistic similarities and a common evolutionary origin.
DOI:10.1126/science.1165322URLPMID:19008417 [本文引用: 1]
Archaea are prokaryotic organisms that lack endomembrane structures. However, a number of hyperthermophilic members of the Kingdom Crenarchaea, including members of the Sulfolobus genus, encode homologs of the eukaryotic endosomal sorting system components Vps4 and ESCRT-III (endosomal sorting complex required for transport-III). We found that Sulfolobus ESCRT-III and Vps4 homologs underwent regulation of their expression during the cell cycle. The proteins interacted and we established the structural basis of this interaction. Furthermore, these proteins specifically localized to the mid-cell during cell division. Overexpression of a catalytically inactive mutant Vps4 in Sulfolobus resulted in the accumulation of enlarged cells, indicative of failed cell division. Thus, the archaeal ESCRT system plays a key role in cell division.
[本文引用: 1]
[本文引用: 1]
DOI:10.1073/pnas.95.11.6239URLPMID:9600949 [本文引用: 1]
Analyses of complete genomes indicate that a massive prokaryotic gene transfer (or transfers) preceded the formation of the eukaryotic cell. In comparisons of the entire set of Methanococcus jannaschii genes with their orthologs from Escherichia coli, Synechocystis 6803, and the yeast Saccharomyces cerevisiae, it is shown that prokaryotic genomes consist of two different groups of genes. The deeper, diverging informational lineage codes for genes which function in translation, transcription, and replication, and also includes GTPases, vacuolar ATPase homologs, and most tRNA synthetases. The more recently diverging operational lineage codes for amino acid synthesis, the biosynthesis of cofactors, the cell envelope, energy metabolism, intermediary metabolism, fatty acid and phospholipid biosynthesis, nucleotide biosynthesis, and regulatory functions. In eukaryotes, the informational genes are most closely related to those of Methanococcus, whereas the majority of operational genes are most closely related to those of Escherichia, but some are closest to Methanococcus or to Synechocystis.
DOI:10.1073/pnas.082112499URLPMID:11983902 [本文引用: 1]
Genes encoding the glycolytic enzymes of the facultative endocellular parasite Bartonella henselae have been analyzed phylogenetically within a very large cohort of homologues from bacteria and eukaryotes. We focus on this relative of Rickettsia prowazekii along with homologues from other alpha-proteobacteria to determine whether there have been systematic transfers of glycolytic genes from the presumed alpha-proteobacterial ancestor of the mitochondrion to the nucleus of the early eukaryote. The alpha-proteobacterial homologues representing the eight glycolytic enzymes studied here tend to cluster in well-supported nodes. Nevertheless, not one of these alpha-proteobacterial enzymes is related as a sister clade to the corresponding eukaryotic homologues. Nor is there a close phylogenetic relationship between glycolytic genes from Eucarya and any other bacterial phylum. In contrast, several of the reconstructions suggest that there may have been systematic transfer of sequences encoding glycolytic enzymes from cyanobacteria to some green plants. Otherwise, surprisingly little exchange between the bacterial and eukaryotic domains is observed. The descent of eukaryotic genes encoding enzymes of intermediary metabolism is reevaluated.
DOI:10.1016/j.tibs.2004.07.002URLPMID:15337120 [本文引用: 1]
Archaea possess unique membrane phospholipids that generally comprise isoprenoid ethers built on sn-glycerol-1-phosphate (G1P). By contrast, bacterial and eukaryal membrane phospholipids are fatty acid esters linked to sn-glycerol-3-phosphate (G3P). The two key dehydrogenase enzymes that produce G1P and G3P, G1PDH and G3PDH, respectively, are not homologous. Various models propose that these enzymes originated during the speciation of the two prokaryotic domains, and the nature (and even the very existence) of lipid membranes in the last universal common ancestor (cenancestor) is subject to debate. G1PDH and G3PDH belong to two separate superfamilies that are universally distributed, suggesting that members of both superfamilies existed in the cenancestor. Furthermore, archaea possess homologues to known bacterial genes involved in fatty acid metabolism and synthesize fatty acid phospholipids. The cenancestor seems likely to have been endowed with membrane lipids whose synthesis was enzymatic but probably non-stereospecific.
DOI:10.1073/pnas.81.12.3786URLPMID:6587394 [本文引用: 3]
Ribosomal large and small subunits are organized in four general structural patterns. The four types are found in ribosomes from eubacteria, archaebacteria, eukaryotes, and a group of sulfur-dependent bacteria ( eocytes ), respectively. All four ribosomal types share a common structural core, but each type also has additional independent structural features. The independent features include the eukaryotic lobes and the archaebacterial bill on the smaller subunit. On the larger subunit, they include the eocytic lobe, eocytic gap, and eocytic bulge and a modified central protuberance. These data are most parsimoniously fit by a single unrooted evolutionary tree. In this tree eocytes are closely related to eukaryotes, while archaebacteria and eubacteria are closest neighbors. The tree is consistent with currently known molecular biological properties and indicates that eocytes have a phylogenetic importance equal to that of the three known kingdoms. When other properties and molecular mechanisms of these organisms are better defined, we suggest that an appropriate kingdom name for this group would be the Eocyta .
URLPMID:1621096 [本文引用: 3]
DOI:10.1038/nature04546URLPMID:16572163 [本文引用: 5]
The idea that some eukaryotes primitively lacked mitochondria and were true intermediates in the prokaryote-to-eukaryote transition was an exciting prospect. It spawned major advances in understanding anaerobic and parasitic eukaryotes and those with previously overlooked mitochondria. But the evolutionary gap between prokaryotes and eukaryotes is now deeper, and the nature of the host that acquired the mitochondrion more obscure, than ever before.
DOI:10.1038/32096URLPMID:9510246 [本文引用: 4]
A new hypothesis for the origin of eukaryotic cells is proposed, based on the comparative biochemistry of energy metabolism. Eukaryotes are suggested to have arisen through symbiotic association of an anaerobic, strictly hydrogen-dependent, strictly autotrophic archaebacterium (the host) with a eubacterium (the symbiont) that was able to respire, but generated molecular hydrogen as a waste product of anaerobic heterotrophic metabolism. The host's dependence upon molecular hydrogen produced by the symbiont is put forward as the selective principle that forged the common ancestor of eukaryotic cells.
DOI:10.1007/pl00006408URLPMID:9797402 [本文引用: 5]
We present a novel hypothesis for the origin of the eukaryotic cell, or eukaryogenesis, based on a metabolic symbiosis (syntrophy) between a methanogenic archaeon (methanobacterial-like) and a delta-proteobacterium (an ancestral sulfate-reducing myxobacterium). This syntrophic symbiosis was originally mediated by interspecies H2 transfer in anaerobic, possibly moderately thermophilic, environments. During eukaryogenesis, progressive cellular and genomic cointegration of both types of prokaryotic partners occurred. Initially, the establishment of permanent consortia, accompanied by extensive membrane development and close cell-cell interactions, led to a highly evolved symbiotic structure already endowed with some primitive eukaryotic features, such as a complex membrane system defining a protonuclear space (corresponding to the archaeal cytoplasm), and a protoplasmic region (derived from fusion of the surrounding bacterial cells). Simultaneously, bacterial-to-archaeal preferential gene transfer and eventual replacement took place. Bacterial genome extinction was thus accomplished by gradual transfer to the archaeal host, where genes adapted to a new genetic environment. Emerging eukaryotes would have inherited archaeal genome organization and dynamics and, consequently, most DNA-processing information systems. Conversely, primordial genes for social and developmental behavior would have been provided by the ancient myxobacterial symbiont. Metabolism would have been issued mainly from the versatile bacterial organotrophy, and progressively, methanogenesis was lost.
DOI:10.1101/gr.652803URLPMID:12618371 [本文引用: 3]
Molecular analysis of conserved sequences in the ribosomal RNAs of modern organisms reveals a three-domain phylogeny that converges in a universal ancestor for all life. We used the Clusters of Orthologous Groups database and information from published genomes to search for other universally conserved genes that have the same phylogenetic pattern as ribosomal RNA, and therefore constitute the ancestral genetic core of cells. Our analyses identified a small set of genes that can be traced back to the universal ancestor and have coevolved since that time. As indicated by earlier studies, almost all of these genes are involved with the transfer of genetic information, and most of them directly interact with the ribosome. Other universal genes have either undergone lateral transfer in the past, or have diverged so much in sequence that their distant past could not be resolved. The nature of the conserved genes suggests innovations that may have been essential to the divergence of the three domains of life. The analysis also identified several genes of unknown function with phylogenies that track with the ribosomal RNA genes. The products of these genes are likely to play fundamental roles in cellular processes.
DOI:10.1126/science.1123061URLPMID:16513982 [本文引用: 3]
We have developed an automatable procedure for reconstructing the tree of life with branch lengths comparable across all three domains. The tree has its basis in a concatenation of 31 orthologs occurring in 191 species with sequenced genomes. It revealed interdomain discrepancies in taxonomic classification. Systematic detection and subsequent exclusion of products of horizontal gene transfer increased phylogenetic resolution, allowing us to confirm accepted relationships and resolve disputed and preliminary classifications. For example, we place the phylum Acidobacteria as a sister group of delta-Proteobacteria, support a Gram-positive origin of Bacteria, and suggest a thermophilic last universal common ancestor.
URLPMID:18463089 [本文引用: 3]
DOI:10.1038/nature02848URLPMID:15356622 [本文引用: 3]
Genomes hold within them the record of the evolution of life on Earth. But genome fusions and horizontal gene transfer seem to have obscured sufficiently the gene sequence record such that it is difficult to reconstruct the phylogenetic tree of life. Here we determine the general outline of the tree using complete genome data from representative prokaryotes and eukaryotes and a new genome analysis method that makes it possible to reconstruct ancient genome fusions and phylogenetic trees. Our analyses indicate that the eukaryotic genome resulted from a fusion of two diverse prokaryotic genomes, and therefore at the deepest levels linking prokaryotes and eukaryotes, the tree of life is actually a ring of life. One fusion partner branches from deep within an ancient photosynthetic clade, and the other is related to the archaeal prokaryotes. The eubacterial organism is either a proteobacterium, or a member of a larger photosynthetic clade that includes the Cyanobacteria and the Proteobacteria.
URLPMID:17504772 [本文引用: 2]
DOI:10.1073/pnas.0810647105URLPMID:19073919 [本文引用: 4]
The origin of the eukaryotic genetic apparatus is thought to be central to understanding the evolution of the eukaryotic cell. Disagreement about the source of the relevant genes has spawned competing hypotheses for the origins of the eukaryote nuclear lineage. The iconic rooted 3-domains tree of life shows eukaryotes and archaebacteria as separate groups that share a common ancestor to the exclusion of eubacteria. By contrast, the eocyte hypothesis has eukaryotes originating within the archaebacteria and sharing a common ancestor with a particular group called the Crenarchaeota or eocytes. Here, we have investigated the relative support for each hypothesis from analysis of 53 genes spanning the 3 domains, including essential components of the eukaryotic nucleic acid replication, transcription, and translation apparatus. As an important component of our analysis, we investigated the fit between model and data with respect to composition. Compositional heterogeneity is a pervasive problem for reconstruction of ancient relationships, which, if ignored, can produce an incorrect tree with strong support. To mitigate its effects, we used phylogenetic models that allow for changing nucleotide or amino acid compositions over the tree and data. Our analyses favor a topology that supports the eocyte hypothesis rather than archaebacterial monophyly and the 3-domains tree of life.
