HTML
--> --> -->The need to better reproduce observed ambient SOA concentrations in models has motivated related research that attempts to distinguish missed SOA sources and unknown SOA formation mechanisms (Li et al., 2017b; Couvidat et al., 2018; Xu et al., 2018). Exploring the SOA formation potential and mechanisms from various anthropogenic and biogenic VOCs (BVOCs) have been the focus of numerous laboratory experiments. Their contribution to the total SOA budget has often been separately parameterized in models (Kelly et al., 2018; Jiang et al., 2019). Globally, the concentration of BVOCs emitted from terrestrial ecosystems was estimated to be 1000 Tg yr?1 (Guenther et al., 2012), which was roughly eight times higher than those from anthropogenic sources (127 Tg yr?1) (Glasius and Goldstein, 2016). BVOCs, including isoprene (C5H8, ~50%), monoterpenes (C10H16, ~15%) and sesquiterpenes (C15H24, ~3%) are important SOA formation precursors owing to their large emissions and high reactivity towards atmospheric oxidants [e.g., hydroxyl radicals (·OH), ozone, nitrate radicals (NO3·)] (Guenther et al., 2012; Jaoui et al., 2013; Ehn et al., 2014; Ng et al., 2017). Consequently, a large fraction of the global SOA (67%–95%) is estimated to derive from biogenic sources (Farina et al., 2010; Hodzic et al., 2016; Kelly et al., 2018).
Separating the anthropogenic SOA from the biogenic contribution in SOA formation is effective to improve model performance but is not sufficient to capture all human-induced SOA formation (Hodzic et al., 2016; Kelly et al., 2018; Jiang et al., 2019). Recently, anthropogenic pollutants have been suggested to indirectly participate in biogenic SOA formation through anthropogenic–biogenic interactions (Hoyle et al., 2011; Xu et al., 2015b; Zhang et al., 2018; Zhao et al., 2018b; Wu et al., 2020). For example, about 80% of biogenic SOA in East Asia was predicted to be influenced by anthropogenic emissions, while in regions with less anthropogenic emissions, like the eastern US, this value is larger than 50% (Carlton et al., 2010; Matsui et al., 2014). This “anthropogenic enhancement” effect on biogenic SOA formation indicates that, although naturally emitted BVOCs dominate over anthropogenic VOCs and cannot be controlled directly, biogenic SOA can to a certain extent be controlled by limiting manmade pollutants through air quality control policies (Edwards et al., 2017; Marais et al., 2017).
Nitrogen oxides (NOx = NO + NO2), sulfur dioxide (SO2), ammonia (NH3), and primary particles are prevalent anthropogenic pollutants. Traditional air quality policies target controlling their emissions for the purpose of mitigating the formation of secondary inorganic aerosols and associated environmental issues (Wang et al., 2013; Liu et al., 2019). Although global SO2 emissions have largely decreased in recent decades, the emissions of NOx and NH3 show increasing trends (Warner et al., 2017; Hoesly et al., 2018), and POA is still a significant component of polluted air in some regions (Zhang et al., 2015a; Li et al., 2017a; Jiang et al., 2019). When anthropogenic emissions–enriched air masses are transported to areas with substantial BVOCs emissions, anthropogenic–biogenic interactions take place, which perturb the oxidation of BVOCs and thus the corresponding SOA formation processes (Zhao et al., 2018b). The key goal of numerous recent studies has therefore been to determine the mechanisms of the anthropogenic–biogenic interactions (Ye et al., 2018; Slade et al., 2019), and the extent to which biogenic SOA can be controlled by eliminating predominant anthropogenic species such as NOx, SO2, NH3 and some primary aerosols (Carlton et al., 2010; Edwards et al., 2017). The ultimate aim is to achieve more reasonable parameterization of SOA budgets and effects, to evaluate models and to formulate more effective policies to alleviate air quality deterioration triggered by aerosol particles (Hettiyadura et al., 2019; Wu et al., 2020).
This review seeks to summarize the recent progress in research related to the interaction between anthropogenic species and natural biogenic emissions. Section 2 reviews the effects of NOx on biogenic SOA formation during daytime and nighttime. Section 3 describes the role of anthropogenic aerosol in gas–particle partitioning and particle-phase reactions. Section 4 discusses the photooxidation and ozonolysis of BVOCs modified by SO2. Current understanding regarding biogenic SOA formation and aging in the presence of NH3/amines is summarized in section 5, and recent field studies focusing on anthropogenic–biogenic interactions in China are discussed in section 6. The final section summarizes the entire review and gives an outlook regarding future studies toward anthropogenic–biogenic interactions. Overall, this review tries to comprehensively summarize recent advances in our understanding of the influence of anthropogenic emissions on biogenic SOA formation, to enlighten future observational and modeling studies in regions influenced by both anthropogenic and natural emissions, and to aid in the better formulation of pollution control strategies.
2
2.1. BVOC photooxidation and SOA formation
BVOC oxidation during daylight hours is dominated by ·OH (Ziemann and Atkinson, 2012). The initial addition or H-abstraction reaction between ·OH and BVOCs results in alkyl-type radicals (R·), most of which react rapidly with O2, leading to organic peroxy radicals (RO2·) (Atkinson, 2000). The general schematic of RO2· chemistry in SOA formation is summarized in Fig. 1. The influence of NOx is derived from its alteration of the fate of RO2·, which can either react with RO2·, hydroperoxy radicals (HO2·) or NOx under certain conditions. The different RO2· branches determine the distribution of oxidized products. For example, the reaction between RO2· and HO2· often produces hydroperoxides with low-volatility, RO2· self-reaction or reactions with other RO2· form alcohol or carbonyls, and the RO2· + NO reaction usually leads to organic nitrates as well as alkoxy radicals (RO·) that either undergo fragmentation or isomerization to form more volatile products (Ziemann and Atkinson, 2012; Sarrafzadeh et al., 2016). Since the fate of RO2· is highly related to the relative concentrations of NOx and VOCs in the urban atmosphere, laboratory chamber experiments often use the ratio of the initial BVOCs and NOx concentration ([BVOC]0/[NOx]0 or [NOx]0/[BVOC]0) to restrict the RO2· chemistry from the interpretation of NOx effects on new particle formation (NPF) and SOA yields (Pandis et al., 1991; Presto et al., 2005; Kim et al., 2012; Wildt et al., 2014; Xu et al., 2014; Stirnweis et al., 2017). It should be noted that attention must be paid to evaluate the O3-induced loss of BVOCs in the photooxidation system because O3 production and its effect would also vary with the [BVOC]0/[NOx]0 ratio, the relative rates of ozonolysis and ·OH oxidation and some other reaction conditions (Griffin et al., 1999). For the biogenic SOA formation in the presence of NOx listed in Table 1, the completely dominant role of ·OH oxidation in BVOC loss was estimated and thus O3 generation would not influence the NOx-dependent SOA yield.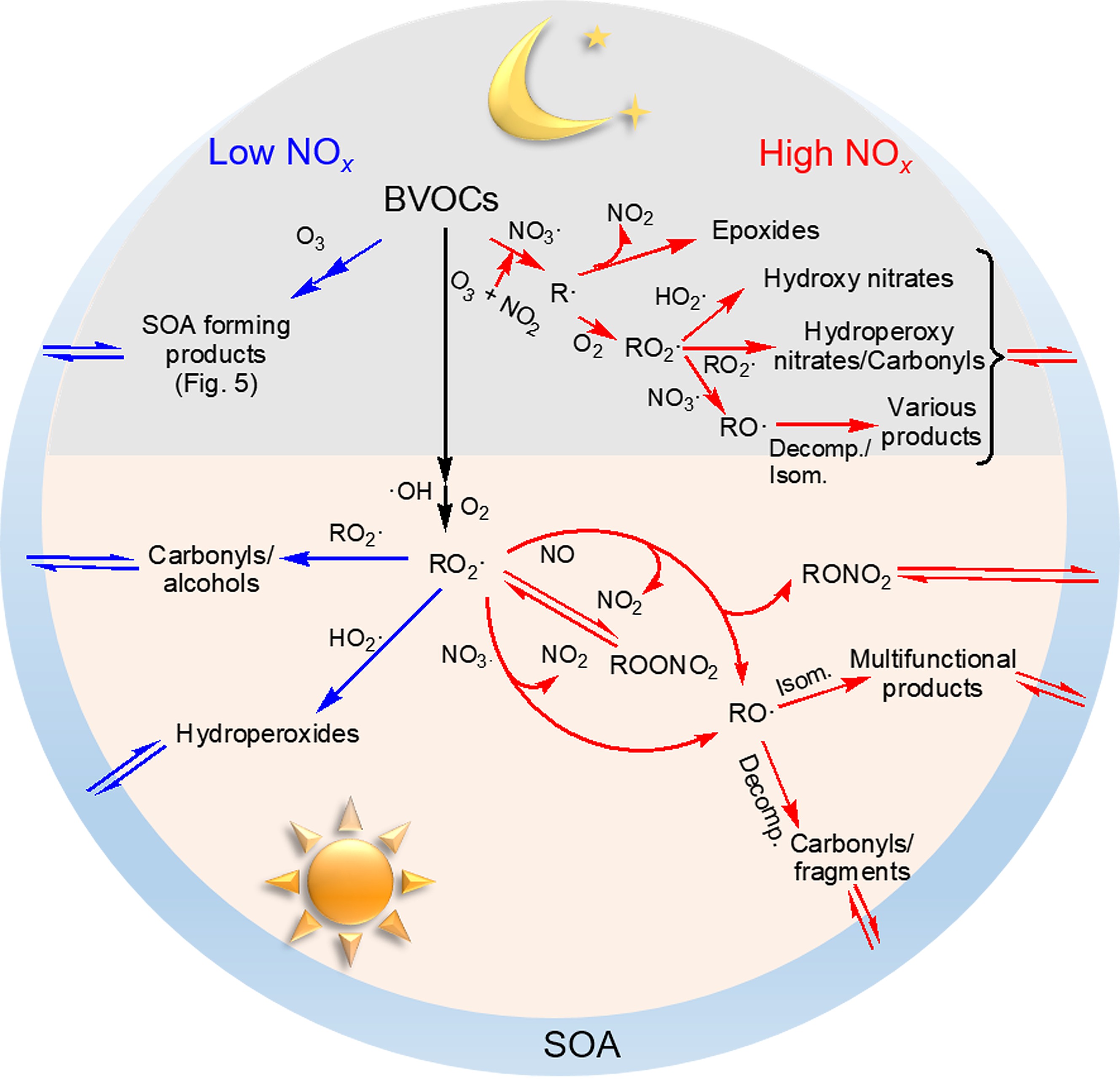 Figure1. General schematic picture of NOx effects on BVOC oxidation during daytime and nighttime. “Decom.” and “Isom.” represent decomposition and isomerization reactions, respectively.
