Advances in assay for transposase-accessible chromatin with high-throughput sequencing
Jie Wu1, Jianping Quan1, Yong Ye1, ZhenFang Wu1, Jie Yang1, Ming Yang,2, Enqin Zheng,1 1. National Engineering Research Center for Breeding Swine Industry, College of Animal Science, South China Agricultural University, Guangzhou 510642, China 2. College of Animal Science and Technology, Zhongkai University of Agriculture and Engineering, Guangzhou 510225, China
Supported by Guangdong YangFan Innovative and Entrepreneurial Research Team Program No.2016YT03H062 Guangdong Modern Agricultural Industry Technology System Pig Innovation Team Project No.2019KJ126 Guangdong Natural Science Foundation.2017A030313213
作者简介 About authors 吴杰,硕士研究生,专业方向:分子遗传与动物育种。E-mail:wujiezi163@163.com。
摘要 染色质转座酶可及性测序(assay for transposase-accessible chromatin with high-throughput sequencing, ATAC-seq)诞生于2013年,具有比脱氧核糖核酸酶I超敏感位点测序(deoxyribonuclease I hypersensitive site sequencing, DNase-seq)和微球菌核酸酶敏感位点测序(micrococcal nuclease sequencing, MNase-seq)更快速、灵敏、简便的优点,是目前分析全基因组范围染色质开放区域的热点技术。通过该技术能获得染色质开放区域的相关信息,从而映射出转录因子等调控蛋白的结合区域和核小体定位等信息,对于研究表观遗传分子机制具有重要意义。本文比较了5种获取染色质开放区域技术的优缺点,重点介绍了ATAC-seq的原理和主要流程,描述了利用ATAC-seq技术研究染色质开放区域的发展概况以及ATAC-seq的相关应用,期望对真核生物全基因组水平的染色质开放区域研究、顺式调控元件鉴定以及遗传调控网络的解析等提供借鉴。 关键词:染色质转座酶可及性测序;染色质开放区域;Tn5转座酶;表观遗传修饰;转录因子
Abstract Assay for transposase accessible chromatin with high-throughput sequencing (ATAC-seq) was developed in 2013. It has the advantages of more convenient operation and higher efficiency for DNA recovery than DNase I hypersensitive site sequencing (DNase-seq) and micrococcal nuclease sequencing (MNase-seq). ATAC-seq currently is the most popular technique of genome-wide mapping for chromatin accessibility. It provides information on binding regions of transcription factors and nucleosome localization on the chromatin. Thus, ATAC-seq is of great significance for studying the epigenetics and molecular mechanisms in chromatin structure. In this review, we compare the advantages and disadvantages of multiple techniques for profiling chromatin accessibility, and summarize the principles, main process, development and applications of ATAC-seq. We hope this review will provide a reference for study of genome-wide mapping for chromatin accessibility, identification of cis-regulatory elements, and dissection of the epigenetic and genetic regulatory networks using the ATAC-seq technology in eukaryotes. Keywords:assay for transposase-accessible chromatin with high-throughput sequencing;open chromatin regions;Tn5 transposase;epigenetic modification;transcription factor