DOI:10.1098/rstb.2009.0034URLPMID:19571240 [本文引用: 3]
The three-domains tree, which depicts eukaryotes and archaebacteria as monophyletic sister groups, is the dominant model for early eukaryotic evolution. By contrast, the 'eocyte hypothesis', where eukaryotes are proposed to have originated from within the archaebacteria as sister to the Crenarchaeota (also called the eocytes), has been largely neglected in the literature. We have investigated support for these two competing hypotheses from molecular sequence data using methods that attempt to accommodate the across-site compositional heterogeneity and across-tree compositional and rate matrix heterogeneity that are manifest features of these data. When ribosomal RNA genes were analysed using standard methods that do not adequately model these kinds of heterogeneity, the three-domains tree was supported. However, this support was eroded or lost when composition-heterogeneous models were used, with concomitant increase in support for the eocyte tree for eukaryotic origins. Analysis of combined amino acid sequences from 41 protein-coding genes supported the eocyte tree, whether or not composition-heterogeneous models were used. The possible effects of substitutional saturation of our data were examined using simulation; these results suggested that saturation is delayed by among-site rate variation in the sequences, and that phylogenetic signal for ancient relationships is plausibly present in these data.
DOI:10.1016/j.tree.2009.09.007URL [本文引用: 1]
Genomes conceal a vast intricate record of their carriers’ descent and evolution. To disclose this information, biologists need phylogenetic models that integrate various levels of organization, ranging from nucleotide sequences to ecological interactions. Rates of duplication and horizontal gene transfer, organism trees and ancestral population sizes can all be inferred through statistical models of gene family evolution and population genetics. Similarly, phylogenomics combined with other fields of natural sciences can reveal the nature of ancient phenotypes and paleoenvironments. These computationally intensive approaches now benefit from progress in statistics and algorithmics. In this article, we review the recent advances and discuss possible developments towards a comprehensive reconstruction of the history of life.
DOI:10.1093/bib/bbu015URLPMID:24872401 [本文引用: 1]
Phylogenetic analysis is used to recover the evolutionary history of species, genes or proteins. Understanding phylogenetic relationships between organisms is a prerequisite of almost any evolutionary study, as contemporary species all share a common history through their ancestry. Moreover, it is important because of its wide applications that include understanding genome organization, epidemiological investigations, predicting protein functions, and deciding the genes to be analyzed in comparative studies. Despite immense progress in recent years, phylogenetic reconstruction involves many challenges that create uncertainty with respect to the true evolutionary relationships of the species or genes analyzed. One of the most notable difficulties is the widespread occurrence of incongruence among methods and also among individual genes or different genomic regions. Presence of widespread incongruence inhibits successful revealing of evolutionary relationships and applications of phylogenetic analysis. In this article, I concisely review the effect of various factors that cause incongruence in molecular phylogenies, the advances in the field that resolved some factors, and explore unresolved factors that cause incongruence along with possible ways for tackling them.
DOI:10.1371/journal.pbio.0030130URLPMID:15799709 [本文引用: 1]
Explaining the diversity of gene repertoires has been a major problem in modern evolutionary biology. In eukaryotes, this diversity is believed to result mainly from gene duplication and loss, but in prokaryotes, lateral gene transfer (LGT) can also contribute substantially to genome contents. To determine the histories of gene inventories, we conducted an exhaustive analysis of gene phylogenies for all gene families in a widely sampled group, the gamma-Proteobacteria. We show that, although these bacterial genomes display striking differences in gene repertoires, most gene families having representatives in several species have congruent histories. Other than the few vast multigene families, gene duplication has contributed relatively little to the contents of these genomes; instead, LGT, over time, provides most of the diversity in genomic repertoires. Most such acquired genes are lost, but the majority of those that persist in genomes are transmitted strictly vertically. Although our analyses are limited to the gamma-Proteobacteria, these results resolve a long-standing paradox-i.e., the ability to make robust phylogenetic inferences in light of substantial LGT.
DOI:10.1038/nrg3962URLPMID:26184597 [本文引用: 1]
Horizontal gene transfer (HGT) is the sharing of genetic material between organisms that are not in a parent-offspring relationship. HGT is a widely recognized mechanism for adaptation in bacteria and archaea. Microbial antibiotic resistance and pathogenicity are often associated with HGT, but the scope of HGT extends far beyond disease-causing organisms. In this Review, we describe how HGT has shaped the web of life using examples of HGT among prokaryotes, between prokaryotes and eukaryotes, and even between multicellular eukaryotes. We discuss replacement and additive HGT, the proposed mechanisms of HGT, selective forces that influence HGT, and the evolutionary impact of HGT on ancestral populations and existing populations such as the human microbiome.
DOI:10.1038/nrmicro.2017.41URLPMID:28502981 [本文引用: 1]
Archaea are diverse, ecologically important, single-celled microorganisms. They have unique functions and features, such as methanogenesis and the composition of their cell envelope, although many characteristics are shared with the other domains of life, either through ancestry or through promiscuous horizontal gene transfer. The exchange of genetic material is a major driving force for genome evolution across the tree of life and has a role in archaeal speciation, adaptation and maintenance of diversity. In this Review, we discuss our current knowledge of archaeal mechanisms of DNA transfer and highlight the role of gene transfer in archaeal evolution.
DOI:10.1098/rstb.2014.0337URLPMID:26323767 [本文引用: 1]
The origin of the eukaryotic cell is considered one of the major evolutionary transitions in the history of life. Current evidence strongly supports a scenario of eukaryotic origin in which two prokaryotes, an archaebacterial host and an alpha-proteobacterium (the free-living ancestor of the mitochondrion), entered a stable symbiotic relationship. The establishment of this relationship was associated with a process of chimerization, whereby a large number of genes from the alpha-proteobacterial symbiont were transferred to the host nucleus. A general framework allowing the conceptualization of eukaryogenesis from a genomic perspective has long been lacking. Recent studies suggest that the origins of several archaebacterial phyla were coincident with massive imports of eubacterial genes. Although this does not indicate that these phyla originated through the same process that led to the origin of Eukaryota, it suggests that Archaebacteria might have had a general propensity to integrate into their genomes large amounts of eubacterial DNA. We suggest that this propensity provides a framework in which eukaryogenesis can be understood and studied in the light of archaebacterial ecology. We applied a recently developed supertree method to a genomic dataset composed of 392 eubacterial and 51 archaebacterial genera to test whether large numbers of genes flowing from Eubacteria are indeed coincident with the origin of major archaebacterial clades. In addition, we identified two potential large-scale transfers of uncertain directionality at the base of the archaebacterial tree. Our results are consistent with previous findings and seem to indicate that eubacterial gene imports (particularly from delta-Proteobacteria, Clostridia and Actinobacteria) were an important factor in archaebacterial history. Archaebacteria seem to have long relied on Eubacteria as a source of genetic diversity, and while the precise mechanism that allowed these imports is unknown, we suggest that our results support the view that processes comparable to those through which eukaryotes emerged might have been common in archaebacterial history.
DOI:10.1073/pnas.1000265107URLPMID:20852068 [本文引用: 1]
The traditional tree of life shows eukaryotes as a distinct lineage of living things, but many studies have suggested that the first eukaryotic cells were chimeric, descended from both Eubacteria (through the mitochondrion) and Archaebacteria. Eukaryote nuclei thus contain genes of both eubacterial and archaebacterial origins, and these genes have different functions within eukaryotic cells. Here we report that archaebacterium-derived genes are significantly more likely to be essential to yeast viability, are more highly expressed, and are significantly more highly connected and more central in the yeast protein interaction network. These findings hold irrespective of whether the genes have an informational or operational function, so that many features of eukaryotic genes with prokaryotic homologs can be explained by their origin, rather than their function. Taken together, our results show that genes of archaebacterial origin are in some senses more important to yeast metabolism than genes of eubacterial origin. This importance reflects these genes' origin as the ancestral nuclear component of the eukaryotic genome.
DOI:10.1093/molbev/msq333URL [本文引用: 1]
Horizontal gene transfer (HGT) is a prevalent and a highly important phenomenon in microbial species evolution. One of the important challenges in HGT research is to better understand the factors that determine the tendency of genes to be successfully transferred and retained in evolution (i.e., transferability). It was previously observed that transferability of genes depends on the cellular process in which they are involved where genes involved in transcription or translation are less likely to be transferred than metabolic genes. It was further shown that gene connectivity in the protein-protein interaction network affects HGT. These two factors were shown to be correlated, and their influence on HGT is collectively termed the "Complexity Hypothesis". In this study, we used a stochastic mapping method utilizing advanced likelihood-based evolutionary models to quantify gene family acquisition events by HGT. We applied our methodology to an extensive across-species genome-wide dataset that enabled us to estimate the overall extent of transfer events in evolution and to study the trends and barriers to gene transferability. Focusing on the biological function and the connectivity of genes, we obtained novel insights regarding the "complexity hypothesis." Specifically, we aimed to disentangle the relationships between protein connectivity, cellular function, and transferability and to quantify the relative contribution of each of these factors in determining transferability. We show that the biological function of a gene family is an insignificant factor in the determination of transferability when proteins with similar levels of connectivity are compared. In contrast, we found that connectivity is an important and a statistically significant factor in determining transferability when proteins with a similar function are compared.
DOI:10.1038/nature12779URL [本文引用: 3]
The discovery of the Archaea and the proposal of the three-domains 'universal' tree, based on ribosomal RNA and core genes mainly involved in protein translation, catalysed new ideas for cellular evolution and eukaryotic origins. However, accumulating evidence suggests that the three-domains tree may be incorrect: evolutionary trees made using newer methods place eukaryotic core genes within the Archaea, supporting hypotheses in which an archaeon participated in eukaryotic origins by founding the host lineage for the mitochondrial endosymbiont. These results provide support for only two primary domains of life-Archaea and Bacteria-because eukaryotes arose through partnership between them.
DOI:10.1038/s41559-019-1040-xURLPMID:31819234 [本文引用: 3]
Hypotheses about the origin of eukaryotic cells are classically framed within the context of a universal 'tree of life' based on conserved core genes. Vigorous ongoing debate about eukaryote origins is based on assertions that the topology of the tree of life depends on the taxa included and the choice and quality of genomic data analysed. Here we have reanalysed the evidence underpinning those claims and apply more data to the question by using supertree and coalescent methods to interrogate >3,000 gene families in archaea and eukaryotes. We find that eukaryotes consistently originate from within the archaea in a two-domains tree when due consideration is given to the fit between model and data. Our analyses support a close relationship between eukaryotes and Asgard archaea and identify the Heimdallarchaeota as the current best candidate for the closest archaeal relatives of the eukaryotic nuclear lineage.
DOI:10.1006/mpev.1999.0675URLPMID:10508549 [本文引用: 1]
Phylogenetic analyses of gene and protein sequences have led to two major competing views of the universal phylogeny, the evolutionary tree relating the three kinds of living organisms, Bacteria, Archaea, and Eukarya. In the first scheme, called
DOI:10.1016/j.tim.2011.09.002URL [本文引用: 1]
Although most hypotheses to explain the emergence of the eukaryotic lineage are conflicting, some consensus exists concerning the requirement of a genomic fusion between archaeal and bacterial components. Recent phylogenomic studies have provided support for eocyte-like scenarios in which the alleged 'archaeal parent' of the eukaryotic cell emerged from the Crenarchaeota/Thaumarchaeota. Here, we provide evidence for a scenario in which this archaeal parent emerged from within the 'TACK' superphylum that comprises the Thaumarchaeota, Crenarchaeota and Korarchaeota, as well as the recently proposed phylum 'Aigarchaeota'. In support of this view, functional and comparative genomics studies have unearthed an increasing number of features that are uniquely shared by the TACK superphylum and eukaryotes, including proteins involved in cytokinesis, membrane remodeling, cell shape determination and protein recycling.