Figure1. General schematic picture of NOx effects on BVOC oxidation during daytime and nighttime. “Decom.” and “Isom.” represent decomposition and isomerization reactions, respectively.| BVOCs | [BVOC]0 (ppb) | [NOx]0/ [BVOC]0 | ·OH precursors | T (K) | RH (%) | Seed | SOA mass (μg m?3) | Yield (%) | Notes | Reference |
| Isoprene | 91.4–114.6 | 0.7–7.3 | H2O2 | ~298 | < 5 | none | 4.2–30.2 | 1.5–8.5 | The SOA yield increased with initial NO/isoprene up to a ratio of 3, beyond which it decreases with increasing initial [NO]0/[isoprene]0 ratio. | (Xu et al., 2014) |
| 45±4 | 0–17 | H2O2 | ~301 | < 10 | ammonium sulfate | 1.7–6.7 | 1.4–5.5 | At high NOx (>200 ppb), the SOA yield decreased with increasing NOx. | (Kroll et al., 2006) | |
| 26 | 0–1.9 | H2O2 | ? | 50 | ammonium sulfate | 1.9–7.6 | 2.7–11.6 | The SOA yield was nearly constant at low NO until the [NO]0/[isoprene]0 ratio reached ~0.38). It further decreased with the increase of NO concentrations. | (Liu et al., 2016) | |
| 50 | 0–0.5 | H2O2 | ~298 | 40±2 | ammonium sulfate | 0.5–1.2 | 0. 4–0.9 | Higher [NOx]0/[isoprene]0 ratios produced lower aerosol yields. | (King et al., 2010) | |
| 33–523 | 1.6–32 | CH3ONO/ HONO | 296–298 | 9–11 | ammonium sulfate | 2.9–65.2 | 3.1–7.4 | SOA yields were relevant to NO2/NO ratio under high NOx conditions. | (Chan et al., 2010) | |
| 25–500 | 0.5–7.6 | HONO | 293–295 | 42–50 | ammonium sulfate | 0.7.–42.6 | 0.9–3 | Higher [NOx]0/[isoprene]0 ratios produced lower aerosol yields. | (Kroll et al., 2005) | |
| 180–2500 | 0.2–0.7 | NOx | 293 | 47–53 | none | 0.7–336 | 0.2–5.3 | SOA yields first increased ([NOx]0/[isoprene]0 < 0.5) and then decreased with [NOx]0/[isoprene]0 ([NOx]0/[isoprene]0 > 0.5). | (Dommen et al., 2006) | |
| α-pinene | ~15 | 0–64.5 | H2O2/HONO | 296–299 | 3.3–6.4 | ammonium sulfate | 4.5–29.3 | 6.6–37.9 | SOA yields were higher at lower initial [NOx]0/[α-pinene]0 ratios. | (Ng et al., 2007b) |
| 18.3–20.3 | 0.1–2.6 | HONO | 294–299 | 27–29 | ammonium hydrogen sulfate and sulfuric acid | 2.1–12 | 1.8–11.6 | The yields at low [NOx]0/[α-pinene]0 ratios were in general higher compared to those at high [NOx]0/[α-pinene]0. | (Stirnweis et al., 2017) | |
| 16.1–20.7 | 1.2–3.8 | HONO | 294–299 | 66–69 | ammonium hydrogen sulfate and sulfuric acid | 8.6–13.4 | 8.1–13.8 | The yields at low [NOx]0/[α-pinene]0 ratios were in general higher compared to those at high [NOx]0/[α-pinene]0. | (Stirnweis et al., 2017) | |
| 45–52.4 | ? | H2O2/HONO/ CH3ONO | 293–298 | < 10 | ammonium sulfate | 37.2–76.6 | 14.4–28.9 | The SOA yield was suppressed under conditions of high NO. | (Eddingsaas et al., 2012) | |
| 65–120 | 0.3–1.2 | NOx | 306–315 | 14–17 | none | 18–136 | 5.3–24 | Aerosol yields should be higher at lower [NOx]0/[α-pinene]0 ratios. | (Kim et al., 2012) | |
| 470–845 | 0.4–0.9 | NOx | 310–316 | 14–17 | none | 830–2100 | 34–68 | SOA yields were higher at lower initial NOx/α-pinene ratios. | (Kim et al., 2010) | |
| ~ 20 | ~ 0–1 | HONO | 291–307 | 29–42 | none | ? | 0–10 | Higher [NOx]0/[α-pinene]0 ratios produced lower aerosol yields. | (Zhao et al., 2018b) | |
| β-pinene | 37 | 0.01–3.9 | HO2/NO | 289±1 | 63±2 | ammonium sulfate | 14.3–38.1 | 8.2–20.0 | SOA yields increased with increasing [NOx] at low-NOx conditions ([NOx]0 < 30 ppb, [NOx]0/[β-pinene]0 < 1 and decreased with [NOx] at high-NOx conditions ([NOx]0 >30 ppb, NOx/β-pinene ~1 to ~3.8). | (Sarrafzadeh et al., 2016) |
| 36–2000 | 0.2–19.6 | NOx | ? | ? | none | ? | ? | Aerosol yields were small when [NOx]0/[β-pinene]0 was larger than 2, increased dramatically and reached maximum for the range of 0.7–1, then decreased slowly as the ratio decrease. | (Pandis et al., 1991) | |
| 405–640 | 0.4–0.9 | NOx | 312–317 | 12–19 | none | 430–900 | 25–37 | Higher [NOx]0/[β-pinene]0 ratios produced lower aerosol yields. | (Kim et al., 2010) | |
| 32.3–96.5a | ~2–10 | NOx | 308–313 | ~5 | ammonium sulfate | 7.2–141.6 | 3.2–27.2 | SOA yields were lower at higher NOx levels than at lower NOx levels.b | (Griffin et al., 1999) | |
| Limonene | 60–75 | 0.3–1.6 | NOx | 304–312 | 14–21 | none | 79.2–136 | 27–40 | Higher [NOx]0/[limonene]0 ratios produced lower aerosol yields. | (Kim et al., 2012) |
| ~ 7 | ~ 0–2.9 | HONO | 293–303 | 28–31 | none | ? | 0–5 | Higher [NOx]0/[limonene]0 ratios produced lower aerosol yields. | (Zhao et al., 2018b) | |
| 20.6–65.1a | ~2–5 | NOx | 309–313 | ~5 | ammonium sulfate | 9.5–120.2 | 8.7–34.4 | SOA yields were lower at higher NOx levels than at lower NOx levels. b | (Griffin et al., 1999) | |
| Sabinene | 13.9–83.3a | ~2–10 | NOx | 310–316 | ~5 | ammonium sulfate | 2.5–14.5 | 1.9–65.2 | SOA yields are lower at higher NOx levels than at lower NOx levels. b | (Griffin et al., 1999) |
| α-humulene | 5–9.2a | ~2–10 | NOx | 309–312 | ~5 | ammonium sulfate | 12.9–59.2 | 31.9–84.5 | The yields dependence on NOx levels is not obvious. b | (Griffin et al., 1999) |
| Longifolene | ~4.3 | 0–131 | H2O2/HONO | 296–299 | 3.3–6.4 | ammonium sulfate | 28.5–51.6 | 84–157 | SOA yields under high-NOx conditions exceed those under low-NOx conditions. | (Ng et al., 2007b) |
| Aromadendrene | ~5 | 0– ~103 | H2O2/HONO | 296–299 | 3.3–6.4 | ammonium sulfate | 19.7–29.3 | 41.7–84.7 | Aerosol yields increase with NOx concentrations. | (Ng et al., 2007b) |
| β-caryophyllene | 3–32 | 0–1.7 | H2O2/HONO | 293±2 | < 10 | ammonium sulfate | 8.4–311 | 19.3–137.8 | SOA yields at low NOx conditions were lower than those at high NOx conditions. | (Tasoglou and Pandis, 2015) |
| 31.1–52.4 | 0.5–1.7 | NOx | ~298 | ~70 | none | 35.6–66.2 | 9.5–19.9 | The yields dependence on NOx levels was not obvious. | (Alfarra et al., 2012) | |
| 5.9–12.9 | ~2–5 | NOx | 309–312 | ~5 | ammonium sulfate | 17.6–82.3 | 13.1–39.0 | The yields dependence on NOx levels was not obvious. | (Griffin et al., 1999) | |
| Notes: a Mixing ratios of BVOCs reacted due to the unavailable initial BVOC concentrations; b Effects of NOx on SOA yields are hypothesized if the reacted BVOCs are equal to the initial ones. | ||||||||||
Table1. SOA formation from BVOC photooxidation in the presence of NOx.
The SOA yield is defined as the formed SOA mass concentration (ΔM, μg m?3) relative to the consumed parent hydrocarbon (ΔBVOC, μg m?3). The impact of NOx on SOA yields depends on the SOA mass production and is also parent hydrocarbon-specific (Table 1). For isoprene, the most abundant BVOC in the atmosphere (Kroll et al., 2006; Chan et al., 2010; Xu et al., 2014), the pathways of its reaction with RO2· under low and high NOx conditions are quite different (Fig. 2). Chamber studies have generally evidenced higher SOA yields at lower [NOx]0/[isoprene]0 ratios, and most of these studies have suggested that SOA yields first increase and then decrease with the increasing [NOx]0/[isoprene]0 ratios (Dommen et al., 2006; Kroll et al., 2006; King et al., 2010; Xu et al., 2014; Liu et al., 2016). The decrease of SOA yield with increasing NOx, more precisely with increasing NO, can generally be explained by the dominance of RO2· + NO reactions over RO2· + HO2· reactions, with the former producing more volatile products (such as organic nitrates) than the latter (hydroperoxides) (Kroll et al., 2006; Xu et al., 2014). Kroll et al. (2006) considered that the decline of the NO/HO2· ratio, which may lead to a switch from high-NOx to low-NOx conditions over the experimental process, might result in the complex SOA yield dependence under lower NOx conditions ([NOx]0/[isoprene]0 < 4.4). Xu et al. (2014) also observed similar nonlinear variation of aerosol volatility and oxidation state level with the [NO]0/[isoprene]0 ratio (0–7.3) as the SOA yield. They proposed that the presence of NO enhances the formation of methacrolein, the first generation product, whose further oxidation forms SOA-forming organics efficiently (Surratt et al., 2010), leading to increased SOA yield and decreased aerosol volatility when [NO]0/[isoprene]0 is lower than 3. In a more recent study focusing on a lower [NO]0/[isoprene]0 range (0–2), the SOA yield was nearly constant when the [NO]0/[isoprene]0 ratio was lower than ~0.38 (Liu et al., 2016). After this NO threshold level, the SOA yield decreased from 12% to 3% with a further increase of NOx, accompanied by a decrease of more highly oxygenated organic nitrates. These observations were explained by the suppression of NO on hydroxy hydroperoxide, which acts as the source of C5H11O6 peroxyl radicals and thus lowers the production of both second-generation multifunctional peroxides and multifunctional organic nitrates (Fig. 2). Similarly, with the composition analysis of isoprene SOA formed under low NOx in laboratory and aerosol samples collected from the isoprene-rich southeastern US environment, the none-IEPOX (isoprene epoxydiols) pathway under low NOx conditions was also suggested to contribute to notable highly oxidized compounds and SOA mass (Riva et al., 2016c).