PDF (739KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文 本文引用格式 吴杰, 全建平, 叶勇, 吴珍芳, 杨杰, 杨明, 郑恩琴. 染色质转座酶可及性测序研究进展. 遗传[J], 2020, 42(4): 333-346 doi:10.16288/j.yczz.19-279 Jie Wu. Advances in assay for transposase-accessible chromatin with high-throughput sequencing. Hereditas(Beijing)[J], 2020, 42(4): 333-346 doi:10.16288/j.yczz.19-279
自然界中的生物根据其细胞核类型可以分为原核生物和真核生物,其中原核生物的细胞核无核膜包被,其遗传物质DNA裸露在外;而真核生物细胞的细胞核DNA并非裸露,而是以左旋超螺旋的方式(约147 bp)绕八聚体结构的组蛋白1.67圈,进而形成核小体[1,2]。相邻核小体的连接区由10~80 bp的游离DNA与组蛋白H1共同构成;核小体通过连接区的连接形成串珠式结构,这种串联结构进一步折叠、凝聚,形成染色质;最终多条染色质以高度螺旋化状态包裹于细胞核中[3]。研究显示,染色质开放区域的基因组占总DNA序列的2%~3%,且超过90%的开放区域均与转录因子(transcription factor, TF)的结合相关[4]。以TF为代表的调控因子可与其他染色质结合蛋白相互作用,从而动态调控和维持染色质稳态,在发育过程的调控中发挥着不可替代的作用[5,6,7]。在DNA复制或转录过程中,DNA的折叠结构被打开,一些染色质区域处于开放状态,调控因子(如转录因子)会与这些裸露的无核小体结合的DNA部位结合,进而调控DNA的复制或转录过程[8]。此外,有研究表明,DNA折叠、凝聚形成的染色质物理结构并不是一成不变的,仍然能够发生动态的表观遗传修饰,如DNA甲基化、组蛋白修饰、染色质重塑等[8,9,10,11]。因此,通过了解相关获取染色质开放信息的技术,学习技术原理和应用,明确了这些技术对于基因组调控元件的鉴定、转录因子结合位点的识别及转录调控机制等研究均具有重要意义。本文主要综述了染色质可及性研究技术的发展概况、以及染色质转座酶可及性测序(assay for transposase accessible chromatin with high-throughput sequencing, ATAC-seq)技术的原理和应用,以期为表观遗传学研究提供重要的参考。
1 染色质开放区域研究技术的发展历程
染色质开放区域的研究源于人们发现某些染色质特定位点表现出对DNase I酶切的高度敏感性[12,13,14,15]。后期研究表明,这些DNase I敏感位点(deoxyribonuclease I hypersensitive site, DHS)通常是顺式调控元件所在区域[16],其染色质裸露、结构疏松,可与转录因子结合,从而便于DNase I与之结合并剪切,进而表现出高度敏感性[17]。基于上述原理,染色质开放区域的鉴定工作也随之展开。最先开展的是DHS鉴定分析工作,该分析依赖DNase I高度敏感性特点,并与 Southern 杂交技术结合,不过很快发现该方法的灵敏性和精确性都较低,并且耗时费力[18,19]。随着高通量测序技术(high-throughput sequencing, HTS)的发展及测序成本不断降低,衍生出一系列研究染色质开放区域的技术与方法,如脱氧核糖核酸酶I超敏感位点测序(deoxyribonuclease I hypersensitive site sequencing, DNase-seq)[20]、微球菌核酸酶敏感位点测序(micrococcal nuclease sequencing, MNase-seq)[11]、甲醛辅助性调控元件分离测序(formaldehyde-assisted isolation of regulatory elements followed by sequencing, FAIRE-seq)[21]、核小体定位和甲基化组测序(nucleosome occupancy and methylome sequencing, NOMe-seq)[22]和ATAC-seq。在上述5种技术中,获取染色质开放信息的方式分为3种:DNase-seq、MNase-seq以及ATAC-seq采用酶切法;FAIRE-seq采用物理断裂法;NOMe-seq技术则利用甲基化修饰。5种技术的具体信息见表1。ATAC-seq与其他4种技术相比表现出更为简便和高效的优势,一经发明就被广泛采用,成为当前染色质开放区域获取的前沿技术。下面将对上述染色质开放区域获取技术的发展历程、作用机理以及研究进程进行描述。
Table 1 表1 表15种染色质可及性研究技术介绍 Table 1Introduction of five chromatin accessibility assays
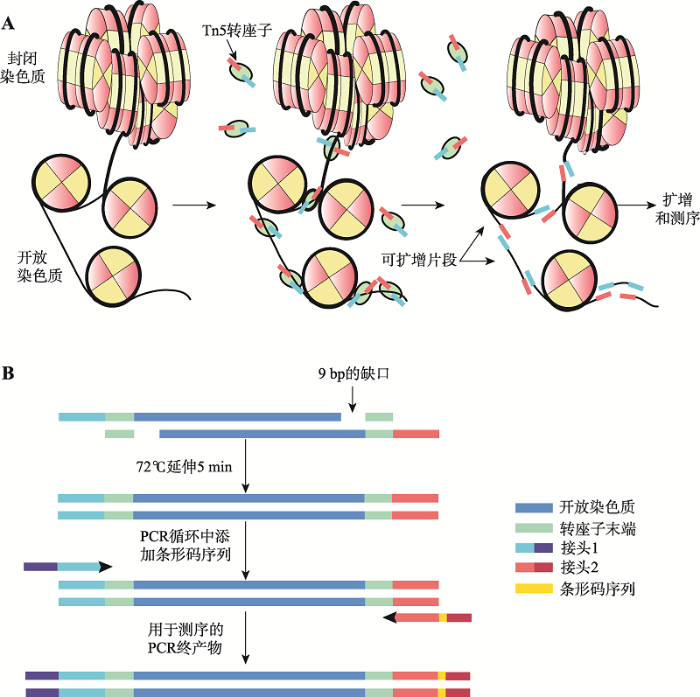
通过上述转座过程不难看出,体外Tn5转座过程仅需4个条件便能完成:Mg2+、转座子末端序列、Tnp和靶DNA[60]。ATAC-seq过程中使用的就是简化后的二聚体转座复合物。复合物仅含有3个部分:转座酶、末端序列和测序接头[61],能够保证在切割DNA的同时连接上接头以便后续的测序工作。同时,简化复合物的Tnp在Tn5主链上携带了特异的点突变体,使Tnp具有了更高的活性[60,62]。另外,之所以需要Mg2+,是由于Mg2+在转座过程中能协同亲核基团,在Mg2+的作用下,转座酶上催化转座子运动的DDE基序(天冬氨酸和谷氨酸)与Mg2+配位发 生突变,使原本不活跃的转座酶变成高度活跃状 态[63,64],是完成转座必不可少的因子之一。目前,Tn5转座子以其转座的随机性好、稳定性高、插入位点容易测序等特点,已经成为分子遗传学研究的热门工具[65,66]。随着高通量测序技术的发展和实验通量的不断增加,Tn5转座酶因其优势被应用得越来越广泛。其中,极速建库、长读长测序技术(single tube long fragment read, stLFR)、单细胞测序、 Mate Pair文库构建、染色质转座酶可及性可视化分析(assay of transposase-accessible chromatin with visualization, ATAC-see),以及近几年发现的Tn5家族对于蛋白结合区域、互作基因片段等研究的帮助都显示Tn5转座酶拥有不可估量的应用潜力[67,68,69]。Buenrostro等[36]建立的ATAC-seq技术,正是充分利用了Tn5酶在测序建库中的巨大优势,能高效、精准的从基因组水平鉴别出染色质开放区域,在生命科学领域的遗传学研究中发挥着至关重要的作用。