DOI:10.1098/rspb.2012.1795URLPMID:23097517 [本文引用: 1]
Determining the relationships among the major groups of cellular life is important for understanding the evolution of biological diversity, but is difficult given the enormous time spans involved. In the textbook 'three domains' tree based on informational genes, eukaryotes and Archaea share a common ancestor to the exclusion of Bacteria. However, some phylogenetic analyses of the same data have placed eukaryotes within the Archaea, as the nearest relatives of different archaeal lineages. We compared the support for these competing hypotheses using sophisticated phylogenetic methods and an improved sampling of archaeal biodiversity. We also employed both new and existing tests of phylogenetic congruence to explore the level of uncertainty and conflict in the data. Our analyses suggested that much of the observed incongruence is weakly supported or associated with poorly fitting evolutionary models. All of our phylogenetic analyses, whether on small subunit and large subunit ribosomal RNA or concatenated protein-coding genes, recovered a monophyletic group containing eukaryotes and the TACK archaeal superphylum comprising the Thaumarchaeota, Aigarchaeota, Crenarchaeota and Korarchaeota. Hence, while our results provide no support for the iconic three-domain tree of life, they are consistent with an extended eocyte hypothesis whereby vital components of the eukaryotic nuclear lineage originated from within the archaeal radiation.
DOI:10.1093/molbev/mst272URL [本文引用: 4]
The evolutionary origin of eukaryotes is a question of great interest for which many different hypotheses have been proposed. These hypotheses predict distinct patterns of evolutionary relationships for individual genes of the ancestral eukaryotic genome. The availability of numerous completely sequenced genomes covering the three domains of life makes it possible to contrast these predictions with empirical data. We performed a systematic analysis of the phylogenetic relationships of ancestral eukaryotic genes with archaeal and bacterial genes. In contrast with previous studies, we emphasize the critical importance of methods accounting for statistical support, horizontal gene transfer, and gene loss, and we disentangle the processes underlying the phylogenomic pattern we observe. We first recover a clear signal indicating that a fraction of the bacteria-like eukaryotic genes are of alphaproteobacterial origin. Then, we show that the majority of bacteria-related eukaryotic genes actually do not point to a relationship with a specific bacterial taxonomic group. We also provide evidence that eukaryotes branch close to the last archaeal common ancestor. Our results demonstrate that there is no phylogenetic support for hypotheses involving a fusion with a bacterium other than the ancestor of mitochondria. Overall, they leave only two possible interpretations, respectively, based on the early-mitochondria hypotheses, which suppose an early endosymbiosis of an alphaproteobacterium in an archaeal host and on the slow-drip autogenous hypothesis, in which early eukaryotic ancestors were particularly prone to horizontal gene transfers.
DOI:10.1126/science.1093857URLPMID:15001713 [本文引用: 1]
We have applied
DOI:10.1038/nrmicro1852URLPMID:18274537 [本文引用: 1]
The archaeal domain is currently divided into two major phyla, the Euryarchaeota and Crenarchaeota. During the past few years, diverse groups of uncultivated mesophilic archaea have been discovered and affiliated with the Crenarchaeota. It was recently recognized that these archaea have a major role in geochemical cycles. Based on the first genome sequence of a crenarchaeote, Cenarchaeum symbiosum, we show that these mesophilic archaea are different from hyperthermophilic Crenarchaeota and branch deeper than was previously assumed. Our results indicate that C. symbiosum and its relatives are not Crenarchaeota, but should be considered as a third archaeal phylum, which we propose to name Thaumarchaeota (from the Greek 'thaumas', meaning wonder).
DOI:10.1073/pnas.0801980105URLPMID:18535141 [本文引用: 1]
The candidate division Korarchaeota comprises a group of uncultivated microorganisms that, by their small subunit rRNA phylogeny, may have diverged early from the major archaeal phyla Crenarchaeota and Euryarchaeota. Here, we report the initial characterization of a member of the Korarchaeota with the proposed name,
DOI:10.1093/nar/gkq1228URLPMID:21169198 [本文引用: 1]
The domain Archaea has historically been divided into two phyla, the Crenarchaeota and Euryarchaeota. Although regarded as members of the Crenarchaeota based on small subunit rRNA phylogeny, environmental genomics and efforts for cultivation have recently revealed two novel phyla/divisions in the Archaea; the 'Thaumarchaeota' and 'Korarchaeota'. Here, we show the genome sequence of Candidatus 'Caldiarchaeum subterraneum' that represents an uncultivated crenarchaeotic group. A composite genome was reconstructed from a metagenomic library previously prepared from a microbial mat at a geothermal water stream of a sub-surface gold mine. The genome was found to be clearly distinct from those of the known phyla/divisions, Crenarchaeota (hyperthermophiles), Euryarchaeota, Thaumarchaeota and Korarchaeota. The unique traits suggest that this crenarchaeotic group can be considered as a novel archaeal phylum/division. Moreover, C. subterraneum harbors an ubiquitin-like protein modifier system consisting of Ub, E1, E2 and small Zn RING finger family protein with structural motifs specific to eukaryotic system proteins, a system clearly distinct from the prokaryote-type system recently identified in Haloferax and Mycobacterium. The presence of such a eukaryote-type system is unprecedented in prokaryotes, and indicates that a prototype of the eukaryotic protein modifier system is present in the Archaea.
DOI:10.1101/cshperspect.a016022URLPMID:24993577 [本文引用: 1]
The origin of the eukaryotic cell can be regarded as one of the hallmarks in the history of life on our planet. The apparent genomic chimerism in eukaryotic genomes is currently best explained by invoking a cellular fusion at the root of the eukaryotes that involves one archaeal and one or more bacterial components. Here, we use a phylogenomics approach to reevaluate the evolutionary affiliation between Archaea and eukaryotes, and provide further support for scenarios in which the nuclear lineage in eukaryotes emerged from within the archaeal radiation, displaying a strong phylogenetic affiliation with, or even within, the archaeal TACK superphylum. Further taxonomic sampling of archaeal genomes in this superphylum will certainly provide a better resolution in the events that have been instrumental for the emergence of the eukaryotic lineage.
DOI:10.1093/gbe/evu274URL [本文引用: 1]
DOI:10.1073/pnas.1420858112URLPMID:25964353 [本文引用: 1]
One of the most fundamental questions in evolutionary biology is the origin of the lineage leading to eukaryotes. Recent phylogenomic analyses have indicated an emergence of eukaryotes from within the radiation of modern Archaea and specifically from a group comprising Thaumarchaeota/
DOI:10.3934/microbiol.2019.1.48URLPMID:31384702 [本文引用: 2]
Elucidating the diversity of the Archaea has many important ecological and evolutionary implications. The Asgard superphylum of the archaea, described recently from metagenomic data, has reignited the decades-old debate surrounding the topology of the tree of life. This review synthesizes recent findings through publicly available genomes and literature to describe the current ecological and evolutionary significance of the Asgard superphylum. Asgard archaea have been found in a diverse range of microbiomes across the globe, primarily from sedimentary environments. Within these environments, positive correlations between specific members of the Asgard archaea and Candidate Division TA06 bacteria have been observed, opening up the possibility of symbiotic interactions between the groupings. Asgard archaeal genomes encode functionally diverse metabolic pathways, including the Wood-Ljungdahl pathway as a carbon-fixation strategy, putative nucleotide salvaging pathways, and novel mechanisms of phototrophy including new rhodopsins. Asgard archaea also appear to be active in nitrogen cycling. Asgard archaea encode genes involved in both dissimilatory nitrate reduction and denitrification, and for the potential to use atmospheric nitrogen or nitrite as nitrogen sources. Asgard archaea also may be involved in the transformation of sulfur compounds, indicating a putative role in sulfur cycling. To date, all Asgard archaeal genomes identified were described as obligately anaerobic. The Asgard archaea also appear to have important evolutionary implications. The presence of eukaryotic signature proteins and the affiliation of Asgard archaea in phylogenetic analyses appears to support two-domain topologies of the tree of life with eukaryotes emerging from within the domain of archaea, as opposed to the eukaryotes being a separate domain of life. Thus far, Heimdallarchaeota appears as the closest archaeal relative of eukaryotes.
DOI:10.1038/ismej.2015.233URLPMID:26824177 [本文引用: 2]
Marine and estuary sediments contain a variety of uncultured archaea whose metabolic and ecological roles are unknown. De novo assembly and binning of high-throughput metagenomic sequences from the sulfate-methane transition zone in estuary sediments resulted in the reconstruction of three partial to near-complete (2.4-3.9 Mb) genomes belonging to a previously unrecognized archaeal group. Phylogenetic analyses of ribosomal RNA genes and ribosomal proteins revealed that this group is distinct from any previously characterized archaea. For this group, found in the White Oak River estuary, and previously registered in sedimentary samples, we propose the name 'Thorarchaeota'. The Thorarchaeota appear to be capable of acetate production from the degradation of proteins. Interestingly, they also have elemental sulfur and thiosulfate reduction genes suggesting they have an important role in intermediate sulfur cycling. The reconstruction of these genomes from a deeply branched, widespread group expands our understanding of sediment biogeochemistry and the evolutionary history of Archaea.
DOI:10.1038/s41564-019-0495-5URLPMID:31222170 [本文引用: 1]
DOI:10.1371/journal.pgen.1006810URLPMID:28604769 [本文引用: 2]
The eocyte hypothesis, in which Eukarya emerged from within Archaea, has been boosted by the description of a new candidate archaeal phylum,
DOI:10.1371/journal.pgen.1007080URL [本文引用: 1]
DOI:10.1371/journal.pgen.1007215URLPMID:29596428 [本文引用: 1]
DOI:10.1093/gbe/evy154URLPMID:30060184 [本文引用: 1]
Diphthamide is a modified histidine residue which is uniquely present in archaeal and eukaryotic elongation factor 2 (EF-2), an essential GTPase responsible for catalyzing the coordinated translocation of tRNA and mRNA through the ribosome. In part due to the role of diphthamide in maintaining translational fidelity, it was previously assumed that diphthamide biosynthesis genes (dph) are conserved across all eukaryotes and archaea. Here, comparative analysis of new and existing genomes reveals that some archaea (i.e., members of the Asgard superphylum, Geoarchaea, and Korarchaeota) and eukaryotes (i.e., parabasalids) lack dph. In addition, while EF-2 was thought to exist as a single copy in archaea, many of these dph-lacking archaeal genomes encode a second EF-2 paralog missing key residues required for diphthamide modification and for normal translocase function, perhaps suggesting functional divergence linked to loss of diphthamide biosynthesis. Interestingly, some Heimdallarchaeota previously suggested to be most closely related to the eukaryotic ancestor maintain dph genes and a single gene encoding canonical EF-2. Our findings reveal that the ability to produce diphthamide, once thought to be a universal feature in archaea and eukaryotes, has been lost multiple times during evolution, and suggest that anticipated compensatory mechanisms evolved independently.
DOI:10.1016/j.biochi.2013.04.016URL [本文引用: 1]
The traditional bacterial rooting of the three superkingdoms in sequence-based gene trees is inconsistent with new phylogenetic reconstructions based on genome content of compact protein domains. We find that protein domains at the level of the SCOP superfamily (SF) from sequenced genomes implement with maximum parsimony fully resolved rooted trees. Such genome content trees identify archaea and bacteria (akaryotes) as sister clades that diverge from an akaryote common ancestor, LACA. Several eukaryote sister clades diverge from a eukaryote common ancestor, LECA. LACA and LECA descend in parallel from the most recent universal common ancestor (MRUCA), which is not a bacterium. Rather, MRUCA presents 75% of the unique SFs encoded by extant genomes of the three superkingdoms, each encoding a proteome that partially overlaps all others. This alone implies that the common ancestor to the superkingdoms was very complex. Such ancestral complexity is confirmed by phylogenetic reconstructions. In addition, the divergence of proteomes from the complex ancestor in each superkingdom is both reductive in numbers of unique SFs as well as cumulative in the abundance of surviving SFs. These data suggest that the common ancestor was not the first cell lineage and that modern global phylogeny is the crown of a "recently" rerooted tree. We suggest that a bottlenecked survivor of an environmental collapse, which preceded the flourishing of the modern crown, seeded the current phylogenetic tree. (C) 2013 Elsevier Masson SAS.