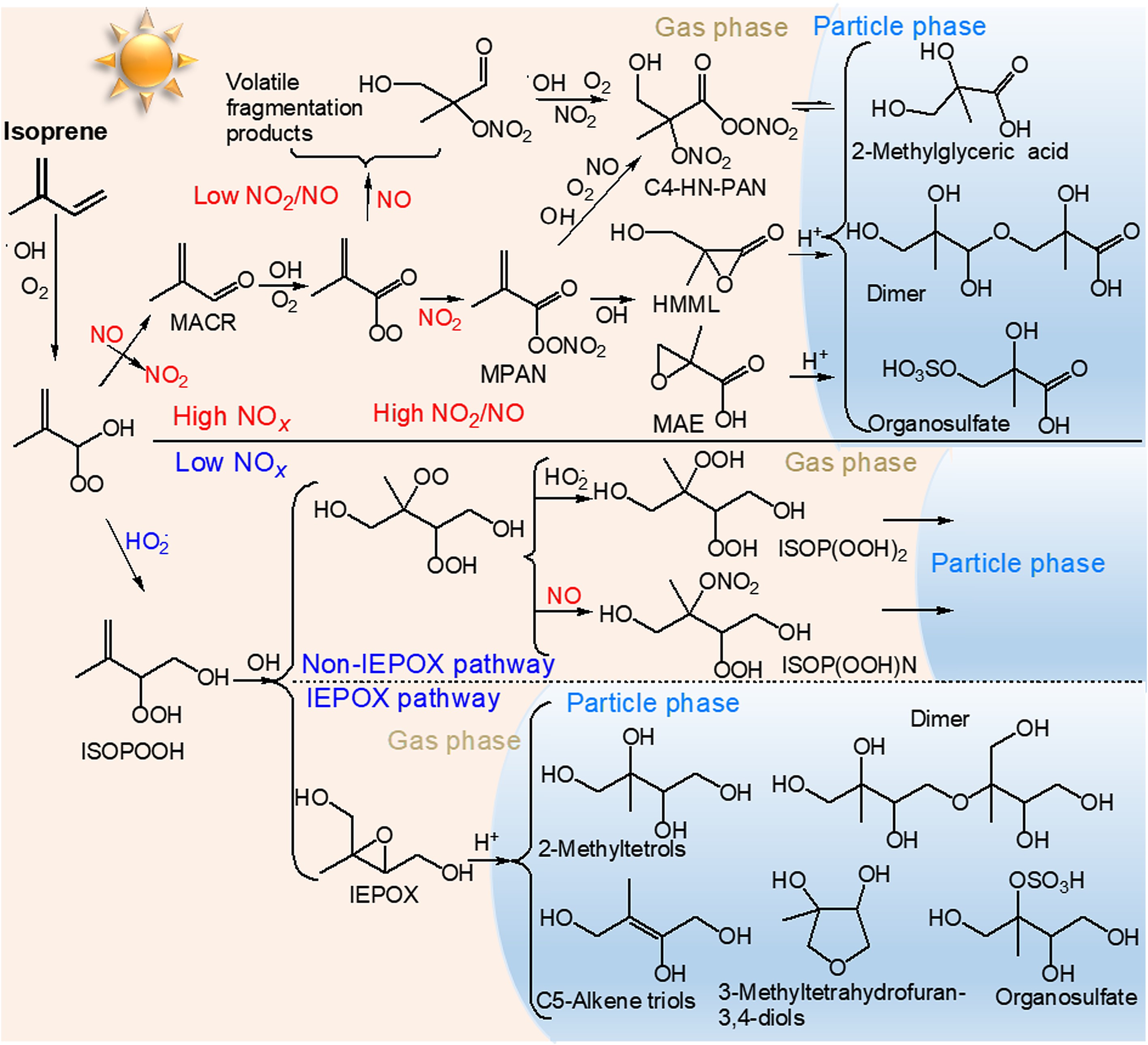 Figure2. Effects of NOx on isoprene SOA formation during daytime. Under high NOx conditions, isoprene RO2· primarily reacts with NO, forming methacrolein (MACR). The oxidation of MACR under high NO2/NO ratios forms methacryloylperoxynitrate (MPAN) while C4-hydroxynitrate peroxyacyl nitrate (C4-HN-PAN) is the main intermediate leading to SOA under high NOx conditions with low NO2/NO ratios. MPAN further reacts with ·OH to form methacrylic epoxide (MAE) and hydroxymethylmethyl-α-lactone (HMML). Acid-catalyzed reactions of MAE in the particle phase produce 2-methylglyceric acid, an organosulfate, and an oligomer. Under low NOx conditions, isoprene RO2· reacts predominantly with HO2·, leading to hydroxy hydroperoxide (ISOPOOH). ISOPOOH-derived epoxydiols (IEPOX) undergo multiphase acid-catalyzed chemistry to give various products in the particle phase. The non-IEPOX pathway that gives dihydroxy dihydroperoxides (ISOP(OOH)2) and organic nitrates (ISOP(OOH)N) is proposed to contribute to SOA formation without reactive aqueous seed particles. References for the non-IEPOX pathways are Liu et al. (2016) and Riva et al. (2016c), while for other pathways they are Lin et al. (2013b), Surratt et al. (2010), Lin et al. (2012) and Lin et al. (2013a).
Figure2. Effects of NOx on isoprene SOA formation during daytime. Under high NOx conditions, isoprene RO2· primarily reacts with NO, forming methacrolein (MACR). The oxidation of MACR under high NO2/NO ratios forms methacryloylperoxynitrate (MPAN) while C4-hydroxynitrate peroxyacyl nitrate (C4-HN-PAN) is the main intermediate leading to SOA under high NOx conditions with low NO2/NO ratios. MPAN further reacts with ·OH to form methacrylic epoxide (MAE) and hydroxymethylmethyl-α-lactone (HMML). Acid-catalyzed reactions of MAE in the particle phase produce 2-methylglyceric acid, an organosulfate, and an oligomer. Under low NOx conditions, isoprene RO2· reacts predominantly with HO2·, leading to hydroxy hydroperoxide (ISOPOOH). ISOPOOH-derived epoxydiols (IEPOX) undergo multiphase acid-catalyzed chemistry to give various products in the particle phase. The non-IEPOX pathway that gives dihydroxy dihydroperoxides (ISOP(OOH)2) and organic nitrates (ISOP(OOH)N) is proposed to contribute to SOA formation without reactive aqueous seed particles. References for the non-IEPOX pathways are Liu et al. (2016) and Riva et al. (2016c), while for other pathways they are Lin et al. (2013b), Surratt et al. (2010), Lin et al. (2012) and Lin et al. (2013a).Note that, although similar trends of the isoprene SOA yield response to NOx levels have been observed among different studies, the critical [NOx]0/[isoprene]0 points for the transition role of NOx are quite different [e.g., 4.4 (Kroll et al., 2006), 0.38 (Liu et al., 2016), and ~3 (Xu et al., 2014)]. It has been shown that, even under the same [NOx]0/[isoprene]0 ratios, the fate of RO2· radicals that are responsible for SOA formation can be quite different (Ng et al., 2007a). Recent studies have suggested that the composition of NOx itself is also a candidate for altering SOA formation pathways (Chan et al., 2010; Surratt et al., 2010). For example, oligoesters of dihydroxycarboxylic acids and hydroxynitrooxycarboxylic acids from isoprene photooxidation increased with increasing NO2/NO ratios (Chan et al., 2010). More recent studies show that SOA yields under high NOx conditions can be as high as those under low-NOx conditions because the NO2 + RO2· reaction can potentially yield substantial SOA mass (e.g., hydroxymethylmethyl-α-lactone, methacrylic acid) via the subsequent oxidation of methacryloylperoxynitrate, which is favorably formed from methacrolein (first-generation products of isoprene photooxidation) oxidation under high NO2/NO ratios (Fig. 2) (Chan et al., 2010; Surratt et al., 2010; Lin et al., 2012, 2013b; Pye et al., 2013; Nguyen et al., 2015). Besides NO2/NO ratios, the ·OH precursors, such as HONO, which strongly suppresses ISOPOOH chemistry and thus the formation of the second-generation organic nitrates, the chamber operation mode (flow or batch mode) and some other reaction conditions (e.g., seed particles), are potential factors that induce the differences in threshold [NOx]0/[isoprene]0 values, thus warranting further studies for more accurate model parametrization (Kroll et al., 2005; Xu et al., 2014; Liu et al., 2016; Shrivastava et al., 2017).
The effects of NOx on SOA formation from the photooxidation of monoterpenes, especially α-pinene, β-pinene and limonene, have also been characterized by chamber studies (Pandis et al., 1991; Zhang et al., 1992; Ng et al., 2007b; Eddingsaas et al., 2012; Kim et al., 2012; Wildt et al., 2014; Sarrafzadeh et al., 2016; Stirnweis et al., 2017; Zhao et al., 2018b). As summarized in Table 1, SOA yields are generally higher under low-NOx than high-NOx conditions when monoterpene ozonolysis is negligible. Besides the perturbation of NOx on RO2· chemistry, recent studies have found that NOx influence the SOA yield by altering the ·OH cycle and NPF (Wildt et al., 2014; Sarrafzadeh et al., 2016; Zhao et al., 2018b). Using realistic BVOC mixtures emitted directly by plants, Wildt et al. (2014) found that NPF was suppressed under high-NOx conditions ([BVOC]0/[NOx]0 < 7, [NOx]0 > 23 ppb). The self-reaction of higher-generation peroxy radical-like intermediates and their reaction with NO commonly limit the rate of NPF. More recently, a study focusing on β-pinene photooxidation showed that under low-NOx conditions ([β-pinene]0/[NOx]0 > 10 ppbC ppb?1) the increase in ·OH radicals through the reaction NO + HO2· → NO2 + ·OH was responsible for the increase in SOA yield with the increase in NOx (Sarrafzadeh et al., 2016). It was also evidenced that the ratio of NO/NO2 was correlated with the ·OH cycle and, thus, probably influenced SOA formation. Under high-NOx conditions ([β-pinene]0/[NOx]0 = ~10 to ~2.6 ppbC ppb?1), the decrease in SOA yield with NOx was attributed to NOx-triggered suppression of low-volatility products (such as hydroperoxides) that participated in NPF. The restrained NPF would further result in limited particle surfaces for the condensation of low-volatility species. Similarly, the suppression effect of NOx on NPF has been evidenced during the photooxidation of α-pinene and limonene (Zhao et al., 2018b).
Sesquiterpenes on a reacted mass basis have much higher SOA formation potential than isoprene and monoterpenes owing to their higher molecular weight and reactivity (Lee et al., 2006; Jaoui et al., 2013). As opposed to NOx effects on SOA formation from isoprene and monoterpenes photooxidation, SOA formed from longifolene, aromadendren and β-caryophyllene photooxidation under high-NOx conditions substantially exceeds that under low-NOx conditions (Ng et al., 2007b; Tasoglou and Pandis, 2015). The formation of less volatile products (e.g., large hydroxycarbonyls, multifunctional species) via isomerization instead of decomposition of large RO· and the relatively low-volatility organic nitrates were proposed to be responsible for this positive NOx effect. However, SOA yields from β-caryophyllene in the works of Griffin et al. (1999) and Alfarra et al. (2012) were less dependent on [NOx]0/[BVOC]0 ratios, probably due to the interference of other experimental conditions (e.g., OH precursors, the initial BVOC mixing ratios). Clearly, if the positive NOx effect on SOA formation observed by Ng et al. (2007b) can be extended to other sesquiterpenes, the contribution of sesquiterpenes to SOA in NOx-polluted air may be much higher (Ng et al., 2007b). A recent modeling study in the southeastern US showed underestimated SOA formation from monoterpenes and sesquiterpenes and argued that anthropogenic emissions would exert complex influences on biogenic SOA formation (Xu et al., 2018). Considering that studies on NOx effects only target a limited number of sesquiterpenes, a thorough evaluation of the effect of NOx on the photooxidation of a complete suite of sesquiterpenes is necessary for better constraint of their oxidation and contribution to ambient SOA.