开展染色质开放区域的表观基因组学研究具有巨大的生物学意义,但过去的研究方法受到了复杂工作流程和大量细胞需求量的限制,从而导致该领域进展相对缓慢。直到ATAC-seq的出现,为注释开放染色质的基因组位置、DNA结合蛋白、转录因子结合位点等基因组功能元件提供了新的契机。ATAC-seq技术摆脱了像DNase-seq需要精确控制酶量以及FAIRE-seq需要确定甲醛交联时长等条件的限制,但依然存在影响其精确性的因素,如线粒体及植物细胞中叶绿体DNA的干扰、冷冻组织细胞DNA提取效率低、接头连接的随机性造成DNA片段的损失,以及大量酶切后的DNA 片段过大而无法富集等[24,36,38,70]。针对上述缺陷,同样产生了一系列改进措施。例如,Lu等[71]开发的FANS-ATAC-seq (fluorescent activated nuclei sorting, FANS)、Roger等[72]开发的与细胞核基因组序列比对能达90%以上的INTACT (isolation of nuclei tagged in specific cell types)系统,以及与INTACT有相似结果的蔗糖沉淀法(crude)确保了在测定中使用高质量的完整细胞核的同时,能最大限度地减少线粒体和叶绿体中DNA的污染[73]。Corces等[74]发明的Omni-ATAC,提高了ATAC-seq对困难细胞系、稀少的原代细胞和临床上相关的冷冻组织中的应用普遍性。此外,针对接头的随机性和剪切后片段过大的问题,Sos等[75]开发了THS-seq技术,具有比传统EzTn5转座酶活性更高的新型Tn5超突变体(Tn5059)以及更优化的反应溶液和条件。同时设计T7启动子加转录引物替换原Tn5转座复合物中的Adapter 1和2。通过转录生成单链RNA,利用与RNA测序相同的原理获得cDNA并加上衔接子,最终完成建库。该技术避免了接头的随机连接,大大提高了转座效率,使得测序数据更为完整。随着ATAC-seq技术被不断改进,ATAC-seq已逐渐成为目前染色质可及性分析的主流实验方法。
3 ATAC-seq的应用和拓展
3.1 ATAC-seq的应用
自ATAC-seq技术诞生起,该技术凭借其稳定性和高灵敏度已广泛应用于表观基因组学研究。除了能用来确定功能基因组调控区域信息、找出组织特异基因以及预测潜在结合蛋白外,还能跟其他分析技术联合,如RNA-seq、ChIP-seq(chromatin immunoprecipitation followed by high throughput sequencing)以及Hi-C (high-through chromosome conformation capture)等,用以发现潜在的关键调控元件、转录因子和理解控制体内复杂过程的基因调控网络。其应用包括如下方面:
KornbergRD . Chromatin structure: a repeating unit of histones and DNA Science, 1974,184(4139):868-871. [本文引用: 1]
RichmondTJ, FinchJT, RushtonB, RhodesD, KlugA . Structure of the nucleosome core particle at 7 A resolution Nature, 1984,311(5986):532-537. [本文引用: 1]
ZhouYB, GerchmanSE, RamakrishnanV, TraversA, MuyldermansS . Position and orientation of the globular domain of linker histone H5 on the nucleosome Nature, 1998,395(6700):402-405. [本文引用: 1]
BellO, TiwariVK, Thom?NH, SchübelerD . Determinants and dynamics of genome accessibility Nat Rev Genet, 2011,12(8):554-564. [本文引用: 2]
KouzaridesT . Chromatin modifications and their function Cell, 2007,128(4):693-705. [本文引用: 1]
JiangC, PughBF . Nucleosome positioning and gene regulation: advances through genomics Nat Rev Genet, 2009,10(3):161-172. [本文引用: 1]
SchonesDE, CuiK, CuddapahS, RohTY, BarskiA, WangZB, WeiG, ZhaoKJ . Dynamic regulation of nucleosome positioning in the human genome Cell, 2008,132(5):887-898. [本文引用: 2]