DOI:10.1016/j.biochi.2017.04.013URLPMID:28461155 [本文引用: 1]
We reconstructed a global tree of life (ToL) with non-reversible and non-stationary models of genome evolution that root trees intrinsically. We implemented Bayesian model selection tests and compared the statistical support for four conflicting ToL hypotheses. We show that reconstructions obtained with a Bayesian implementation (Klopfstein et al., 2015) are consistent with reconstructions obtained with an empirical Sankoff parsimony (ESP) implementation (Harish et al., 2013). Both are based on the genome contents of coding sequences for protein domains (superfamilies) from hundreds of genomes. Thus, we conclude that the independent descent of Eukaryotes and Akaryotes (archaea and bacteria) from the universal common ancestor (UCA) is the most probable as well as the most parsimonious hypothesis for the evolutionary origins of extant genomes. Reconstructions of ancestral proteomes by both Bayesian and ESP methods suggest that at least 70% of unique domain-superfamilies known in extant species were present in the UCA. In addition, identification of a vast majority (96%) of the mitochondrial superfamilies in the UCA proteome precludes a symbiotic hypothesis for the origin of eukaryotes. Accordingly, neither the archaeal origin of eukaryotes nor the bacterial origin of mitochondria is supported by the data. The proteomic complexity of the UCA suggests that the evolution of cellular phenotypes in the two primordial lineages, Akaryotes and Eukaryotes, was driven largely by duplication of common superfamilies as well as by loss of unique superfamilies. Finally, innovation of novel superfamilies has played a surprisingly small role in the evolution of Akaryotes and only a marginal role in the evolution of Eukaryotes.
DOI:10.7717/peerj.5770URLPMID:30357005 [本文引用: 1]
The recognition of the group Archaea as a major branch of the tree of life (ToL) prompted a new view of the evolution of biodiversity. The genomic representation of archaeal biodiversity has since significantly increased. In addition, advances in phylogenetic modeling of multi-locus datasets have resolved many recalcitrant branches of the ToL. Despite the technical advances and an expanded taxonomic representation, two important aspects of the origins and evolution of the Archaea remain controversial, even as we celebrate the 40th anniversary of the monumental discovery. These issues concern (i) the uniqueness (monophyly) of the Archaea, and (ii) the evolutionary relationships of the Archaea to the Bacteria and the Eukarya; both of these are relevant to the deep structure of the ToL. To explore the causes for this persistent ambiguity, I examine multiple datasets and different phylogenetic approaches that support contradicting conclusions. I find that the uncertainty is primarily due to a scarcity of information in standard datasets-universal core-genes datasets-to reliably resolve the conflicts. These conflicts can be resolved efficiently by comparing patterns of variation in the distribution of functional genomic signatures, which are less diffused unlike patterns of primary sequence variation. Relatively lower heterogeneity in distribution patterns minimizes uncertainties and supports statistically robust phylogenetic inferences, especially of the earliest divergences of life. This case study further highlights the limitations of primary sequence data in resolving difficult phylogenetic problems, and raises questions about evolutionary inferences drawn from the analyses of sequence alignments of a small set of core genes. In particular, the findings of this study corroborate the growing consensus that reversible substitution mutations may not be optimal phylogenetic markers for resolving early divergences in the ToL, nor for determining the polarity of evolutionary transitions across the ToL.
URLPMID:12840035 [本文引用: 1]
DOI:10.1073/pnas.0408810102URLPMID:15630082 [本文引用: 1]
A simple classification scheme that uses only the presence or absence of a protein domain architecture has been used to determine the phylogeny of 174 complete genomes. The method correctly divides the 174 taxa into Archaea, Bacteria, and Eukarya and satisfactorily sorts most of the major groups within these superkingdoms. The most challenging problem involved 119 Bacteria, many of which have reduced genomes. When a weighting factor was used that takes account of difference in genome size (number of considered folds), small-genome taxa were mostly grouped with their full-sized counterparts. Although not every organism appears exactly at its classical phylogenetic position in these trees, the agreement appears comparable with the efforts of others by using sophisticated sequence analysis and/or combinations of gene content and gene order. During the course of the study, it emerged that there is a core set of approximately 50 folds that is found in all 174 genomes and a single fold diagnostic of all Archaea.
DOI:10.12688/f1000researchURL [本文引用: 2]
DOI:10.3389/fmicb.2018.01896URLPMID:30158917 [本文引用: 1]
The recent discovery of the Lokiarchaeota and other members of the Asgard superphylum suggests that closer analysis of the cell biology and evolution of these groups may help shed light on the origin of the eukaryote cell. Asgard lineages often appear in molecular phylogenies as closely related to eukaryotes, and possess
URLPMID:30602773 [本文引用: 2]
[本文引用: 1]
[本文引用: 1]
DOI:10.1186/s12866-020-1707-0URLPMID:32013868 [本文引用: 1]
BACKGROUND: Amplification of small subunit (SSU) rRNA genes with universal primers is a common method used to assess microbial populations in various environmental samples. However, owing to limitations in coverage of these universal primers, some microorganisms remain unidentified. The present study aimed to establish a method for amplifying nearly full-length SSU rRNA gene sequences of previously unidentified prokaryotes, using newly designed targeted primers via primer evaluation in meta-transcriptomic datasets. METHODS: Primer binding regions of universal primer 8F/Arch21F for bacteria or archaea were used for primer evaluation of SSU rRNA sequences in meta-transcriptomic datasets. Furthermore, targeted forward primers were designed based on SSU rRNA reads from unclassified groups unmatched with the universal primer 8F/Arch21F, and these primers were used to amplify nearly full-length special SSU rRNA gene sequences along with universal reverse primer 1492R. Similarity and phylogenetic analysis were used to confirm their novel status. RESULTS: Using this method, we identified unclassified SSU rRNA sequences that were not matched with universal primer 8F and Arch21F. A new group within the Asgard superphylum was amplified by the newly designed specific primer based on these unclassified SSU rRNA sequences by using mudflat samples. CONCLUSION: We showed that using specific primers designed based on universal primer evaluation from meta-transcriptomic datasets, identification of novel taxonomic groups from a specific environment is possible.
[本文引用: 1]
DOI:10.1186/gb-2010-11-5-209URLPMID:20441612 [本文引用: 2]
Phylogenomics of eukaryote supergroups suggest a highly complex last common ancestor of eukaryotes and a key role of mitochondrial endosymbiosis in the origin of eukaryotes.
DOI:10.3109/10409238.2013.821444URLPMID:23895660 [本文引用: 2]
Eukaryogenesis, the origin of the eukaryotic cell, represents one of the fundamental evolutionary transitions in the history of life on earth. This event, which is estimated to have occurred over one billion years ago, remains rather poorly understood. While some well-validated examples of fossil microbial eukaryotes for this time frame have been described, these can provide only basic morphology and the molecular machinery present in these organisms has remained unknown. Complete and partial genomic information has begun to fill this gap, and is being used to trace proteins and cellular traits to their roots and to provide unprecedented levels of resolution of structures, metabolic pathways and capabilities of organisms at these earliest points within the eukaryotic lineage. This is essentially allowing a molecular paleontology. What has emerged from these studies is spectacular cellular complexity prior to expansion of the eukaryotic lineages. Multiple reconstructed cellular systems indicate a very sophisticated biology, which by implication arose following the initial eukaryogenesis event but prior to eukaryotic radiation and provides a challenge in terms of explaining how these early eukaryotes arose and in understanding how they lived. Here, we provide brief overviews of several cellular systems and the major emerging conclusions, together with predictions for subsequent directions in evolution leading to extant taxa. We also consider what these reconstructions suggest about the life styles and capabilities of these earliest eukaryotes and the period of evolution between the radiation of eukaryotes and the eukaryogenesis event itself.
DOI:10.1146/annurev-biochem-061516-044643URL [本文引用: 1]
DOI:10.1016/j.cub.2018.01.086URLPMID:29689219 [本文引用: 1]
Biological membranes are thin amphiphilic sheaths, only a few nanometres thick, that define both the boundaries of all cells as well as the diversity of the internal compartments in eukaryotes. The plasma membrane of a typical prokaryote houses about 20-30% of the cell's expressed proteins, and its lipids account for approximately 10% of the cell's dry mass. The numbers for eukaryotic cells are comparable - the difference in surface area to volume ratio is overall compensated by the eukaryotic endomembrane system. Roughly a fourth of the protein encoded by the human genome carries at least one stretch of sequence predicted to serve as a transmembrane domain. Membranes host substrate exchange, sensing and communication, and life-giving energy conservation via chemiosmotic ATP synthesis.
DOI:10.1016/j.tree.2015.09.005URLPMID:26455774 [本文引用: 2]
Despite recent progress, the origin of the eukaryotic cell remains enigmatic. It is now known that the last eukaryotic common ancestor was complex and that endosymbiosis played a crucial role in eukaryogenesis at least via the acquisition of the alphaproteobacterial ancestor of mitochondria. However, the nature of the mitochondrial host is controversial, although the recent discovery of an archaeal lineage phylogenetically close to eukaryotes reinforces models proposing archaea-derived hosts. We argue that, in addition to improved phylogenomic analyses with more comprehensive taxon sampling to pinpoint the closest prokaryotic relatives of eukaryotes, determining plausible mechanisms and selective forces at the origin of key eukaryotic features, such as the nucleus or the bacterial-like eukaryotic membrane system, is essential to constrain existing models.
DOI:10.1073/pnas.1421379112URLPMID:25848019 [本文引用: 1]
Comparative studies of the mitochondrial proteome have identified a conserved core of proteins descended from the alpha-proteobacterial endosymbiont that gave rise to the mitochondrion and was the source of the mitochondrial genome in contemporary eukaryotes. A surprising result of phylogenetic analyses is the relatively small proportion (10-20%) of the mitochondrial proteome displaying a clear alpha-proteobacterial ancestry. A large fraction of mitochondrial proteins typically has detectable homologs only in other eukaryotes and is presumed to represent proteins that emerged specifically within eukaryotes. A further significant fraction of the mitochondrial proteome consists of proteins with homologs in prokaryotes, but without a robust phylogenetic signal affiliating them with specific prokaryotic lineages. The presumptive evolutionary source of these proteins is quite different in contending models of mitochondrial origin.
DOI:10.1016/j.pt.2018.08.008URLPMID:30201278 [本文引用: 1]
Mitochondria originated from the endosymbiotic event commencing from the engulfment of an ancestral alpha-proteobacterium by the first eukaryotic ancestor. Establishment of niches has led to various adaptations among eukaryotes. In anaerobic parasitic protists, the mitochondria have undergone modifications by combining features shared from the aerobic mitochondria with lineage-specific components and mechanisms; a diversified class of organelles emerged and are generally called mitochondrion-related organelles (MROs). In this review we summarize and discuss the recent advances in the knowledge of MROs from parasitic protists, particularly the themes such as metabolic functions, contribution to parasitism, dynamics, protein targeting, and novel lineage- specific proteins, with emphasis on the diversity among these organelles.
[本文引用: 1]
DOI:10.1016/j.cub.2016.03.053URLPMID:27185558 [本文引用: 1]
The presence of mitochondria and related organelles in every studied eukaryote supports the view that mitochondria are essential cellular components. Here, we report the genome sequence of a microbial eukaryote, the oxymonad Monocercomonoides sp., which revealed that this organism lacks all hallmark mitochondrial proteins. Crucially, the mitochondrial iron-sulfur cluster assembly pathway, thought to be conserved in virtually all eukaryotic cells, has been replaced by a cytosolic sulfur mobilization system (SUF) acquired by lateral gene transfer from bacteria. In the context of eukaryotic phylogeny, our data suggest that Monocercomonoides is not primitively amitochondrial but has lost the mitochondrion secondarily. This is the first example of a eukaryote lacking any form of a mitochondrion, demonstrating that this organelle is not absolutely essential for the viability of a eukaryotic cell.