2
2.2. Biogenic SOA formation under dark conditions
Nighttime biogenic SOA formation in the atmosphere is sensitive to NOx levels because of the changed radical (e.g., RO2·, HO2·, NO3·) chemistry and the oxidation capacity (Brown and Stutz, 2012; Ng et al., 2017). While ·OH dominates daytime BVOC oxidation, NO3·, which is mainly produced via the reaction between O3 and NO2, becomes one of the main oxidants at night (Fig. 1) (Wayne et al., 1991; Rollins et al., 2012; Edwards et al., 2017). The unsaturated and non-aromatic nature of BVOCs makes them particularly susceptible to oxidation by NO3· and O3 (Atkinson and Arey, 1998; Ayres et al., 2015). The competition between these two BVOC sinks is closely associated with the NOx level and composition because of the loss of NO3· through its reaction of NO and a decrease in its production through the reaction of NO2 and O3, as O3 is decreased by the reaction of O3 and NO (Rollins et al., 2012; Qin et al., 2018b; Wang et al., 2020a). The oxidation of BVOCs by NO3· occurs mainly via the addition of NO3· to the unsaturated bonds (another pathway is hydrogen abstraction, favored for aldehydic species), forming alkyl radicals that would either lose NO2 to form epoxides or further react with O2 to form RO2· (Fig. 1) (Ng et al., 2017; Fouqueau et al., 2020). RO2· would isomerize or react with HO2·, NO3· or RO2· to form various products such as organic nitrates that potentially generate SOA. NO3·–BVOCs chemistry is thus regarded as a prominent candidate for the generation of biogenic SOA and organic nitrates that are correlated with anthropogenic tracers (Fry et al., 2009; Kiendler-Scharr et al., 2016; Huang et al., 2019).Such correlations have been evidenced in recent field observations around the world (Rollins et al., 2012; Brown et al., 2013; Kiendler-Scharr et al., 2016; Edwards et al., 2017; Fry et al., 2018; Yu et al., 2019). In a rural area in Southwest Germany, the contribution of organic nitrates to the increase of newly formed particles after sunset was observed to be 18%–25%. Considering both high BVOCs and NOx emissions in this area, the reactions between NO3· and BVOCs, especially monoterpenes, are responsible for organic nitrates and SOA formation (Huang et al., 2019). In some forest regions of the US, the concentration of organic nitrates was found to peak at night and its contribution to the total OA was up to 40% in Bakersfield owing to nighttime oxidation of BVOCs by NO3· (Rollins et al., 2012; Fry et al., 2013; Xu et al., 2015a). A substantial contribution of organic nitrates formed via nocturnal NO3·–BVOCs chemistry to particulate organic mass has also been observed in Europe and China (Kiendler-Scharr et al., 2016; Yu et al., 2019). Interestingly, the observation in the forest region of the western US showed that the concentration of nighttime aerosol organic nitrates was positively correlated with the product of the mixing ratios of NO2 and O3 instead of that of O3 alone (Fry et al., 2013). This indicates that NO3·-initiated oxidation of monoterpenes is related to the NOx level and is an important source of particle-phase organic nitrates at night.
The SOA formation potential of various BVOCs oxidized by NO3· has been investigated in many chamber studies [Ng et al. (2017) and references therein]. The reported SOA yields vary among different BVOCs, from nearly 0 for α-pinene, to 0.12 for isoprene, 0.33–0.44 for β-pinene, 0.44–0.57 for limonene, and up to 0.86 for β-caryophyllene at an atmospheric relevant aerosol mass loading of 10 μg m?3 (Fry et al., 2014). Except for α-pinene, these yield values are much higher than those from the ozonolysis of corresponding BVOCs (Song et al., 2007; von Hessberg et al., 2009; Saathoff et al., 2009; Tasoglou and Pandis, 2015). The relative importance of NO3· oxidation versus O3 is connected with the ratio of NO3· production to BVOC ozonolysis (Griffin et al., 1999). Considering, for example, 10 ppt NO3· and 30 ppb O3, the oxidation of these monoterpenes by NO3· proceeds 20–90 times faster than their ozonolysis, due to the much higher rate constants of the former reactions (Fry et al., 2014). The accelerated BVOC consumption by NO3· here is somewhat consistent with the field observations, which found NO3· + monoterpenes chemistry to be a significant nighttime aerosol source in regions with a high NOx level.
While most chamber studies have directly investigated NO3·-induced SOA under purified NO3· conditions (Griffin et al., 1999; Hallquist et al., 1999; Fry et al., 2014), some recent works have examined the biogenic SOA formation in the presence of Ox (O3 + NO2) (Table 2) (Presto et al., 2005; Perraud et al., 2012; Draper et al., 2015; Chen et al., 2017; Xu et al., 2020). The effects of NO2 on the dark ozonolysis of β-pinene, Δ3-carene, and limonene were examined by keeping the O3 mixing ratio constant while varying the NO2 mixing ratios ([O3]0/[NO2]0 = 2–0.5, [NO2]0/[BVOCs]0 = 0.5–1). It was found that, for β-pinene and Δ3-carene, SOA yields were comparable over the range of oxidation conditions. An increase of limonene SOA yield with increasing NO2 mixing ratio was observed and attributed to the increased fraction of oligomers and multifunctional organic nitrates in SOA through NO3· chemistry (Draper et al., 2015). More recently, the γ-terpinene SOA yield, as well as the contribution of organic nitrates to particle mass, were found to have both increased with increasing NO2 levels ([NO2]0/[O3]0 = 0–0.7, [NO2]0/[γ-terpinene]0 = 0–3), due to the change from O3-dominant to NO3·-dominant γ-terpinene oxidation, which yields organic nitrates as significant SOA components (Xu et al., 2020). Among the studied monoterpenes, α-pinene exhibited quite a different NO2 response during ozonolysis. Several studies have consistently found that SOA yields, as well as particle number concentrations, decreased with increasing NOx (Presto et al., 2005; N?jgaard et al., 2006; Perraud et al., 2012; Draper et al., 2015). This is expected because the SOA yield from α-pinene ozonolysis is higher than that from NO3· oxidation, the latter process forming organic nitrates that have relative high volatility and are thus inefficient to nucleate (Perraud et al., 2012). In the real atmosphere, a good correlation between Ox and biogenic SOA tracers was also observed in a field campaign carried out in the Pearl River Delta, South China (Zhang et al., 2019b). With the elevation of Ox in the atmosphere, more observations focusing on the linkage between Ox and biogenic SOA are necessary but still limited. Altogether, these studies suggest that models should carefully handle the Ox effects on nocturnal SOA formation by capturing the detailed spatial distribution of BVOCs and Ox in order to reduce the uncertainty in the estimation of regional or global SOA budgets (Fry et al., 2014, 2018).
| BVOCs | [BVOC]0 (ppb) | [NOx]0/ [BVOC]0 | [NOx]0/ [O3]0 | T(K) | RH(%) | Seed | SOA mass (μg m?3) | Yield (%) | Notes | Reference |
| α-pinene | 15–200 | 0.7–70 | ~0.03–4 | 288–313 | ? | none | 1–346 | 0–0.29 | The yields increase as NO2 concentrations decrease and reach an asymptote near [NOx]0/[BVOC]0 = 0.7. | (Presto et al., 2005) |
| 300–960 | ~0–4.7 | 0–2.9 | 294–295 | 22–30 | none | ? | ? | The increase of [NO2]0 substantially depletes SOA formation. | (Draper et al., 2015) | |
| 1000 | 0–6.3 | ~0–4.5 | ? | < 3 | none | ? | ? | Fewer particles are formed at higher NO2 conditions. | (Perraud et al., 2012) | |
| 47±3 | 0–9.6 | ~0–8.7 | 294±2 | < 1 | none | ? | ? | Particle number concentration and volume were substantially reduced in the presence of NO2. | (N?jgaard et al., 2006) | |
| β-pinene | 300–1100 | ~0–6.7 | 0–4.2 | 295 | 23–40 | none | ? | ? | SOA yields are comparable over oxidant conditions studied. | (Draper et al., 2015) |
| Δ3-carene | 220–650 | ~0–3 | 0–1.9 | 294–295 | 27–38 | none | ? | ? | SOA yields are comparable over oxidant conditions studied. | (Draper et al., 2015) |
| limonene | 150–159 | 0.2–0.4 | 0.5–75.9 | 295–297 | 9.2–9.9 | none | 30.3–157.3 | 0.27–0.73 | The highest SOA yield occurred when [O3]/[NO] is around 1. | (Chen et al., 2017) |
| 300–560 | ~0–3.3 | 0–2.2 | 295 | 20–31 | none | ? | ? | SOA formation was enhanced at higher NO2. | (Draper et al., 2015) | |
| 51±3 | 0–6.9 | 0–7.1 | 294±2 | < 1 | none | ? | ? | Particle number concentrations were lower at higher NOx conditions. | (N?jgaard et al., 2006) | |
| γ-terpene | 152–154 | 0–2.9 | 0–0.7 | 297–301 | 24–30 | none | ? | 0.38–0.77 | NOx enhance SOA yields and decrease particle number concentrations. | (Xu et al., 2020) |
Table2. SOA formation from BVOC ozonolysis in the presence of NOx.
2
3.1. Gas–particle partitioning
BVOC oxidation can form semi-volatile organic compounds (SVOCs) that undergo partitioning between the gas and particle phases. The SOA yield (Y) that is defined as the ratio of the OA mass concentration (M0) to the BVOC consumption, can be modeled by the gas–particle partitioning absorption model (Pankow, 1994b; Odum et al., 1996),where αi is the mass-based stoichiometric coefficient of product i, and Kom,i (m3 μg?1) is the partitioning coefficient of the gas–particle partitioning defined by the ratio of absorption equilibrium constant (Kp,i) to the mass fraction of species i in the aerosol phase (fom) (Pankow, 1994a, b; Odum et al., 1996).
Kp,i can be calculated by the following equation (Donahue et al., 2006; Zhang et al., 2015b):
where




Anthropogenic POA makes up a significant fraction of the total OA in the atmosphere, especially under severe air pollution in winter (Li et al., 2017a; Zhang et al., 2017a). Based on the gas–particle partitioning mechanism, it has been predicted that biogenic SOA would be largely enhanced by POA emissions (Heald et al., 2008; Hoyle et al., 2009; Carlton et al., 2010, 2018). Some recent studies have argued that the enhanced biogenic SOA formation would be overestimated probably because the assumption of a well-mixed OA phase between POA and SOA in models is not always the case for the real atmosphere (Loza et al., 2013; Robinson et al., 2015), though this assumption is reasonable in SOA formation because most of the SOA components are oxygenated polar organic species that are miscible with one another (Song et al., 2007). Ambient POA contains a large fraction of hydrophobic non-polar species and phase separation often occurs. The existence of such morphologies would affect the gas–particle partitioning of SVOCs, therefore affecting SOA formation and its optical properties (Song et al., 2007; George et al., 2015).
Several chamber experiments have been conducted to examine the mixing behavior of POA and biogenic SOA, and thus the applicability of the single-phase assumption in models (Robinson et al., 2013). For example, using dioctyl phthalate (DOP) and lubricating oil as surrogates for urban hydrophobic POA, the SOA mass from α-pinene ozonolysis (can also be applied to other BVOCs) was insensitive to these seed aerosols [Fig. 3a (Song et al., 2007)]. Implying the no seed parameters and the sum of seed and aerosol masses as M0, the seeded SOA mass was 13%–44% higher than the observed value and phase separation could therefore appear. This is reasonable because multifunctional species formed from α-pinene ozonolysis have polar properties, which are exactly opposite to those of DOP and lubricating oil components. The layered phase between α-pinene SOA and DOP POA was further confirmed by a single-particle mass spectrometer, which could distinguish whether SOA and DOP were homogeneously mixed by changing the laser power (Vaden et al., 2010). A high MS intensity of the surface material at low laser power instead of the constant relative MS intensities with changing laser powers was observed, supporting the phase separation (Fig. 3b). On the contrary, the temporal evolution of the aerodynamic vacuum diameter of diesel POA (DL) and α-pinene SOA mixture showed transformation from bimodal distribution to single modal distribution (Fig. 3c), indicating the formation of a single phase between α-pinene SOA and DL (Asa-Awuku et al., 2009). SOA from β-caryophyllene ozonolysis also formed a well-mixed phase with DL, but these SOA and α-pinene SOA were immiscible with motor oil and diesel fuel POA. This study supports the use of the single-phase assumption in atmospheric models because diesel exhaust POA is the most atmospherically relevant case. Anthropogenic SOA formed from the photooxidation of aromatic hydrocarbons enhanced SOA formation from α-pinene oxidation, indicating that the interaction between these different types of SOA formed an ideal mixing state (Hildebrandt et al., 2011; Emanuelsson et al., 2013; Robinson et al., 2013). Clearly, the polarity of anthropogenic OA reflects its mixing behavior with biogenic SOA. The incorporation of the distribution of different types of anthropogenic OA in the real atmosphere into regional or global models is crucial in evaluating the effects of anthropogenic OA on biogenic SOA formation.