HewishDR, BurgoyneLA . Chromatin sub-structure. The digestion of chromatin DNA at regularly spaced sites by a nuclear deoxyribonuclease Biochem Biophys Res Commun, 1973,52(2):504-510. [本文引用: 1]
ScottWA, WigmoreDJ . Sites in simian virus 40 chromatin which are preferentially cleaved by endonucleases Cell, 1978,15(4):1511-1518. [本文引用: 1]
WuC, BinghamPM, LivakKJ, HolmgrenR, ElginSCR . The chromatin structure of specific genes: I. Evidence for higher order domains of defined DNA sequence Cell, 1979,16(4):797-806. [本文引用: 1]
StalderJ, LarsenA, EngelJD, DolanM, GroudineM, WeintraubH . Tissue-specific DNA cleavages in the globin chromatin domain introduced by DNase I Cell, 1980,20(2):451-460. [本文引用: 1]
McgheeJD, WoodWI, DolanM, EngelJD, FelsenfeldG . A 200 base pair region at the 5’ end of the chicken adult β-globin gene is accessible to nuclease digestion Cell, 1981,27(1, Part 2):45-55. [本文引用: 1]
GrossDS, GarrardWT . Nuclease hypersensitive sites in chromatin Annu Rev Biochem, 2003,57(57):159-197. [本文引用: 1]
WuC . The 5’ ends of Drosophila heat shock genes in chromatin are hypersensitive to DNase I Nature, 1980,286(5776):854-860. [本文引用: 1]
CrawfordGE, HoltIE, MullikinJC, TaiD, BlakesleyR, BouffardG, YoungA, MasielloC, GreenED, WolfsbergTG, CollinsFS . Identifying gene regulatory elements by genome-wide recovery of DNase hypersensitive sites Proc Natl Acad Sci USA, 2004,101(4):992-997. [本文引用: 1]
BoyleAP, DavisS, ShulhaHP, MeltzerP, MarguliesEH, WengZ, FureyTS, CrawfordGE . High-resolution mapping and characterization of open chromatin across the genome Cell, 2008,132(2):311-322. [本文引用: 2]
GiresiPG, KimJ, McdaniellRM, IyerVR, LiebJD . FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin Genome Res, 2007,17(6):877-885. [本文引用: 3]
KellyTK, LiuYP, LayFD, LiangGN, BermanBP, JonesPA . Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules Genome Res, 2012,22(12):2497-2506. [本文引用: 3]
TsompanaM, BuckMJ . Chromatin accessibility: a window into the genome Epigenetics Chromatin, 2014,7(1):33. [本文引用: 4]
CumbieJS, FilichkinSA, MegrawM . Improved DNase-seq protocol facilitates high resolution mapping of DNase I hypersensitive sites in roots in Arabidopsis thaliana Plant Methods, 2015,11(1):42. [本文引用: 1]
RizzoJM, SinhaS . Analyzing the global chromatin structure of keratinocytes by MNase-seq Methods Mol Biol, 2014,1195:49-59. [本文引用: 1]
TelfordDJ, StewartBW . Micrococcal nuclease: its specificity and use for chromatin analysis Int J Biochem, 1989,21(2):127-137. [本文引用: 1]
ChungHR, DunkelI, HeiseF, LinkeC, KrobitschS, Ehrenhofer-MurrayAE, SperlingSR, VingronM . The effect of micrococcal nuclease digestion on nucleosome positioning data PLoS One, 2010,5(12):e15754. [本文引用: 1]