DOI:10.1016/j.cub.2017.09.015URLPMID:29112874 [本文引用: 3]
Mitochondria are best known for their role in the generation of ATP by aerobic respiration. Yet, research in the past half century has shown that they perform a much larger suite of functions and that these functions can vary substantially among diverse eukaryotic lineages. Despite this diversity, all mitochondria derive from a common ancestral organelle that originated from the integration of an endosymbiotic alphaproteobacterium into a host cell related to Asgard Archaea. The transition from endosymbiotic bacterium to permanent organelle entailed a massive number of evolutionary changes including the origins of hundreds of new genes and a protein import system, insertion of membrane transporters, integration of metabolism and reproduction, genome reduction, endosymbiotic gene transfer, lateral gene transfer and the retargeting of proteins. These changes occurred incrementally as the endosymbiont and the host became integrated. Although many insights into this transition have been gained, controversy persists regarding the nature of the original endosymbiont, its initial interactions with the host and the timing of its integration relative to the origin of other features of eukaryote cells. Since the establishment of the organelle, proteins have been gained, lost, transferred and retargeted as mitochondria have specialized into the spectrum of functional types seen across the eukaryotic tree of life.
DOI:10.1016/j.jtbi.2017.02.031URLPMID:28254477 [本文引用: 3]
Fifty years ago, Lynn Margulis, inspiring in early twentieth-century ideas that put forward a symbiotic origin for some eukaryotic organelles, proposed a unified theory for the origin of the eukaryotic cell based on symbiosis as evolutionary mechanism. Margulis was profoundly aware of the importance of symbiosis in the natural microbial world and anticipated the evolutionary significance that integrated cooperative interactions might have as mechanism to increase cellular complexity. Today, we have started fully appreciating the vast extent of microbial diversity and the importance of syntrophic metabolic cooperation in natural ecosystems, especially in sediments and microbial mats. Also, not only the symbiogenetic origin of mitochondria and chloroplasts has been clearly demonstrated, but improvement in phylogenomic methods combined with recent discoveries of archaeal lineages more closely related to eukaryotes further support the symbiogenetic origin of the eukaryotic cell. Margulis left us in legacy the idea of 'eukaryogenesis by symbiogenesis'. Although this has been largely verified, when, where, and specifically how eukaryotic cells evolved are yet unclear. Here, we shortly review current knowledge about symbiotic interactions in the microbial world and their evolutionary impact, the status of eukaryogenetic models and the current challenges and perspectives ahead to reconstruct the evolutionary path to eukaryotes.
DOI:10.1016/0022-5193(67)90079-3URLPMID:11541392 [本文引用: 1]
A theory of the origin of eukaryotic cells (
DOI:10.1093/nar/4.3.663URLPMID:866186 [本文引用: 1]
We present a catalog of sequences of oligonucleotides produced by T1 ribonuclease digestion of 32P-labeled small-ribosomal-subunit RNA (
DOI:10.1126/science.202030URLPMID:202030 [本文引用: 2]
DOI:10.1073/pnas.82.13.4443URLPMID:3892535 [本文引用: 1]
The 16S ribosomal RNA sequences from Agrobacterium tumefaciens and Pseudomonas testosteroni have been determined to further delimit the origin of the endosymbiont that gave rise to the mitochondrion. These two prokaryotes represent the alpha and beta subdivisions, respectively, of the so-called purple bacteria. The endosymbiont that gave rise to the mitochondrion belonged to the alpha subdivision, a group that also contains the rhizobacteria, the agrobacteria, and the rickettsias--all prokaryotes that have developed intracellular or other close relationships with eukaryotic cells.
DOI:10.1038/326332a0URLPMID:3561476 [本文引用: 1]
DOI:10.1038/339100a0URL [本文引用: 1]
DOI:10.1099/00207713-52-2-297URL [本文引用: 3]
DOI:10.1186/1745-6150-5-7URL [本文引用: 2]
DOI:10.1098/rspb.2004.2705URLPMID:15306349 [本文引用: 2]
There are many more phyla of microbes than of macro-organisms, but microbial biodiversity is poorly understood because most microbes are uncultured. Phylogenetic analysis of rDNA sequences cloned after PCR amplification of DNA extracted directly from environmental samples is a powerful way of exploring our degree of ignorance of major groups. As there are only five eukaryotic kingdoms, two claims using such methods for numerous novel 'kingdom-level' lineages among anaerobic eukaryotes would be remarkable, if true. By reanalysing those data with 167 known species (not merely 8-37), I identified relatives for all 8-10 'mysterious' lineages. All probably belong to one of five already recognized phyla (Amoebozoa, Cercozoa, Apusozoa, Myzozoa, Loukozoa) within the basal kingdom Protozoa, mostly in known classes, sometimes even in known orders, families or genera. This strengthens the idea that the ancestral eukaryote was a mitochondrial aerobe. Analogous claims of novel bacterial divisions or kingdoms may reflect the weak resolution and grossly non-clock-like evolution of ribosomal rRNA, not genuine phylum-level biological disparity. Critical interpretation of environmental DNA sequences suggests that our overall picture of microbial biodiversity at phylum or division level is already rather good and comprehensive and that there are no uncharacterized kingdoms of life. However, immense lower-level diversity remains to be mapped, as does the root of the tree of life.
DOI:10.1666/0094-8373(2005)031[0175:IAOITO]2.0.CO;2URL [本文引用: 3]
[本文引用: 3]
DOI:10.1073/pnas.97.13.6954URLPMID:10860956 [本文引用: 1]
We present a testable model for the origin of the nucleus, the membrane-bounded organelle that defines eukaryotes. A chimeric cell evolved via symbiogenesis by syntrophic merger between an archaebacterium and a eubacterium. The archaebacterium, a thermoacidophil resembling extant Thermoplasma, generated hydrogen sulfide to protect the eubacterium, a heterotrophic swimmer comparable to Spirochaeta or Hollandina that oxidized sulfide to sulfur. Selection pressure for speed swimming and oxygen avoidance led to an ancient analogue of the extant cosmopolitan bacterial consortium
DOI:10.1007/pl00006331URLPMID:9545461 [本文引用: 2]
One of the most important omissions in recent evolutionary theory concerns how eukaryotes could emerge and evolve. According to the currently accepted views, the first eukaryotic cell possessed a nucleus, an endomembrane system, and a cytoskeleton but had an inefficient prokaryotic-like metabolism. In contrast, one of the most ancient eukaryotes, the metamonada Giardia lamblia, was found to have formerly possessed mitochondria. In sharp contrast with the traditional views, this paper suggests, based on the energetic aspect of genome organization, that the emergence of eukaryotes was promoted by the establishment of an efficient energy-converting organelle, such as the mitochondrion. Mitochondria were acquired by the endosymbiosis of ancient alpha-purple photosynthetic Gram-negative eubacteria that reorganized the prokaryotic metabolism of the archaebacterial-like ancestral host cells. The presence of an ATP pool in the cytoplasm provided by this cell organelle allowed a major increase in genome size. This evolutionary change, the remarkable increase both in genome size and complexity, explains the origin of the eukaryotic cell itself. The loss of cell wall and the appearance of multicellularity can also be explained by the acquisition of mitochondria. All bacteria use chemiosmotic mechanisms to harness energy; therefore the periplasm bounded by the cell wall is an essential part of prokaryotic cells. Following the establishment of mitochondria, the original plasma membrane-bound metabolism of prokaryotes, as well as the funcion of the periplasm providing a compartment for the formation of different ion gradients, has been transferred into the inner mitochondrial membrane and intermembrane space. After the loss of the essential function of periplasm, the bacterial cell wall could also be lost, which enabled the naked cells to establish direct connections among themselves. The relatively late emergence of mitochondria may be the reason why multicellularity evolved so slowly.
[本文引用: 2]
[本文引用: 1]
DOI:10.1038/35086563URLPMID:11473316 [本文引用: 1]
Some insects have cultivated intimate relationships with mutualistic bacteria since their early evolutionary history. Most ancient 'primary' endosymbionts live within the cytoplasm of large, polyploid host cells of a specialized organ (bacteriome). Within their large, ovoid bacteriomes, mealybugs (Pseudococcidae) package the intracellular endosymbionts into 'mucus-filled' spheres, which surround the host cell nucleus and occupy most of the cytoplasm. The genesis of symbiotic spheres has not been determined, and they are structurally unlike eukaryotic cell vesicles. Recent molecular phylogenetic and fluorescent in situ hybridization (FISH) studies suggested that two unrelated bacterial species may share individual host cells, and that bacteria within spheres comprise these two species. Here we show that mealybug host cells do indeed harbour both beta- and gamma-subdivision Proteobacteria, but they are not co-inhabitants of the spheres. Rather, we show that the symbiotic spheres themselves are beta-proteobacterial cells. Thus, gamma-Proteobacteria live symbiotically inside beta-Proteobacteria. This is the first report, to our knowledge, of an intracellular symbiosis involving two species of bacteria.
DOI:10.1016/s0168-9525(01)02312-5URLPMID:11418217 [本文引用: 2]
It is well known that genes from chloroplasts and mitochondria were transferred to the nucleus many times during plant evolution. But in what form do the transferred genes physically make that intracellular journey--as RNA, as cDNA, as pieces of organelle DNA, or as whole organelle chromosomes? Current views focus upon cDNA as the vehicle, based upon some examples from plants. But other mechanisms, involving direct transfer of DNA from organelle chromosomes, could also account for the available data. Direct DNA transfer, rather than cDNA-mediated transfer, does occur today, and it probably prevailed during the early phases of organelle evolution.
URLPMID:26323761 [本文引用: 1]
DOI:10.1038/nature09486URLPMID:20962839 [本文引用: 1]
All complex life is composed of eukaryotic (nucleated) cells. The eukaryotic cell arose from prokaryotes just once in four billion years, and otherwise prokaryotes show no tendency to evolve greater complexity. Why not? Prokaryotic genome size is constrained by bioenergetics. The endosymbiosis that gave rise to mitochondria restructured the distribution of DNA in relation to bioenergetic membranes, permitting a remarkable 200,000-fold expansion in the number of genes expressed. This vast leap in genomic capacity was strictly dependent on mitochondrial power, and prerequisite to eukaryote complexity: the key innovation en route to multicellular life.
DOI:10.1093/gbe/evw136URLPMID:27345956 [本文引用: 2]
Theories for the origin of sex traditionally start with an asexual mitosing cell and add recombination, thereby deriving meiosis from mitosis. Though sex was clearly present in the eukaryote common ancestor, the order of events linking the origin of sex and the origin of mitosis is unknown. Here, we present an evolutionary inference for the origin of sex starting with a bacterial ancestor of mitochondria in the cytosol of its archaeal host. We posit that symbiotic association led to the origin of mitochondria and gene transfer to host's genome, generating a nucleus and a dedicated translational compartment, the eukaryotic cytosol, in which-by virtue of mitochondria-metabolic energy was not limiting. Spontaneous protein aggregation (monomer polymerization) and Adenosine Tri-phosphate (ATP)-dependent macromolecular movement in the cytosol thereby became selectable, giving rise to continuous microtubule-dependent chromosome separation (reduction division). We propose that eukaryotic chromosome division arose in a filamentous, syncytial, multinucleated ancestor, in which nuclei with insufficient chromosome numbers could complement each other through mRNA in the cytosol and generate new chromosome combinations through karyogamy. A syncytial (or coenocytic, a synonym) eukaryote ancestor, or Coeca, would account for the observation that the process of eukaryotic chromosome separation is more conserved than the process of eukaryotic cell division. The first progeny of such a syncytial ancestor were likely equivalent to meiospores, released into the environment by the host's vesicle secretion machinery. The natural ability of archaea (the host) to fuse and recombine brought forth reciprocal recombination among fusing (syngamy and karyogamy) progeny-sex-in an ancestrally meiotic cell cycle, from which the simpler haploid and diploid mitotic cell cycles arose. The origin of eukaryotes was the origin of vertical lineage inheritance, and sex was required to keep vertically evolving lineages viable by rescuing the incipient eukaryotic lineage from Muller's ratchet. The origin of mitochondria was, in this view, the decisive incident that precipitated symbiosis-specific cell biological problems, the solutions to which were the salient features that distinguish eukaryotes from prokaryotes: A nuclear membrane, energetically affordable ATP-dependent protein-protein interactions in the cytosol, and a cell cycle involving reduction division and reciprocal recombination (sex).