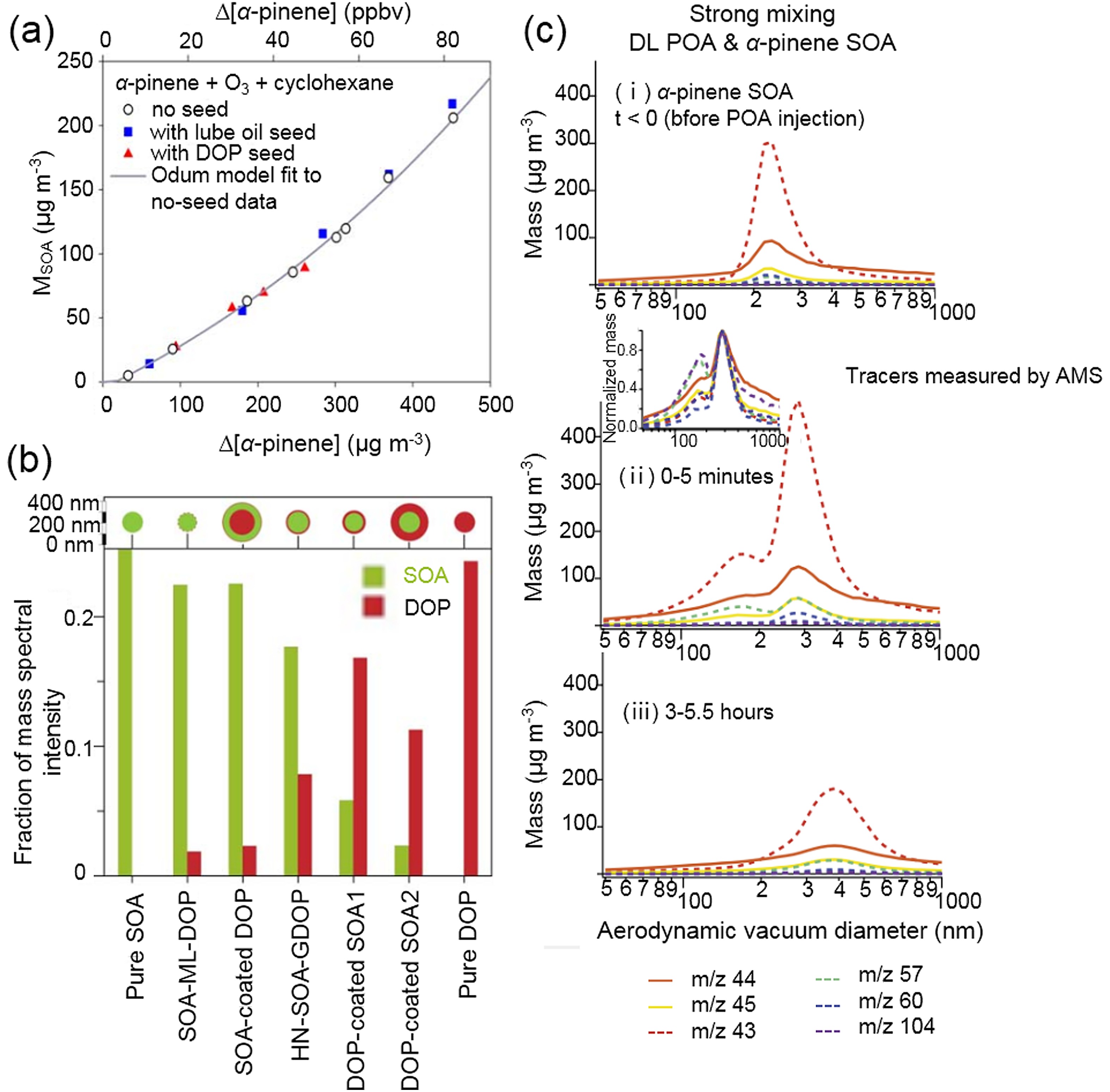 Figure3. The mixing behavior of α-pinene SOA with anthropogenic POA. (a) DOP and lubricating oil seeds exhibited no influence on SOA mass formation from α-pinene ozonolysis [adapted with permission from Song et al. (2007). Copyright 2007 John Wiley & Sons, Inc.]. (b) Relative MS intensity of DOP and SOA for different types of particles under the same laser power. High MS intensity of surface material was observed, indicating the phase-separation between α-pinene and DOP [adapted with permission from Vaden et al. (2010). Copyright 2010 National Academy of Sciences.]. (c) A single-phase mixture formed between DOP seed and α-pinene SOA. POA tracers and SOA tracers measured by aerosol mass spectrometer (AMS) are represented by solid and dashed lines, respectively. m/z 43 signal is prevalent in both SOA and POA [adapted with permission from Asa-Awuku et al. (2009). Copyright 2009 John Wiley & Sons, Inc.].
Figure3. The mixing behavior of α-pinene SOA with anthropogenic POA. (a) DOP and lubricating oil seeds exhibited no influence on SOA mass formation from α-pinene ozonolysis [adapted with permission from Song et al. (2007). Copyright 2007 John Wiley & Sons, Inc.]. (b) Relative MS intensity of DOP and SOA for different types of particles under the same laser power. High MS intensity of surface material was observed, indicating the phase-separation between α-pinene and DOP [adapted with permission from Vaden et al. (2010). Copyright 2010 National Academy of Sciences.]. (c) A single-phase mixture formed between DOP seed and α-pinene SOA. POA tracers and SOA tracers measured by aerosol mass spectrometer (AMS) are represented by solid and dashed lines, respectively. m/z 43 signal is prevalent in both SOA and POA [adapted with permission from Asa-Awuku et al. (2009). Copyright 2009 John Wiley & Sons, Inc.].2
3.2. Particle-phase reactions
Particle-phase reactions, including both heterogeneous and multiphase reactions, are significant in biogenic SOA formation because of their ability to form lower-volatility compounds (Kroll and Seinfeld, 2008). Reactive uptake of gaseous products via accretion reactions, such as hydration, polymerization, esterification, hemiacetal/acetal formation, and aldol condensation are often acid catalyzed (Fig. 4) (Jang et al., 2002; Hallquist et al., 2009; Darer et al., 2011; Ziemann and Atkinson, 2012; Couvidat et al., 2018). Isomerization of highly reactive species in the presence of acidic sulfate particles is also a potential pathway to induce acid-catalyzed enhancement on SOA formation (Fig. 4h) (Lin et al., 2012; Iinuma et al., 2013). Combining field measurements of concentrations of water-soluble ions (K+, Ca2+, Mg2+, NO3?, NH4+, Na+, SO42?, Cl?) and indirect estimation (e.g., thermodynamic equilibrium models and ion balance method), the acidity of ambient particles was determined. For instance, the mean pH values in urban Beijing and rural Gucheng of China were determined to be 5.0 and 5.3, respectively (Hennigan et al., 2015; Shi et al., 2017; Chi et al., 2018). More acidic particles (pH ranges from 0 to 2) were observed in the southeastern US (Guo et al., 2015; Weber et al., 2016). Sources of particle acidity have been resolved to be secondary nitrate and sulfate associated with gaseous NOx, SO2 and NH3, coal combustion, vehicle exhaust, and mineral dust (Weber et al., 2016; Shi et al., 2017). Thus, human activities link to biogenic SOA formation via these particle-phase reactions that are correlated with particle acidity (Surratt et al., 2010; Qin et al., 2018a).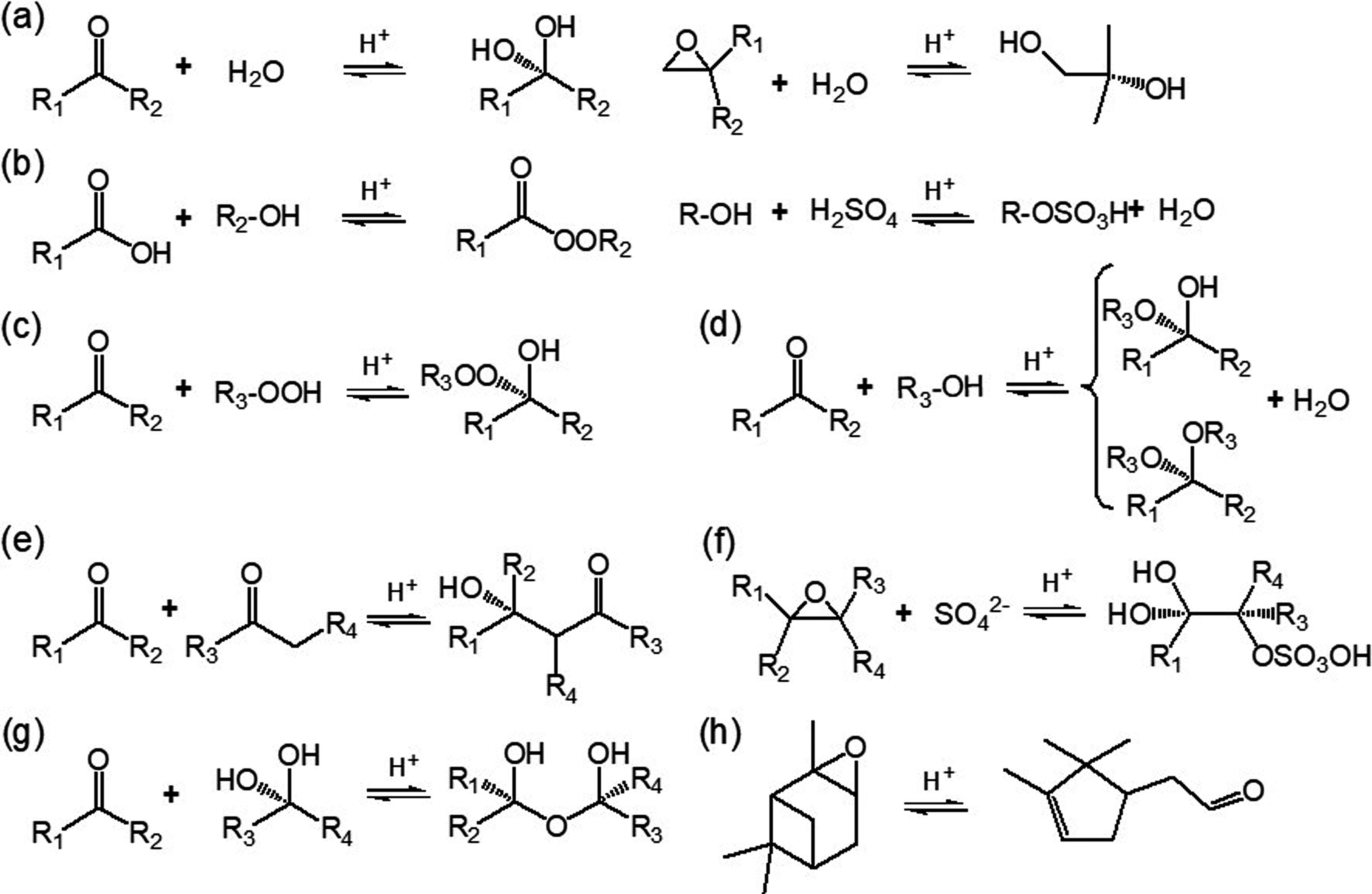 Figure4. Acid-catalyzed particle-phase reactions that might affect the volatility of organics from BVOC oxidation. (a) Hydration reactions of carbonyl and epoxide. (b) Esterification between alcohol and carboxylic acid and/or sulfuric acid. (c) Peroxyhemiacetal formation via the reaction between hydroperoxide and aldehyde. (d) Hemiacetal or acetal formation via the reaction between aldehyde and alcohol. (e) Aldol condensation reaction between two carbonyls. (f) Organosulfates formation via nucleophilic addition reaction. (g) Polymerization. (h) Isomerization. References for these reactions include Kroll and Seinfeld (2008), Hallquist et al. (2009), Darer et al. (2011), Ziemann and Atkinson (2012) and Iinuma et al. (2013).