Clark DJ. Nucleosome Positioning , Nucleosome spacing and the nucleosome code J Biomol Struct Dyn, 2010,27(6):781-793. [本文引用: 1]
ZentnerGE, HenikoffS . Surveying the epigenomic landscape, one base at a time Genome Biol, 2012,13(10):250. [本文引用: 1]
KumarV, MurataniM, RayanNA, KrausP, LufkinT, NgHH, PrabhakarS . Uniform, optimal signal processing of mapped deep-sequencing data Nat Biotechnol, 2013,31(7):615-622. [本文引用: 1]
SimonJM, GiresiPG, DavisIJ, LiebJD . Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA Nat Protoc, 2012,7(2):256-267. [本文引用: 1]
AuerbachRK, EuskirchenG, RozowskyJ, Lamarre- VincentN, MoqtaderiZ, LefrancoisP, StruhlK, GersteinM, SnyderM . Mapping accessible chromatin regions using Sono-Seq Proc Natl Acad Sci USA, 2009,106(35):14926-14931. [本文引用: 1]
RhieSK, SchreinerS, FarnhamPJ . Defining regulatory elements in the human genome using nucleosome occupancy and methylome sequencing (NOMe-Seq) Methods Mol Biol, 2018,1766:209-229. [本文引用: 1]
KlemmSL, ShiponyZ, GreenleafWJ . Chromatin accessibility and the regulatory epigenome Nat Rev Genet, 2019,20(4):207-220. [本文引用: 1]
BuenrostroJD, GiresiPG, ZabaLC, ChangHY, GreenleafWJ . Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position Nat Met, 2013,10(12):1213-1218. [本文引用: 5]
BuenrostroJD, WuB, ChangHY, GreenleafWJ . ATAC-seq: A method for assaying chromatin accessibility genome-wide Curr Protoc Mol Biol, 2015,109:21-29. [本文引用: 2]
McclintockB . The origin and behavior of mutable loci in maize Proc Natl Acad Sci USA, 1950,36(6):344-355. [本文引用: 1]
BucherE, ReindersJ, MirouzeM . Epigenetic control of transposon transcription and mobility in Arabidopsis Curr Opin Plant Biol, 2012,15(5):503-510. [本文引用: 1]
HuangCR, BurnsKH, BoekeJD . Active transposition in genomes Annu Rev Genet, 2012,46:651-675. [本文引用: 1]
BergDE, DaviesJ, AlletB, RochaixJD . Transposition of R factor genes to bacteriophage lambda Proc Natl Acad Sci USA, 1975,72(9):3628-3632. [本文引用: 1]
RothsteinSJ, JorgensenRA, PostleK, ReznikoffWS . The inverted repeats of Tn5 are functionally different Cell, 1980,19(3):795-805. [本文引用: 2]
LovellS, GoryshinIY, ReznikoffWR, RaymentI . Two-metal active site binding of a Tn5 transposase synaptic complex Nat Struct Biol, 2002,9(4):278-281. [本文引用: 1]
AuerswaldEA, LudwigG, Schaller H. Structural analysis of Tn5 Cold Spring Harb Symp Quant Biol, 1981, 45 Pt1:107. [本文引用: 1]
BergDE . Julian Davies and the discovery of kanamycin resistance transposon Tn5 J Antibiot, 2017,70(4):339-346. [本文引用: 1]
ReznikoffWS . The TN5 transposon Annu Rev Microbiol, 1993,47(1):945-963. [本文引用: 1]