DOI:10.1016/j.tim.2016.03.005URLPMID:27040918 [本文引用: 1]
Eukaryotes possess an elaborate endomembrane system with endoplasmic reticulum, nucleus, Golgi, lysosomes, peroxisomes, autophagosomes, and dynamic vesicle traffic. Theories addressing the evolutionary origin of eukaryotic endomembranes have overlooked the outer membrane vesicles (OMVs) that bacteria, archaea, and mitochondria secrete into their surroundings. We propose that the eukaryotic endomembrane system originated from bacterial OMVs released by the mitochondrial ancestor within the cytosol of its archaeal host at eukaryote origin. Confined within the host's cytosol, OMVs accumulated naturally, fusing either with each other or with the host's plasma membrane. This matched the host's archaeal secretory pathway for cotranslational protein insertion with outward bound mitochondrial-derived vesicles consisting of bacterial lipids, forging a primordial, secretory endoplasmic reticulum as the cornerstone of the eukaryotic endomembrane system. VIDEO ABSTRACT.
URLPMID:29689215 [本文引用: 1]
DOI:10.1186/1745-6150-4-9URL [本文引用: 1]
DOI:10.1038/nature14522URLPMID:25945740 [本文引用: 1]
DOI:10.1038/s41559-018-0477-7URLPMID:29459706 [本文引用: 2]
With the current explosion of genomic data, there is a greater need to draw inference on phenotypic information based on DNA sequence alone. We considered complete genomes from 35 diverse eukaryotic lineages, and discovered sets of proteins predictive of trophic mode, including a set of 485 proteins that are enriched among phagocytotic eukaryotes (organisms that internalize large particles). Our model is also predictive of other aspects of trophic mode, including photosynthesis and the ability to synthesize a set of organic compounds needed for growth (prototrophy for those molecules). We applied our model to the Asgard archaea, a group of uncultured microorganisms that show close affinities to eukaryotes, to test whether the organisms are capable of phagocytosis, a phenotypic trait often considered a prerequisite for mitochondrial acquisition. Our analyses suggest that members of the Asgard archaea-despite having some eukaryote-specific protein families not found in other prokaryotes-do not use phagocytosis. Moreover, our data suggest that the process of phagocytosis arose from a combination of both archaeal and bacterial components, but also required additional eukaryote-specific innovations.
DOI:10.1016/j.tim.2018.10.005URL [本文引用: 1]
DOI:10.1038/nature16941URLPMID:26840490 [本文引用: 1]
The origin of eukaryotes stands as a major conundrum in biology. Current evidence indicates that the last eukaryotic common ancestor already possessed many eukaryotic hallmarks, including a complex subcellular organization. In addition, the lack of evolutionary intermediates challenges the elucidation of the relative order of emergence of eukaryotic traits. Mitochondria are ubiquitous organelles derived from an alphaproteobacterial endosymbiont. Different hypotheses disagree on whether mitochondria were acquired early or late during eukaryogenesis. Similarly, the nature and complexity of the receiving host are debated, with models ranging from a simple prokaryotic host to an already complex proto-eukaryote. Most competing scenarios can be roughly grouped into either mito-early, which consider the driving force of eukaryogenesis to be mitochondrial endosymbiosis into a simple host, or mito-late, which postulate that a significant complexity predated mitochondrial endosymbiosis. Here we provide evidence for late mitochondrial endosymbiosis. We use phylogenomics to directly test whether proto-mitochondrial proteins were acquired earlier or later than other proteins of the last eukaryotic common ancestor. We find that last eukaryotic common ancestor protein families of alphaproteobacterial ancestry and of mitochondrial localization show the shortest phylogenetic distances to their closest prokaryotic relatives, compared with proteins of different prokaryotic origin or cellular localization. Altogether, our results shed new light on a long-standing question and provide compelling support for the late acquisition of mitochondria into a host that already had a proteome of chimaeric phylogenetic origin. We argue that mitochondrial endosymbiosis was one of the ultimate steps in eukaryogenesis and that it provided the definitive selective advantage to mitochondria-bearing eukaryotes over less complex forms.
DOI:10.1038/nature16876URLPMID:26840482 [本文引用: 1]
DOI:10.1093/gbe/evx027URLPMID:28199635 [本文引用: 1]
The origin of mitochondria was a crucial event in eukaryote evolution. A recent report claimed to provide evidence, based on branch length variation in phylogenetic trees, that the mitochondrion came late in eukaryotic evolution. Here, we reinvestigate their claim with a reanalysis of the published data. We show that the analyses underpinning a late mitochondrial origin suffer from multiple fatal flaws founded in inappropriate statistical methods and analyses, in addition to erroneous interpretations.
[本文引用: 7]
DOI:10.1038/nature14963URLPMID:26287458 [本文引用: 1]
Chloroplasts arose from cyanobacteria, mitochondria arose from proteobacteria. Both organelles have conserved their prokaryotic biochemistry, but their genomes are reduced, and most organelle proteins are encoded in the nucleus. Endosymbiotic theory posits that bacterial genes in eukaryotic genomes entered the eukaryotic lineage via organelle ancestors. It predicts episodic influx of prokaryotic genes into the eukaryotic lineage, with acquisition corresponding to endosymbiotic events. Eukaryotic genome sequences, however, increasingly implicate lateral gene transfer, both from prokaryotes to eukaryotes and among eukaryotes, as a source of gene content variation in eukaryotic genomes, which predicts continuous, lineage-specific acquisition of prokaryotic genes in divergent eukaryotic groups. Here we discriminate between these two alternatives by clustering and phylogenetic analysis of eukaryotic gene families having prokaryotic homologues. Our results indicate (1) that gene transfer from bacteria to eukaryotes is episodic, as revealed by gene distributions, and coincides with major evolutionary transitions at the origin of chloroplasts and mitochondria; (2) that gene inheritance in eukaryotes is vertical, as revealed by extensive topological comparison, sparse gene distributions stemming from differential loss; and (3) that continuous, lineage-specific lateral gene transfer, although it sometimes occurs, does not contribute to long-term gene content evolution in eukaryotic genomes.
DOI:10.1038/24094URLPMID:9823893 [本文引用: 1]
We describe here the complete genome sequence (1,111,523 base pairs) of the obligate intracellular parasite Rickettsia prowazekii, the causative agent of epidemic typhus. This genome contains 834 protein-coding genes. The functional profiles of these genes show similarities to those of mitochondrial genes: no genes required for anaerobic glycolysis are found in either R. prowazekii or mitochondrial genomes, but a complete set of genes encoding components of the tricarboxylic acid cycle and the respiratory-chain complex is found in R. prowazekii. In effect, ATP production in Rickettsia is the same as that in mitochondria. Many genes involved in the biosynthesis and regulation of biosynthesis of amino acids and nucleosides in free-living bacteria are absent from R. prowazekii and mitochondria. Such genes seem to have been replaced by homologues in the nuclear (host) genome. The R. prowazekii genome contains the highest proportion of non-coding DNA (24%) detected so far in a microbial genome. Such non-coding sequences may be degraded remnants of 'neutralized' genes that await elimination from the genome. Phylogenetic analyses indicate that R. prowazekii is more closely related to mitochondria than is any other microbe studied so far.
DOI:10.1093/molbev/msj009URLPMID:16151187 [本文引用: 1]
Placement of the mitochondrial branch on the tree of life has been problematic. Sparse sampling, the uncertainty of how lateral gene transfer might overwrite phylogenetic signals, and the uncertainty of phylogenetic inference have all contributed to the issue. Here we address this issue using a supertree approach and completed genomic sequences. We first determine that a sensible alpha-proteobacterial phylogenetic tree exists and that it can confidently be inferred using orthologous genes. We show that congruence across these orthologous gene trees is significantly better than might be expected by random chance. There is some evidence of horizontal gene transfer within the alpha-proteobacteria, but it appears to be restricted to a minority of genes ( approximately 23%) most of whom ( approximately 74%) can be categorized as operational. This means that placement of the mitochondrion should not be excessively hampered by interspecies gene transfer. We then show that there is a consistently strong signal for placement of the mitochondrion on this tree and that this placement is relatively insensitive to methodological approach or data set. A concatenated alignment was created consisting of 15 mitochondrion-encoded proteins that are unlikely to have undergone any lateral gene transfer in the timeline under consideration. This alignment infers that the sister group of the mitochondria, for the taxa that have been sampled, is the order Rickettsiales.
DOI:10.1128/JB.00269-07URLPMID:17483224 [本文引用: 1]
The branching order and coherence of the alphaproteobacterial orders have not been well established, and not all studies have agreed that mitochondria arose from within the Rickettsiales. A species tree for 72 alphaproteobacteria was produced from a concatenation of alignments for 104 well-behaved protein families. Coherence was upheld for four of the five orders with current standing that were represented here by more than one species. However, the family Hyphomonadaceae was split from the other Rhodobacterales, forming an expanded group with Caulobacterales that also included Parvularcula. The three earliest-branching alphaproteobacterial orders were the Rickettsiales, followed by the Rhodospirillales and then the Sphingomonadales. The principal uncertainty is whether the expanded Caulobacterales group is more closely associated with the Rhodobacterales or the Rhizobiales. The mitochondrial branch was placed within the Rickettsiales as a sister to the combined Anaplasmataceae and Rickettsiaceae, all subtended by the Pelagibacter branch. Pelagibacter genes will serve as useful additions to the bacterial outgroup in future evolutionary studies of mitochondrial genes, including those that have transferred to the eukaryotic nucleus.
URLPMID:22355532 [本文引用: 1]
DOI:10.1371/journal.pone.0024457URLPMID:21935411 [本文引用: 1]
BACKGROUND: According to the endosymbiont hypothesis, the mitochondrial system for aerobic respiration was derived from an ancestral Alphaproteobacterium. Phylogenetic studies indicate that the mitochondrial ancestor is most closely related to the Rickettsiales. Recently, it was suggested that Candidatus Pelagibacter ubique, a member of the SAR11 clade that is highly abundant in the oceans, is a sister taxon to the mitochondrial-Rickettsiales clade. The availability of ocean metagenome data substantially increases the sampling of Alphaproteobacteria inhabiting the oxygen-containing waters of the oceans that likely resemble the originating environment of mitochondria. METHODOLOGY/PRINCIPAL FINDINGS: We present a phylogenetic study of the origin of mitochondria that incorporates metagenome data from the Global Ocean Sampling (GOS) expedition. We identify mitochondrially related sequences in the GOS dataset that represent a rare group of Alphaproteobacteria, designated OMAC (Oceanic Mitochondria Affiliated Clade) as the closest free-living relatives to mitochondria in the oceans. In addition, our analyses reject the hypothesis that the mitochondrial system for aerobic respiration is affiliated with that of the SAR11 clade. CONCLUSIONS/SIGNIFICANCE: Our results allude to the existence of an alphaproteobacterial clade in the oxygen-rich surface waters of the oceans that represents the closest free-living relative to mitochondria identified thus far. In addition, our findings underscore the importance of expanding the taxonomic diversity in phylogenetic analyses beyond that represented by cultivated bacteria to study the origin of mitochondria.