Figure4. Acid-catalyzed particle-phase reactions that might affect the volatility of organics from BVOC oxidation. (a) Hydration reactions of carbonyl and epoxide. (b) Esterification between alcohol and carboxylic acid and/or sulfuric acid. (c) Peroxyhemiacetal formation via the reaction between hydroperoxide and aldehyde. (d) Hemiacetal or acetal formation via the reaction between aldehyde and alcohol. (e) Aldol condensation reaction between two carbonyls. (f) Organosulfates formation via nucleophilic addition reaction. (g) Polymerization. (h) Isomerization. References for these reactions include Kroll and Seinfeld (2008), Hallquist et al. (2009), Darer et al. (2011), Ziemann and Atkinson (2012) and Iinuma et al. (2013).Numerous laboratory experiments have been conducted with acidic seed particles to investigate the acidity effects on biogenic SOA formation. Improved SOA yields in the presence of acidic seeds have been observed for a series of BVOCs owing to the reactive uptake of the oxidation products (Gao et al., 2004; Iinuma et al., 2004; Offenberg et al., 2009; Han et al., 2016; Riva et al., 2016a), but this dependence is not always the case for all BVOCs because of the varied experimental conditions. Taking α-pinene as an example, while organic carbon from its pure ozonolysis in the presence of sulfuric acid was 40% higher than that of ammonium sulfate (Iinuma et al., 2004), another ozonolysis study under low-NOx conditions found a negligible effect of increasing particle acidity on α-pinene SOA formation (Kristensen et al., 2014). In α-pinene photooxidation, the SOA yield increased almost linearly with particle acidity under high-NOx conditions, which was significantly different from the negligible acidity effect under low-NOx conditions (Han et al., 2016). In another study, it was observed that seed acidity only enhanced particle yields under high-NO conditions but not under high-NO2 conditions, because increased nitric acid and peroxyacyl nitrates in the latter case would make the aerosol acidic enough even in the presence of neutral seeds (Eddingsaas et al., 2012). These inconsistent results suggest that the effect of acidity on biogenic SOA formation might be mediated by other conditions, such as the initial hydrocarbon concentration, oxidant type, and NOx levels. Exploring the effect of acidity on biogenic SOA formation under more relevant atmospheric conditions is needed.
Isoprene, as the most abundant biogenic hydrocarbon and largest SOA source, has gained particular concern (Hallquist et al., 2009). Collectively, acidic seeds enhance isoprene SOA yields from both photooxidation and ozonolysis through acid-catalyzed particle-phase reactions (Jang et al., 2002; Czoschke et al., 2003; Surratt et al., 2007; Zhang et al., 2011; Riva et al., 2016a). This was evidenced by increased 2-methyltetrol, organosulfates, and high molecular weight oligomers with aerosol acidity. Further studies underlined the importance of the reactive uptake of IEPOX, which are formed through isoprene photooxidation under low-NOx conditions (Fig. 2) (Lin et al., 2012; Riva et al., 2016b). The acid-catalyzed nucleophilic addition of sulfate to the epoxide ring of IEPOX contributed substantially to SOA formation (Fig. 4f) (Surratt et al., 2010; Darer et al., 2011; Gaston et al., 2014). Organosulfates formed through this low-NOx channel accounted for ~97%, ~55% and 62%–83% of SOA mass in the Amazon, the southeastern US, and southwestern China, respectively (Qin et al., 2018a; Yee et al., 2020). It has been shown that the reaction probability of IEPOX on ammonium bisulfate is more than 500 times greater than on ammonium sulfate, and low NOx isoprene SOA yields increased from 1.3% in the presence of neutral particle to 28.6% in the presence of acidic particles (Surratt et al., 2010; Gaston et al., 2014). However, the reactive uptake of IEPOX was also observed to increase the OA mass when base hydrated ammonium sulfate was used (Nguyen et al., 2014). Hydrated seed particles here promoted not only the dissolution of water-soluble compounds but also hydrolysis reactions in the aqueous phase.
The weak correlations between particle acidity and IEPOX-derived SOA here are somewhat consistent with field observations (Budisulistiorini et al., 2013, 2015; Worton et al., 2013; Xu et al., 2015b). For example, in southwestern and eastern China and the southeastern US, isoprene SOA was found to be more strongly correlated with sulfate than with particle acidity or water mass concentration, partially because the surface area provided by sulfate particles promoted IEPOX reactive uptake and sulfate as a nucleophile and/or the salting-in effect accelerated the ring-opening reactions of IEPOX (Xu et al., 2015b; Rattanavaraha et al., 2016; Zhang et al., 2017c; Qin et al., 2018a). The insignificant correlation of IEPOX SOA with pH was ascribed to the small pH range and the regional transportation–caused gap between the calculated and real particle pH at the time/site where acidity-dependent chemistry occurred (Yee et al., 2020). With a more detailed interpretation of the observed relationship between isoprene SOA tracers and pH, the particle acidity was found to negatively correlated with the ratio of 2-methyltetrols to C5-alkene triols (IEPOX pathway in Fig. 2), indicating that the formation of C5-alkene triols was favored with increasing particle acidity (Yee et al., 2020). However, it might be easy to misinterpret the effect of particle acidity and water on isoprene SOA formation because they are driven by sulfate (Xu et al., 2015b). The much more complex atmospheric environments than experimental conditions may partially lead to the gap between these two kinds of studies. The difference in particle acidity in laboratory experiments and the real atmosphere, as well as the accurate manner and degree to which relative humidity (RH) and the liquid water content, seed particle composition, and acidity influence isoprene and other BVOC-derived SOA formation remain elusive and are deserving of further systemic exploration under more atmospherically relevant conditions (Lin et al., 2013a; Budisulistiorini et al., 2015; Riva et al., 2016b; Faust et al., 2017; Stirnweis et al., 2017).
Dicarboxylic acids (DCAs), predominantly oxalic acid, are major components of atmospheric OAs. They have gained considerable attention in recent years owing to their contribution to OA budgets via SOA formation and the potential impacts on climate via changing the solar radiation and acting as cloud condensation nuclei (Bikkina et al., 2014; Kawamura and Bikkina, 2016). While their emissions from primary sources such as biomass burning and vehicle exhausts, cooking and natural marine sources are relatively low, those in the atmosphere originate largely from the photochemistry of biogenic unsaturated fatty acids and VOCs such as isoprene and intermediates (Lim et al., 2005; Carlton et al., 2007; Ervens et al., 2011). These processes are discussed to help understand how anthropogenic factors would influence DCA SOA formation.
Unsaturated fatty acids such as oleic acid are abundant in marine phytoplankton and terrestrial higher-plant leaves (Ho et al., 2010). For SOA formation from unsaturated fatty acids, the ozonolysis of oleic acid particles under dry conditions showed a pronounced mass loss of oleic acid particles due to the evaporation of volatile oxidation products such as nonanal (Lee et al., 2012). However, azelaic acid in the particulate phase can further generate low molecular weight DCAs such as oxalic acid, which is a major class of SOA. In marine regions, azelaic acid and DCA concentrations were found to be higher in more biologically influenced aerosols than in less biologically influenced ones, suggesting the contribution of biogenic unsaturated fatty acids to DCA formation (Bikkina et al., 2014).
The ability of isoprene as a precursor of DCAs, especially oxalic acid, has been evidenced in various regions. In marine regions, isoprene was proposed to be one source of DCAs through aqueous-phase reactions (Bikkina et al., 2014, 2015). Pyruvic and glyoxylic acids and methylglyoxal in the aerosol phase are key precursors for the final formation of oxalic acid. In continental regions, oxalic acid and glyoxylic acid derived from glyoxal and methylglyoxal (important isoprene oxidation products) oxidation were observed to have a robust linear correlation and both acids showed close correlation with sulfate, indicating that oxalic acid may be largely produced by aqueous-phase oxidation of glyoxylic acid in aerosols (Yu et al., 2005; Fu et al., 2008; Wang et al., 2012). More recently, a field observation in Xi’an, China, focusing on the formation mechanism of SOA on dust surfaces, investigated the concentrations and compositions of DCAs during the dust storm episodes (Wang et al., 2015). According to the strong correlation of oxalic acid with NO3?, Ca(NO3)2, which strongly absorbs water vapor, was proposed to be produced via the heterogeneous reaction of nitric acid and/or NOx with dust (Wang et al., 2015). Gas-phase water-soluble organic precursors (e.g., glyoxal and methylglyoxal) that partitioned into the aqueous phase on the surface of dust aerosols can be subsequently oxidized into oxalic acid and thus contribute to SOA formation. It seems that liquid water in particles favors organic acid formation (Lim et al., 2005). However, no correlation between oxalic acid concentration and particle liquid water content was observed in aerosols collected from Huashan Mountain in central China and the western North Pacific (Meng et al., 2014; Bikkina et al., 2015). At Huashan Mountain, the oxalic acid concentration was observed to instead correlate with particle acidity. An acidic condition was suggested to be favorable for oxalic acid formation from isoprene and monoterpene oxidation products in the aqueous phase (Meng et al., 2014). Based on these results, it is therefore speculated that anthropogenic species like sulfate and nitrate, which would influence the particle liquid water content and acidity in particles, may affect the fate of intermediates from isoprene/monoterpenes/unsaturated fatty acids oxidation and thus the formation of DCAs in SOA (Kawamura and Bikkina, 2016). Considering the wide distribution of DCAs in aerosols and their effects on climate, such anthropogenic–biogenic interaction needs further exploration.
2
4.1. SO2 effects on BVOC photooxidation
The primary sink of SO2 in the atmosphere is the reaction with ·OH, forming HSO3 (R1) that can further react with O2 to produce SO3 and HO2· (R2). The reaction between SO3 and water vapor ultimately gives H2SO4 (R3):Though numerous chamber experiments have shown enhanced SOA formation with particle acidity, as described in section 3.2, the effect of SO2 on SOA formation is not only limited to H2SO4 formation and corresponding acid-catalyzed reactions but also to its perturbation on the radical fate in the chamber. When the addition of SO2 was disabled to change the radical level and thus gas chemistry, the generation of H2SO4 increased linearly with initial SO2 concentrations (Kleindienst et al., 2006). The enhanced SOA yield from isoprene and α-pinene photooxidation in the presence of SO2 can be attributed to the acid-catalyzed reactions involving carbonyl compounds. Similarly, when excluding gas-phase chemistry during the photooxidation of limonene and α-pinene, the presence of SO2 increased the SOA yield for these two hydrocarbons under both low and high NOx conditions (Zhao et al., 2018b). This was primarily because NPF induced by SO2 oxidation could act as seeds to provide more surface and volume for the condensation of product vapors, though the effect of particle acidity may also exist. However, H2SO4-induced enhancement on SOA formation is not always the case for all VOC precursors. For example, in the photooxidation of cyclohexene under atmospheric relevant conditions, SO2 was observed to suppress the SOA yield (Liu et al., 2017). Despite the oxidation of SO2 by ·OH forming H2SO4 that can exert an enhancing effect on SOA formation, this effect is insufficient to compensate the simultaneously reduced ·OH reactivity towards cyclohexene so that the net SO2 effect is to weaken SOA formation. Another study focusing on the effects of SO2 on the α- and β-pinene photooxidation proposed that the presence of SO2 leads to enhanced products with a lesser degree of oxygenation but the increased RH dampens this enhancement (Friedman et al., 2016). Here, the SO2-induced change in the ·OH/HO2· ratio and/or SO3 reacting directly with organic molecules were suggested to be responsible for the SO2 perturbations. These results indicate that, altogether, the perturbation of SO2 on both particle- and gas-phase reactions determine the extent to which SO2 influences SOA formation. More BVOC photooxidation processes under atmospheric-related conditions deserve continued focus.