ReznikoffWS, BhasinA, DaviesDR, GoryshinIY, MahnkeLA, NaumannT, RaymentI, Steiniger-WhiteM, TwiningSS . Tn5: A molecular window on transposition Biochem Biophys Res Commun, 1999,266(3):729-734. [本文引用: 1]
ZhouM, ReznikoffWS . Tn5 transposase mutants that alter DNA binding specificity J Mol Biol, 1997,271(3):362-373. [本文引用: 1]
YorkD, ReznikoffWS . DNA binding and phasing analyses of Tn5 transposase and a monomeric variant Nucleic Acids Res, 1997,25(11):2153-2160. [本文引用: 1]
Steiniger-WhiteM, ReznikoffWS . The C-terminal alpha helix of Tn5 transposase is required for synaptic complex formation J Biol Chem, 2000,275(30):23127-23133. [本文引用: 1]
BhasinA, GoryshinIY, Steiniger-WhiteM, YorkD, ReznikoffWS . Characterization of a Tn5 pre-cleavage synaptic complex J Mol Biol, 2000,302(1):49-63. [本文引用: 1]
MizuuchiK, AdzumaK . Inversion of the phosphate chirality at the target site of Mu DNA strand transfer: Evidence for a one-step transesterification mechanism Cell, 1991,66(1):129-140. [本文引用: 1]
MizuuchiK . Transpositional recombination: mechanistic insights from studies of mu and other elements Annu Rev Biochem, 1992,61:1011-1051. [本文引用: 1]
CrellinP, ChalmersR . Protein-DNA contacts and conformational changes in the Tn10 transpososome during assembly and activation for cleavage EMBO J, 2001,20(14):3882-3891. [本文引用: 1]
AdeyA, MorrisonHG, Asan, XunX, Kitzman JO, TurnerEH, StackhouseB, MackenzieAP, CaruccioNC, ZhangX, ShendureJ . Rapid, low-input, low-bias construction of shotgun fragment libraries by high- density in vitro transposition Genome Biol, 2010,11(12):R119. [本文引用: 2]
ReznikoffWS . Transposon Tn5 Annu Rev Genet, 2008,42:269-286. [本文引用: 1]
DaviesDR, Mahnke BraamL, ReznikoffWS, RaymentI . The three-dimensional structure of a Tn5 transposase- related protein determined to 2.9-A resolution J Biol Chem, 1999,274(17):11904-11913. [本文引用: 1]
PetersonG, ReznikoffW . Tn5 transposase active site mutations suggest position of donor backbone DNA in synaptic complex J Biol Chem, 2003,278(3):1904-1909. [本文引用: 1]
ReznikoffWS . Tn5 as a model for understanding DNA transposition Mol Microbiol, 2003,47(5):1199-1206. [本文引用: 1]
SakamotoH, ThibergeS, AkermanS, JanseCJ, CarvalhoTG, MénardR . Towards systematic identification of Plasmodium essential genes by transposon shuttle mutagenesis Nucleic Acids Res, 2005,33(20):e174. [本文引用: 1]
CaruccioN . Preparation of next-generation sequencing libraries using Nextera TM technology: simultaneous DNA fragmentation and adaptor tagging by in vitro transposition Methods Mol Biol, 2011,733:241-255. [本文引用: 1]