DOI:10.1371/journal.pone.0030520URLPMID:22291975 [本文引用: 2]
Although free living, members of the successful SAR11 group of marine alpha-proteobacteria contain a very small and A+T rich genome, two features that are typical of mitochondria and related obligate intracellular parasites such as the Rickettsiales. Previous phylogenetic analyses have suggested that Candidatus Pelagibacter ubique, the first cultured member of this group, is related to the Rickettsiales+mitochondria clade whereas others disagree with this conclusion. In order to determine the evolutionary position of the SAR11 group and its relationship to the origin of mitochondria, we have performed phylogenetic analyses on the concatenation of 24 proteins from 5 mitochondria and 71 proteobacteria. Our results support that SAR11 group is not the sistergroup of the Rickettsiales+mitochondria clade and confirm that the position of this group in the alpha-proteobacterial tree is strongly affected by tree reconstruction artefacts due to compositional bias. As a consequence, genome reduction and bias toward a high A+T content may have evolved independently in the SAR11 species, which points to a different direction in the quest for the closest relatives to mitochondria and Rickettsiales. In addition, our analyses raise doubts about the monophyly of the newly proposed Pelagibacteraceae family.
DOI:10.1038/srep07949URLPMID:25609566 [本文引用: 3]
Overwhelming evidence supports the endosymbiosis theory that mitochondria originated once from the Alphaproteobacteria. However, its exact position in the tree of life remains highly debated. This is because systematic errors, including biased taxonomic sampling, high evolutionary rates and sequence composition bias have long plagued the mitochondrial phylogenetics. In this study, we address this issue by 1) increasing the taxonomic representation of alphaproteobacterial genomes by sequencing 18 phylogenetically novel species. They include 5 Rickettsiales and 4 Rhodospirillales, two orders that have shown close affiliations with mitochondria previously, 2) using a set of 29 slowly evolving mitochondria-derived nuclear genes that are less biased than mitochondria-encoded genes as the alternative
DOI:10.1371/journal.pone.0110685URLPMID:25333787 [本文引用: 1]
Reconstruction of mitochondrial ancestor has great impact on our understanding of the origin of mitochondria. Previous studies have largely focused on reconstructing the last common ancestor of all contemporary mitochondria (proto-mitochondria), but not on the more informative pre-mitochondria (the last common ancestor of mitochondria and their alphaproteobacterial sister clade). Using a phylogenomic approach and leveraging on the increased taxonomic sampling of alphaproteobacterial and eukaryotic genomes, we reconstructed the metabolisms of both proto-mitochondria and pre-mitochondria. Our reconstruction depicts a more streamlined proto-mitochondrion than these predicted by previous studies, and revealed several novel insights into the mitochondria-derived eukaryotic metabolisms including the lipid metabolism. Most strikingly, pre-mitochondrion was predicted to possess a plastid/parasite type of ATP/ADP translocase that imports ATP from the host, which posits pre-mitochondrion as an energy parasite that directly contrasts with the current role of mitochondria as the cell's energy producer. In addition, pre-mitochondrion was predicted to encode a large number of flagellar genes and several cytochrome oxidases functioning under low oxygen level, strongly supporting the previous finding that the mitochondrial ancestor was likely motile and capable of oxidative phosphorylation under microoxic condition.
URLPMID:29695865 [本文引用: 2]
[本文引用: 1]
DOI:10.1016/j.tim.2013.12.006URL [本文引用: 1]
Alphaproteobacterial genomes show a remarkable genome plasticity linked with different lifestyles (intracellular, facultative, and free-living). They represent the major source of the genome repertoire of mitochondria, and their genes (specifically those of Wolbachia) have been massively transferred into their modern eukaryotic hosts, such as arthropods and nematodes. Conversely, other organisms (bacteria, viruses, archaea, and eukaryotes) and selfish DNA have contributed to their genomes. This bidirectional lateral sequence transfer explains the mosaic nature of their genomes. In contrast to those living in allopatry, alphaproteobacteria living in sympatry (in protist cells such as in the environment) favor lateral sequence transfer. Evidence shows that intracellular transfer of the type IV secretion system might have played a critical role in the evolution of these alphaproteobacteria.
DOI:10.1016/j.cub.2017.10.051URLPMID:29174886 [本文引用: 1]
The origin of eukaryotic cells represents a key transition in cellular evolution and is closely tied to outstanding questions about mitochondrial endosymbiosis [1, 2]. For example, gene-rich mitochondrial genomes are thought to be indicative of an ancient divergence, but this relies on unexamined assumptions about endosymbiont-to-host gene transfer [3-5]. Here, we characterize Ancoracysta twista, a new predatory flagellate that is not closely related to any known lineage in 201-protein phylogenomic trees and has a unique morphology, including a novel type of extrusome (ancoracyst). The Ancoracysta mitochondrion has a gene-rich genome with a coding capacity exceeding that of all other eukaryotes except the distantly related jakobids and Diphylleia, and it uniquely possesses heterologous, nucleus-, and mitochondrion-encoded cytochrome c maturase systems. To comprehensively examine mitochondrial genome reduction, we also assembled mitochondrial genomes from picozoans and colponemids and re-annotated existing mitochondrial genomes using hidden Markov model gene profiles. This revealed over a dozen previously overlooked mitochondrial genes at the level of eukaryotic supergroups. Analysis of trends over evolutionary time demonstrates that gene transfer to the nucleus was non-linear, that it occurred in waves of exponential decrease, and that much of it took place comparatively early, massively independently, and with lineage-specific rates. This process has led to differential gene retention, suggesting that gene-rich mitochondrial genomes are not a product of their early divergence. Parallel transfer of mitochondrial genes and their functional replacement by new nuclear factors are important in models for the origin of eukaryotes, especially as major gaps in our knowledge of eukaryotic diversity at the deepest level remain unfilled.
DOI:10.7554/eLife.42535URLPMID:30789345 [本文引用: 1]
The Alphaproteobacteria is an extraordinarily diverse and ancient group of bacteria. Previous attempts to infer its deep phylogeny have been plagued with methodological artefacts. To overcome this, we analyzed a dataset of 200 single-copy and conserved genes and employed diverse strategies to reduce compositional artefacts. Such strategies include using novel dataset-specific profile mixture models and recoding schemes, and removing sites, genes and taxa that are compositionally biased. We show that the Rickettsiales and Holosporales (both groups of intracellular parasites of eukaryotes) are not sisters to each other, but instead, the Holosporales has a derived position within the Rhodospirillales. A synthesis of our results also leads to an updated proposal for the higher-level taxonomy of the Alphaproteobacteria. Our robust consensus phylogeny will serve as a framework for future studies that aim to place mitochondria, and novel environmental diversity, within the Alphaproteobacteria.
DOI:10.1038/sj.cr.7290168URLPMID:12974613 [本文引用: 1]
Although mitochondria provide eukaryotic cells with certain metabolic advantages, in other ways they may be disadvantageous. For example, mitochondria produce reactive oxygen species that damage both nucleocytoplasm and mitochondria, resulting in mutations, diseases, and aging. The relationship of mitochondria to the cytoplasm is best understood in the context of evolutionary history. Although it is clear that mitochondria evolved from symbiotic bacteria, the exact nature of the initial symbiosis is a matter of continuing debate. The exchange of nutrients between host and symbiont may have differed from that between the cytoplasm and mitochondria in modern cells. Speculations about the initial relationships include the following. (1) The pre-mitochondrion may have been an invasive, parasitic bacterium. The host did not benefit. (2) The relationship was a nutritional syntrophy based upon transfer of organic acids from host to symbiont. (3) The relationship was a syntrophy based upon H2 transfer from symbiont to host, where the host was a methanogen. (4) There was a syntrophy based upon reciprocal exchange of sulfur compounds. The last conjecture receives support from our detection in eukaryotic cells of substantial H2S-oxidizing activity in mitochondria, and sulfur-reducing activity in the cytoplasm.
DOI:10.1073/pnas.91.8.2880URLPMID:8159671 [本文引用: 1]
DOI:10.1186/2045-3701-2-29URLPMID:22913376 [本文引用: 1]
For over a century, the origin of eukaryotes has been a topic of intense debate among scientists. Although it has become widely accepted that organelles such as the mitochondria and chloroplasts arose via endosymbiosis, the origin of the eukaryotic nucleus remains enigmatic. Numerous models for the origin of the nucleus have been proposed over the years, many of which use endosymbiosis to explain its existence. Proposals of microbes whose ancestors may have served as either a host or a guest in various endosymbiotic scenarios abound, none of which have been able to sufficiently incorporate the cell biological as well as phylogenetic data which links these organisms to the nucleus. While it is generally agreed that eukaryotic nuclei share more features in common with archaea rather than with bacteria, different studies have identified either one or the other of the two major groups of archaea as potential ancestors, leading to somewhat of a stalemate. This paper seeks to resolve this impasse by presenting evidence that not just one, but a pair of archaea might have served as host to the bacterial ancestor of the mitochondria. This pair may have consisted of ancestors of both Ignicoccus hospitalis as well as its ectosymbiont/ectoparasite 'Nanoarchaeum equitans'.
DOI:10.1016/j.mib.2016.02.001URLPMID:26894379 [本文引用: 1]
Several authors have suggested that viruses from the NucleoCytoplasmic Large DNA Viruses group (NCLDV) have played an important role in the origin of modern eukaryotes. Notably, the viral eukaryogenesis theory posits that the nucleus originated from an ancient NCLDV-related virus. Focusing on the viral factory instead of the virion adds credit to this hypothesis, but also suggests alternative scenarios. Beside a role in the emergence of the nucleus, ancient NCLDV may have provided new genes and/or chromosomes to the proto-eukaryotic lineage. Phylogenetic analyses suggest that NCLDV informational proteins, related to those of Archaea and Eukarya, were either recruited by ancient NCLDV from proto-eukaryotes and/or transferred to proto-eukaryotes, in agreement with the antiquity of NCLDV and their possible role in eukaryogenesis.
DOI:10.1111/j.1749-6632.2009.04994.xURL [本文引用: 1]
[本文引用: 1]
DOI:10.1038/s41396-018-0060-xURLPMID:29445130 [本文引用: 1]
Thorarchaeota are a new archaeal phylum within the Asgard superphylum, whose ancestors have been proposed to play possible ecological roles in cellular evolution. However, little is known about the lifestyles of these uncultured archaea. To provide a better resolution of the ecological roles and metabolic capacity of Thorarchaeota, we obtained Thorarchaeota genomes reconstructed from metagenomes of different depth layers in mangrove and mudflat sediments. These genomes from deep anoxic layers suggest the presence of Thorarchaeota with the potential to degrade organic matter, fix inorganic carbon, reduce sulfur/sulfate and produce acetate. In particular, Thorarchaeota may be involved in ethanol production, nitrogen fixation, nitrite reduction, and arsenic detoxification. Interestingly, these Thorarchaeotal genomes are inferred to contain the tetrahydromethanopterin and tetrahydrofolate Wood-Ljungdahl (WL) pathways for CO2 reduction, and the latter WL pathway appears to have originated from bacteria. These archaea are predicted to be able to use various inorganic and organic carbon sources, possessing genes inferred to encode ribulose bisphosphate carboxylase-like proteins (normally without RuBisCO activity) and a near-complete Calvin-Benson-Bassham cycle. The existence of eukaryotic selenocysteine insertion sequences and many genes for proteins previously considered eukaryote-specific in Thorarchaeota genomes provide new insights into their evolutionary roles in the origin of eukaryotic cellular complexity. Resolving the metabolic capacities of these enigmatic archaea and their origins will enhance our understanding of the origins of eukaryotes and their roles in ecosystems.