2
4.2. SO2 effects on BVOC ozonolysis
Besides reacting with ·OH, another important way for the transformation of SO2 to H2SO4 is by reacting with the stabilized Criegee Intermediates (sCIs) that are formed during alkene ozonolysis (Mauldin III et al., 2012; Boy et al., 2013; Sipila et al., 2014). The reaction between SO2 and sCIs forms SO3, which further reacts efficiently with water to produce H2SO4, as shown in Fig. 5. This non-·OH SO2 oxidation pathway is potentially responsible for the missing H2SO4 source in both boreal forest and coastal sites (Mauldin III et al., 2012; Berresheim et al., 2014). Modeling results showed that SO2 oxidation by sCIs from monoterpenes ozonolysis accounted for about 60% of the gas-phase SO2 removal in tropical forest regions (Newland et al., 2018).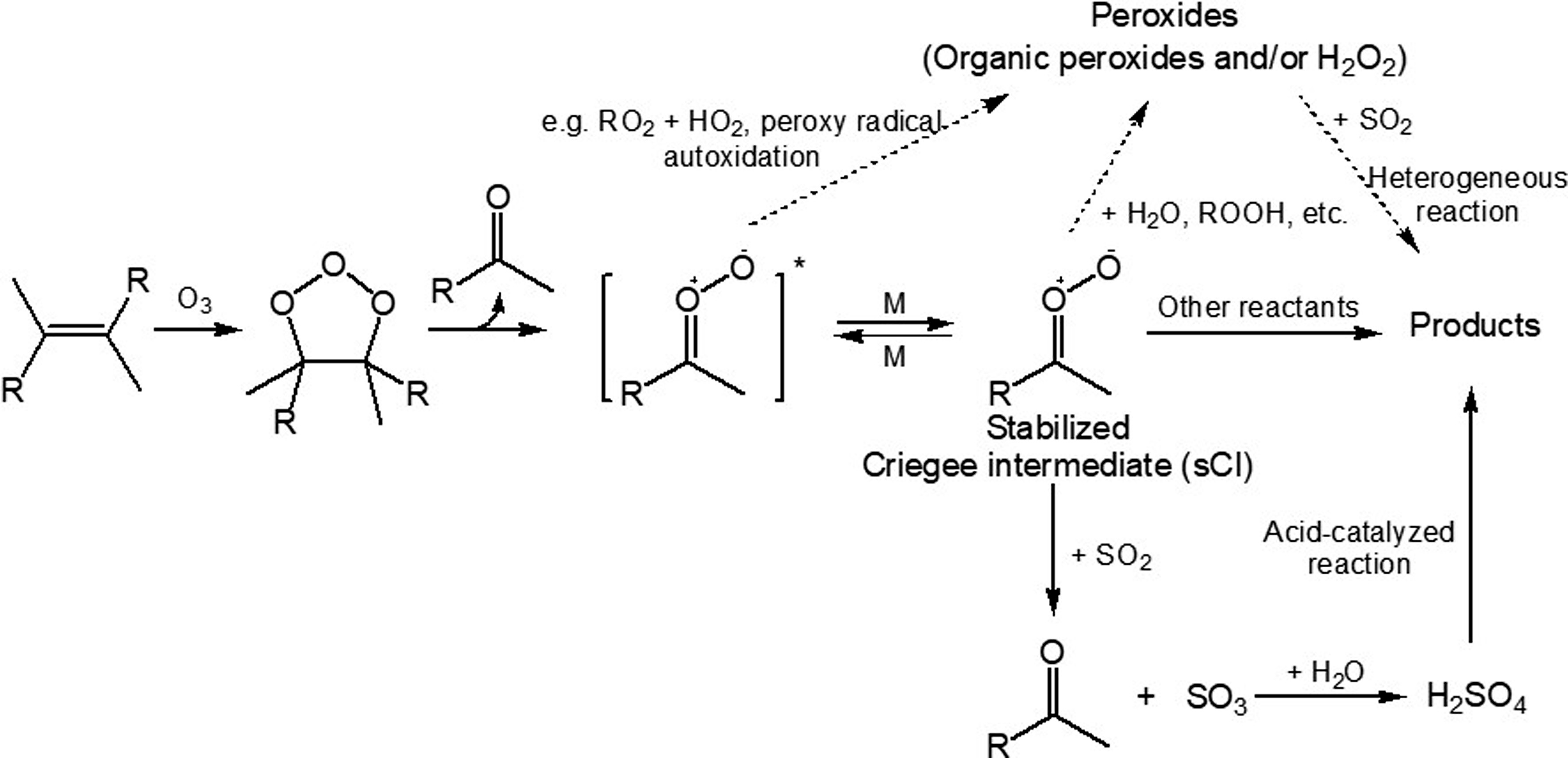 Figure5. SO2 effects on the formation of SOA from monoterpene ozonolysis: sCIs + SO2, sCIs + H2O, and SO2 + peroxides reactions [adapted with permission from Ye et al. (2018)].
Figure5. SO2 effects on the formation of SOA from monoterpene ozonolysis: sCIs + SO2, sCIs + H2O, and SO2 + peroxides reactions [adapted with permission from Ye et al. (2018)].sCIs are key precursors to the formation of condensable species (Mackenzie-Rae et al., 2018), such as carboxylic acid formed from sCIs isomerization, α-acyloxyalkyl hydroperoxides formed from carboxylic acids + sCIs reactions and secondary ozonides formed from carbonyl + sCIs reactions (Sipila et al., 2014; Chhantyal-Pun et al., 2018; Zhao et al., 2019). SO2 may influence SOA formation by altering sCIs chemistry and H2SO4-related enhancement effects (Sipila et al., 2014). For example, it was found that SOA formation from limonene ozonolysis was enhanced by the presence of SO2, regardless of dry (RH < 16%) or humid (RH = ~ 50%) conditions (Ye et al., 2018). Under dry conditions, the formation and condensation of H2SO4 from the SO2 + sCIs reaction and further acid-catalyzed reactions (Fig. 5) were expected for the enhanced SOA yields. The composition analysis showed reduced oligomers but enhanced organosulfates and oxidation state, suggesting that the H2SO4-related enhancement outweighed the reduction of condensable species directly from sCI reactions. However, under humid conditions, the dominant SO2 sink was proposed to be its heterogeneous reaction with condensed-phase organic peroxides. A similar pathway for the transformation of SO2 to organosulfates was also characterized in the case of α-pinene, although SO2 exhibited a minor effect on the SOA yield, likely because the enhanced functionalization was offset by reduced oligomerization (Wang et al., 2019). In addition, SO2 also influences NPF during BVOC ozonolysis. In the absence of SO2, NPF was not observed in the ozonolysis of isoprene, α-pinene, β-pinene, and limonene under dry conditions. However, with the addition of SO2, NPF emerged and the amount of nucleation was correlated with the sCI yield (Stangl et al., 2019).
Water vapor is a potential competitor to SO2 for sCIs to change the overall effect of SO2 on gas- and particle-phase reactions (Fig. 5), such as the masked SO2 enhancing effect on the SOA yield from butyl vinyl ether ozonolysis when RH > 40% (Huang et al., 2015; Zhang et al., 2019a). This competition, however, highly depends on the structure of the hydrocarbon itself (Vereecken et al., 2015). For monoterpene-derived sCIs, their reactions with SO2 are nearly independent of RH, implying minor competitiveness of water than SO2 even under high RH conditions (Sipila et al., 2014). Nevertheless, observations for α-pinene and limonene ozonolysis still showed smaller enhancement on particle volume concentration under humid (RH = ~50%) rather than dry (RH = ~10%) conditions (Ye et al., 2018). Besides the suppressed formation of high molecular weight species and organosulfates caused by water uptake and thus diluted particle acidity, a novel way for SO2 to form organosulfates was proposed to be its heterogeneous reaction with organic (hydro-) peroxides (Fig. 5). It should be noted that this mechanism is still linked to uncertainties and needs continued focus. Besides, the effects of SO2 on SOA formation from the ozonolysis of other BVOCs such as isoprene and sesquiterpenes are still scarcely studied and warrant more attention to better evaluate SO2-involved anthropogenic–biogenic interactions at night.
2
5.1. NH3 effects on SOA formation
The potential role of NH3 in SOA formation was first investigated in the styrene ozonolysis system (Na et al., 2006). The addition of excessive NH3 into the chamber where SOA formation had ceased resulted in the decreased aerosol volume concentration, which was attributed to the rapid decomposition of the main SOA-forming species (3,5-diphenyl-1,2,4-trioxolane and the hydroxyl-substituted ester) caused by the nucleophilic attack of NH3. When styrene ozonolysis was further studied in the presence of NH3, the SOA yield was significantly reduced (Ma et al., 2018). Quantum chemical calculations revealed that the reaction between NH3 and sCIs suppressed the formation of condensable secondary ozonide (3,5-diphenyl-1,2,4-trioxolane) that was formed via the sCIs + aldehyde reaction. Different from styrene, NH3 exhibited an enhancement effect on the particle growth and SOA yield from α-pinene ozonolysis regardless of whether NH3 was added at the beginning or at the end of the reaction (Na et al., 2007; Babar et al., 2017). Ammonium salts generated via the gas-phase reaction between NH3 and organic acids (such as pinic acid and pinonic acid) nucleated and contributed to the increased SOA formation. In the photooxidation of α-pinene in the presence of NH3, particle-phase ammonium correlated well with organic mono- and di-carboxylic acids in the gas-phase, highlighting the central role of ammonium salts formed via acid–base reactions between NH3 and organic acids in SOA formation (Hao et al., 2020). Similarly, the ozonolysis of limonene and mixed BVOCs (emitted from cleaning products) in the presence of NH3 yielded 60% and 35% higher maximum total particle number concentrations than those in the absence of NH3, respectively (Huang et al., 2012; Niu et al., 2017). Both nuclei coagulation and condensation caused by acid–base reactions were responsible for the SOA growth. However, for SOA from isoprene ozonolysis, its reaction with NH3 did not significantly change particle number and volume concentrations, suggesting that not all gas-phase organic acids (e.g., 2-methylglyceric acid, pyruvic acid) could experience gas-to-particle conversion through acid–base reactions (Na et al., 2007).2
5.2. NH3 effects on SOA aging
Heterogeneous uptake of NH3 by SOA is an important way to complex SOA composition and optical properties by forming N-containing organic compounds (NOC). NOC are regarded as a significant class of heteroatom-containing brown carbon (BrC) compounds that absorb light with a strong wavelength dependence [Liu et al. (2015) and reference therein]. SOA from limonene ozonolysis was the first biogenic SOA that had been found to turn to being more light-absorbing when an aqueous extract of SOA was aged by ammonium ions (Bones et al., 2010). The key aging reactions involve firstly the acid-catalyzed transformation of carbonyls to primary imines (Fig. 6a). Particularly, imines formed via the reaction between NH4+ and 1,5-dicarbonyl compounds from limonene SOA may undergo cyclization to give the dihydropyridinium ion (Fig. 6b). The combined product of two dihydropyridinium ions further disproportionates, finally leading to conjugated NOC that are responsible for the enhanced light absorption. A similar NH3 effect on the light absorption of SOA has been further observed when exposing NH3 directly to SOA from the photooxidation or ozonolysis of various biogenic as well as anthropogenic VOCs (Laskin et al., 2010; Updyke et al., 2012; Lee et al., 2013; Babar et al., 2017). The light absorption of aged SOA from ozonolysis was generally stronger than that from ·OH oxidation, confirming the role of the carbonyl + NH3 reaction in NOC formation as alkene ozonolysis yields more carbonyl than ·OH-initiated oxidation (Updyke et al., 2012). Besides the heterocyclic NOC formation through the intramolecular cyclization of the primary imine, the reaction between primary imines with another carbonyl that leads to a more stable secondary imine (Schiff base formation) (Fig. 6c) and the 1,2-dicarbonyls + aldehydes reaction in the presence of NH3 that gives imidazoles (Fig. 6d) are likely to induce light-absorbing products in aged SOA (Laskin et al., 2010; Updyke et al., 2012; Laskin et al., 2014). Very recently, uptake coefficients of NH3 onto SOA from α-pinene ozonolysis or m-xylene ·OH-oxidation were observed to be positively correlated with the acidity of aerosol and negatively correlated with the concentration of NH3, kinetically confirming that NOCs were formed via the heterogeneous reaction of NH3 with SOA (Liu et al., 2015). It should be noted that some BrC formed via the mechanism discussed above may be unstable towards sunlight or oxidants, but this needs further exploration (Sareen et al., 2013; Lee et al., 2014). Regardless, considering the increasing trend of NH3 emissions, NH3 is of great significance to mediate the components and physical properties of biogenic SOA. Hence, more relevant studies are warranted. Figure6. Aging pathways of biogenic SOA by NH3. (a) Acid-catalyzed reaction of carbonyls with NH3 that results in the formation of primary imines [Moise et al. (2015) and reference therein]. (b) The reaction between NH3 and 1,5-dicarbonyl compounds: the primary imine can further react with the second carbonyl group present in the same molecule through nucleophilic addition, resulting in nitrogen-containing heterocyclic compounds [Moise et al. (2015) and reference therein]. (c) Reactions between the primary imine with another carbonyl group, leading to a more stable secondary imine (Schiff base) [Moise et al. (2015) and reference therein]. (d) The reaction between NH3 and 1,2-dicarbonyls through a Debus reaction, yielding substituted imidazoles (Updyke et al., 2012).