ChenCY, XingD, TanLZ, LiH, ZhouGY, HuangL, XieXS . Single-cell whole-genome analyses by linear amplification via transposon insertion (LIANTI) Science, 2017,356(6334):189-194. [本文引用: 1]
ChenXQ, ShenY, DraperW, BuenrostroJD, LitzenburgerU, ChoSW, SatpathyAT, CarterAC, GhoshRP, East-SeletskyA, DoudnaJA, GreenleafWJ, LiphardtJT, ChangHY . ATAC-see reveals the accessible genome by transposase-mediated imaging and sequencing Nat Methods, 2016,13(12):1013-1020. [本文引用: 1]
Buenrostro JD Giresi PG ZabaLC, ChangHY, GreenleatW . Transposition of native chromatin for multimodal regulatory analysis and personal epigenomics Nat Methods, 2013,10(12):1213-1218. [本文引用: 1]
LuZ, HofmeisterBT, VollmersC, DuboisRM, SchmitzRJ . Combining ATAC-seq with nuclei sorting for discovery of cis-regulatory regions in plant genomes Nucleic Acids Res, 2017,45(6):e41. [本文引用: 1]
DealRB, HenikoffS . A simple method for gene expression and chromatin profiling of individual cell types within a tissue Dev Cell, 2010,18(6):1030-1040. [本文引用: 1]
MaherKA, BajicM, KajalaK, ReynosoM, PauluzziG, WestDA, ZumsteinK, WoodhouseM, BubbK, DorrityMW, QueitschC, Bailey-SerresJ, SinhaN, BradySM, DealRB . Profiling of accessible chromatin regions across multiple plant species and cell types reveals common gene regulatory principles and new control modules Plant Cell, 2018,30(1):15-36. [本文引用: 1]
SosBC, FungHL, GaoDR, OsothpraropTF, KiaA, HeMM, ZhangK . Characterization of chromatin accessibility with a transposome hypersensitive sites sequencing (THS-seq) assay Genome Biol, 2016,17(1):20. [本文引用: 1]
TanL, KeZ, TomblineG, MacorettaN, HayesK, TianX, LvR, AblaevaJ, GilbertM, BhanuNV, Yuan ZF, GarciaBA, ShiYG, ShiY, SeluanovA, GorbunovaV . Naked mole rat cells have a stable epigenome that resists iPSC reprogramming Stem Cell Reports, 2017,9(5):1721-1734. [本文引用: 1]
ScharerCD, BlalockEL, BarwickBG, HainesRR, WeiC, SanzI, BossJM . ATAC-seq on biobanked specimens defines a unique chromatin accessibility structure in na?ve SLE B cells Sci Rep, 2016,6(1):27030. [本文引用: 1]
DavieK, JacobsJ, AtkinsM, PotierD, ChristiaensV, HalderG, AertsS . Discovery of transcription factors and regulatory regions driving in vivo tumor development by ATAC-seq and FAIRE-seq open chromatin profiling PLoS Genet, 2015,11(2):e1004994. [本文引用: 1]
LiuYJ, ZhangF, LiuHD, SunX . The application of next-generation sequencing techniques in studying transcriptional regulation in embryonic stem cells Hereditas(Beijing), 2017,39(8):717-725. [本文引用: 1]
 ,2, 郑恩琴
,2, 郑恩琴
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT
{kind=link}
{kind=link}