DOI:10.1128/mBio.02039-19URLPMID:31506313 [本文引用: 2]
The genomes of Asgard Archaea, a novel archaeal proposed superphylum, share an enriched repertoire of eukaryotic signature genes and thus promise to provide insights into early eukaryote evolution. However, the distribution, metabolisms, cellular structures, and ecology of the members within this superphylum are not well understood. Here we provide a meta-analysis of the environmental distribution of the Asgard archaea, based on available 16S rRNA gene sequences. Metagenome sequencing of samples from a salt-crusted lagoon on the Baja California Peninsula of Mexico allowed the assembly of a new Thorarchaeota and three Lokiarchaeota genomes. Comparative analyses of all known Lokiarchaeota and Thorarchaeota genomes revealed overlapping genome content, including central carbon metabolism. Members of both groups contained putative reductive dehalogenase genes, suggesting that these organisms might be able to metabolize halogenated organic compounds. Unlike the first report on Lokiarchaeota, we identified genes encoding glycerol-1-phosphate dehydrogenase in all Loki- and Thorarchaeota genomes, suggesting that these organisms are able to synthesize bona fide archaeal lipids with their characteristic glycerol stereochemistry.IMPORTANCE Microorganisms of the superphylum Asgard Archaea are considered to be the closest living prokaryotic relatives of eukaryotes (including plants and animals) and thus promise to give insights into the early evolution of more complex life forms. However, very little is known about their biology as none of the organisms has yet been cultivated in the laboratory. Here we report on the ecological distribution of Asgard Archaea and on four newly sequenced genomes of the Lokiarchaeota and Thorarchaeota lineages that give insight into possible metabolic features that might eventually help to identify these enigmatic groups of archaea in the environment and to culture them.
DOI:10.1038/s41586-018-0225-9URLPMID:29925949 [本文引用: 1]
Many organisms capture or sense sunlight using rhodopsin pigments(1,2), which are integral membrane proteins that bind retinal chromophores. Rhodopsins comprise two distinct protein families (1) , type-1 (microbial rhodopsins) and type-2 (animal rhodopsins). The two families share similar topologies and contain seven transmembrane helices that form a pocket in which retinal is linked covalently as a protonated Schiff base to a lysine at the seventh transmembrane helix(2,3). Type-1 and type-2 rhodopsins show little or no sequence similarity to each other, as a consequence of extensive divergence from a common ancestor or convergent evolution of similar structures (1) . Here we report a previously unknown and diverse family of rhodopsins-which we term the heliorhodopsins-that we identified using functional metagenomics and that are distantly related to type-1 rhodopsins. Heliorhodopsins are embedded in the membrane with their N termini facing the cell cytoplasm, an orientation that is opposite to that of type-1 or type-2 rhodopsins. Heliorhodopsins show photocycles that are longer than one second, which is suggestive of light-sensory activity. Heliorhodopsin photocycles accompany retinal isomerization and proton transfer, as in type-1 and type-2 rhodopsins, but protons are never released from the protein, even transiently. Heliorhodopsins are abundant and distributed globally; we detected them in Archaea, Bacteria, Eukarya and their viruses. Our findings reveal a previously unknown family of light-sensing rhodopsins that are widespread in the microbial world.
DOI:10.1038/s41564-019-0404-yURLPMID:30936485 [本文引用: 1]
Recent advances in phylogenomic analyses and increased genomic sampling of uncultured prokaryotic lineages have brought compelling evidence in support of the emergence of eukaryotes from within the archaeal domain of life (eocyte hypothesis)(1,2). The discovery of Asgardarchaeota and its supposed position at the base of the eukaryotic tree of life(3,4) provided cues about the long-awaited identity of the eocytic lineage from which the nucleated cells (Eukaryota) emerged. While it is apparent that Asgardarchaeota encode a plethora of eukaryotic-specific proteins (the highest number identified yet in prokaryotes)(5), the lack of genomic information and metabolic characterization has precluded inferences about their lifestyles and the metabolic landscape that favoured the emergence of the protoeukaryote ancestor. Here, we use advanced phylogenetic analyses for inferring the deep ancestry of eukaryotes, and genome-scale metabolic reconstructions for shedding light on the metabolic milieu of Asgardarchaeota. In doing so, we: (1) show that Heimdallarchaeia (the closest eocytic lineage to eukaryotes to date) are likely to have a microoxic niche, based on their genomic potential, with aerobic metabolic pathways that are unique among Archaea (that is, the kynurenine pathway); (2) provide evidence of mixotrophy within Asgardarchaeota; and (3) describe a previously unknown family of rhodopsins encoded within the recovered genomes.
DOI:10.1038/s41564-019-0406-9URL [本文引用: 2]
DOI:10.1111/1462-2920.13754URLPMID:28489281 [本文引用: 1]
Modern phototrophic microbial mats are complex communities often used as analogs of major Precambrian ecosystems. Characterizing biotic, notably metabolic, interactions among different microbial mat members is essential to gain insights into the ecology and biogeochemistry of these systems. We applied 16S/18S rRNA metabarcoding approaches to characterize the structure of archaea, bacteria and protist communities from microbial mats collected along strong physicochemical (oxygen, salinity, temperature, depth) gradients in a shallow pond at the salar de Llamara (Chile). All mats were highly diverse, including members of virtually all known high-rank eukaryotic and prokaryotic taxa but also many novel lineages. Bacterial candidate divisions accounted for almost 50% of sequences in deeper mats, while Archaea represented up to 40% of sequences in some mat layers. Molecular phylogenetic analyses revealed six novel deeply divergent archaeal groups, along abundant and diverse Pacearchaeota and Woesearchaeota. Multivariate statistical analyses showed that local environmental conditions strongly influenced community composition. Co-occurrence network structure was markedly different between surface mats located in the oxygenated zone and mats located in transition and anoxic water layers. We identified potential biotic interactions between various high- and low-rank taxa. Notably, a strong positive correlation was observed between Lokiarchaeota and the poorly known candidate bacterial division TA06.
DOI:10.1038/s41586-018-0548-6URLPMID:30283132 [本文引用: 1]
The origin of the eukaryotic cell is unresolved(1,2). Metagenomics sequencing has recently identified several potential eukaryotic gene homologues in Asgard archaea(3,4), consistent with the hypothesis that the eukaryotic cell evolved from within the Archaea domain. However, many of these eukaryotic-like sequences are highly divergent and the organisms have yet to be imaged or cultivated, which brings into question the extent to which these archaeal proteins represent functional equivalents of their eukaryotic counterparts. Here we show that Asgard archaea encode functional profilins and thereby establish that this archaeal superphylum has a regulated actin cytoskeleton, one of the hallmarks of the eukaryotic cell(5). Loki profilin-1, Loki profilin-2 and Odin profilin adopt the typical profilin fold and are able to interact with rabbit actin-an interaction that involves proteins from species that diverged more than 1.2 billion years ago(6). Biochemical experiments reveal that mammalian actin polymerizes in the presence of Asgard profilins; however, Loki, Odin and Heimdall profilins impede pointed-end elongation. These archaeal profilins also retard the spontaneous nucleation of actin filaments, an effect that is reduced in the presence of phospholipids. Asgard profilins do not interact with polyproline motifs and the profilin-polyproline interaction therefore probably evolved later in the Eukarya lineage. These results suggest that Asgard archaea possess a primordial, polar, profilin-regulated actin system, which may be localized to membranes owing to the sensitivity of Asgard profilins to phospholipids. Because Asgard archaea are also predicted to encode potential eukaryotic-like genes involved in membrane-trafficking and endocytosis(3,4), imaging is now necessary to elucidate whether these organisms are capable of generating eukaryotic-like membrane dynamics that are regulated by actin, such as are observed in eukaryotic cell movement, podosomes and endocytosis.
DOI:10.1038/ismej.2011.64URL [本文引用: 1]
Microbial methanogenesis in subseafloor sediments is a key process in the carbon cycle on the Earth. However, the cultivation-dependent evidences have been poorly demonstrated. Here we report the cultivation of a methanogenic microbial consortium from subseafloor sediments using a continuous-flow-type bioreactor with polyurethane sponges as microbial habitats, called down-flow hanging sponge (DHS) reactor. We anaerobically incubated methane-rich core sediments collected from off Shimokita Peninsula, Japan, for 826 days in the reactor at 10 degrees C. Synthetic seawater supplemented with glucose, yeast extract, acetate and propionate as potential energy sources was provided into the reactor. After 289 days of operation, microbiological methane production became evident. Fluorescence in situ hybridization analysis revealed the presence of metabolically active microbial cells with various morphologies in the reactor. DNA-and RNA-based phylogenetic analyses targeting 16S rRNA indicated the successful growth of phylogenetically diverse microbial components during cultivation in the reactor. Most of the phylotypes in the reactor, once it made methane, were more closely related to culture sequences than to the subsurface environmental sequence. Potentially methanogenic phylotypes related to the genera Methanobacterium, Methanococcoides and Methanosarcina were predominantly detected concomitantly with methane production, while uncultured archaeal phylotypes were also detected. Using the methanogenic community enrichment as subsequent inocula, traditional batch-type cultivations led to the successful isolation of several anaerobic microbes including those methanogens. Our results substantiate that the DHS bioreactor is a useful system for the enrichment of numerous fastidious microbes from subseafloor sediments and will enable the physiological and ecological characterization of pure cultures of previously uncultivated subseafloor microbial life. The ISME Journal (2011) 5, 1913-1925; doi: 10.1038/ismej.2011.64; published online 9 June 2011
DOI:10.1093/sysbio/syu048URLPMID:25070970 [本文引用: 1]
This article reviews the various models that have been used to describe the relationships between gene trees and species trees. Molecular phylogeny has focused mainly on improving models for the reconstruction of gene trees based on sequence alignments. Yet, most phylogeneticists seek to reveal the history of species. Although the histories of genes and species are tightly linked, they are seldom identical, because genes duplicate, are lost or horizontally transferred, and because alleles can coexist in populations for periods that may span several speciation events. Building models describing the relationship between gene and species trees can thus improve the reconstruction of gene trees when a species tree is known, and vice versa. Several approaches have been proposed to solve the problem in one direction or the other, but in general neither gene trees nor species trees are known. Only a few studies have attempted to jointly infer gene trees and species trees. These models account for gene duplication and loss, transfer or incomplete lineage sorting. Some of them consider several types of events together, but none exists currently that considers the full repertoire of processes that generate gene trees along the species tree. Simulations as well as empirical studies on genomic data show that combining gene tree-species tree models with models of sequence evolution improves gene tree reconstruction. In turn, these better gene trees provide a more reliable basis for studying genome evolution or reconstructing ancestral chromosomes and ancestral gene sequences. We predict that gene tree-species tree methods that can deal with genomic data sets will be instrumental to advancing our understanding of genomic evolution.
DOI:10.1017/S1431927614001317URL [本文引用: 1]
Eukaryotes rely on mitochondrial division to guarantee that each new generation of cells acquires an adequate number of mitochondria. Mitochondrial division has long been thought to occur by binary fission and, more recently, evidence has supported the idea that binary fission is mediated by dynamin-related protein (Drp1) and the endoplasmic reticulum. However, studies to date have depended on fluorescence microscopy and conventional electron microscopy. Here, we utilize whole cell cryo-electron tomography to visualize mitochondrial division in frozen hydrated intact HeLa cells. We observe a large number of relatively small mitochondria protruding from and connected to large mitochondria or mitochondrial networks. Therefore, this study provides evidence that mitochondria divide by budding.
DOI:10.1073/pnas.1813143115URLPMID:30373839 [本文引用: 1]
It has been hypothesized that mitochondria evolved from a bacterial ancestor that initially became established in an archaeal host cell as an endosymbiont. Here we model this first stage of mitochondrial evolution by engineering endosymbiosis between Escherichia coli and Saccharomyces cerevisiae An ADP/ATP translocase-expressing E. coli provided ATP to a respiration-deficient cox2 yeast mutant and enabled growth of a yeast-E. coli chimera on a nonfermentable carbon source. In a reciprocal fashion, yeast provided thiamin to an endosymbiotic E. coli thiamin auxotroph. Expression of several SNARE-like proteins in E. coli was also required, likely to block lysosomal degradation of intracellular bacteria. This chimeric system was stable for more than 40 doublings, and GFP-expressing E. coli endosymbionts could be observed in the yeast by fluorescence microscopy and X-ray tomography. This readily manipulated system should allow experimental delineation of host-endosymbiont adaptations that occurred during evolution of the current, highly reduced mitochondrial genome.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}