Figure6. Aging pathways of biogenic SOA by NH3. (a) Acid-catalyzed reaction of carbonyls with NH3 that results in the formation of primary imines [Moise et al. (2015) and reference therein]. (b) The reaction between NH3 and 1,5-dicarbonyl compounds: the primary imine can further react with the second carbonyl group present in the same molecule through nucleophilic addition, resulting in nitrogen-containing heterocyclic compounds [Moise et al. (2015) and reference therein]. (c) Reactions between the primary imine with another carbonyl group, leading to a more stable secondary imine (Schiff base) [Moise et al. (2015) and reference therein]. (d) The reaction between NH3 and 1,2-dicarbonyls through a Debus reaction, yielding substituted imidazoles (Updyke et al., 2012).2
5.3. Amine-involved particle-phase reactions
As derivatives of NH3, amines have been observed in both gas and particle phases (Ge et al., 2011). Although they can participate in SOA formation via various pathways, here we only focus on those likely occurring during biogenic SOA formation. Amine-epoxide reactions were proposed to be kinetically feasible for isoprene-derived epoxides and high amine SOA concentrations (Stropoli and Elrod, 2015). However, it should be noted that such reactions can only be favored when the pH values of the reaction environment are higher than the pKa (Ka is the acid dissociation constant) values of particular amines. The prevalent acidic SOA in the atmosphere may not be conducive to such reactions.Similar to NH3, amines could also engage in the heterogeneous reactions with carbonyls to form imine/enamine compounds (Zhang et al., 2015b). The particle-phase reaction between methylamine and glyoxal that is mainly derived from biogenic sources showed that glyoxal could irreversibly trap amines in the aerosol phase and convert them into oligomers (De Haan et al., 2009). SOAs formed through this pathway were estimated to be up to 11 Tg yr?1 globally if glyoxal was consumed exclusively in this path. To explain the formation of high molecular weight NOC observed in ambient aerosols, the Mannich reaction among amines (or NH3), aldehydes, and carbonyls with an adjacent, acidic proton was proposed (Wang et al., 2010).
Acid–base reactions are another class of amine-involved reactions of interest. The heterogeneous uptake of methylamine, dimethylamine, and trimethylamine onto citric acid and humic acid confirmed acid–base reactions between amines and carboxylic acids (Liu et al., 2012). The aminium salts formed would enhance the water uptake of particles and thus alter the particle properties. Based on the equilibrium partitioning of dimethylamine, NH3, acetic acid, pinic acid and their salts, amines were suggested to contribute significantly to the formation of organic salts that might have a potential contribution to new particle growth (Barsanti et al., 2009). Theoretical calculations for the thermodynamics of accretion reactions between organic acids (malic, maleic, and pinic acids) and amines showed that such interactions could contribute to SOA formation via the kinetically favored formation of ester and amide (Barsanti and Pankow, 2006). Additionally, NPF in a flow tube was also considerably enhanced when amines reacted with methanesulfonic acid in the presence of water (Dawson et al., 2012). Considering that epoxides, carbonyls and organic acids are important BVOC oxidation products, it is plausible that the reactions between amines and epoxides/carbonyls/acids from BVOC oxidation may influence biogenic SOA formation, but current studies on this process are still limited and thus need further attention.
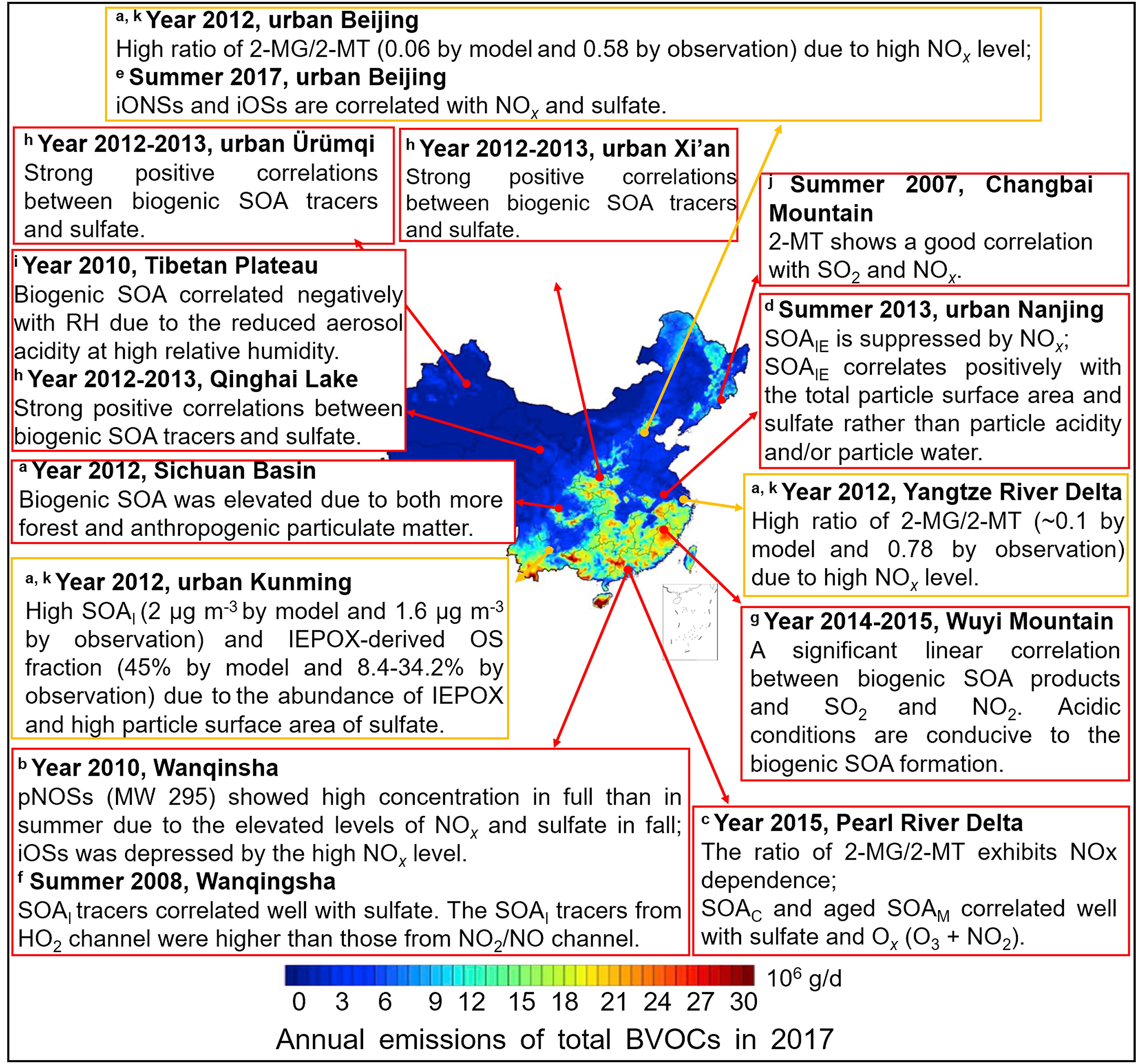 Figure7. Anthropogenic-biogenic interactions in China. The color-mapped annual emissions of total BVOCs in China, 2017, are adapted with permission from Wu et al. (2020). Copyright 2020 Elsevier. The observed correlations between anthropogenic pollutants and biogenic SOA are shown in red boxes and the modeled results are shown in yellow boxes. The pONSs, iONSs, iOSs, SOAI, SOAIE, SOAM, and SOAC refer to pinene-derived nitrooxyorganosulfates, isoprene-derived nitrooxyorganosulfates, isoprene-derived organosulfates, isoprene-derived SOA, IEPOX-derived SOA, monoterpene-derived SOA and β-caryophyllene-derived SOA, respectively; 2-MG and 2-MT are 2-methylglyceric acid and 2-methyltetrols derived from isoprene oxidation under high- and low-NOx conditions, respectively. a The modeled anthropogenic–biogenic interactions are taken from Qin et al. (2018). b–j The field-observed anthropogenic–biogenic interactions are taken from He et al. (2014), Zhang et al. (2019b), Zhang et al. (2017), Bryant et al. (2020), He et al. (2018), Ren et al. (2019), Ren et al. (2018), Li et al. (2013), and Wang et al. (2008), respectively.
Figure7. Anthropogenic-biogenic interactions in China. The color-mapped annual emissions of total BVOCs in China, 2017, are adapted with permission from Wu et al. (2020). Copyright 2020 Elsevier. The observed correlations between anthropogenic pollutants and biogenic SOA are shown in red boxes and the modeled results are shown in yellow boxes. The pONSs, iONSs, iOSs, SOAI, SOAIE, SOAM, and SOAC refer to pinene-derived nitrooxyorganosulfates, isoprene-derived nitrooxyorganosulfates, isoprene-derived organosulfates, isoprene-derived SOA, IEPOX-derived SOA, monoterpene-derived SOA and β-caryophyllene-derived SOA, respectively; 2-MG and 2-MT are 2-methylglyceric acid and 2-methyltetrols derived from isoprene oxidation under high- and low-NOx conditions, respectively. a The modeled anthropogenic–biogenic interactions are taken from Qin et al. (2018). b–j The field-observed anthropogenic–biogenic interactions are taken from He et al. (2014), Zhang et al. (2019b), Zhang et al. (2017), Bryant et al. (2020), He et al. (2018), Ren et al. (2019), Ren et al. (2018), Li et al. (2013), and Wang et al. (2008), respectively.| Location | Period | T (°C) | RH (%) | SO2 a (μg m?3) | NOx a (μg m?3) | NO3? a (μg m?3) | SO42? a (μg m?3) | NH4+ a (μg m?3) | SOAI Tracers b | ∑SOAI c (ng m?3) | SOAM Tracers d | ∑SOAM e (ng m?3) | LWC f (μg m?3) | pHg | References |
| Guangzhou (urban) | Year 2015 | 24.0 | 58 | 15.1 | 76.8 | 3.2 | 8.4 | 4.0 | 2-MT, 2-MG, C5-alkene triols, and 3-MeTHF-3,4-diols | 22.6 | cis-pinonic acid, pinic acid, 3-HGA, HDMGA, MBTCA | 50.0 | ? | ? | (Zhang et al., 2019b) |
| Zhaoqing (urban) | Year 2015 | 22.7 | 59 | 25.5 | 40.3 | 4.2 | 10.0 | 5.0 | 2-MT, 2-MG, C5-alkene triols, and 3-MeTHF-3,4-diols | 49.3 | cis-pinonic acid, pinic acid, 3-HGA, HDMGA, MBTCA | 54.3 | ? | ? | (Zhang et al., 2019b) |
| Dongguan (urban) | Year 2015 | 24.9 | 61 | 16.2 | 49.0 | 2.9 | 8.5 | 3.4 | 2-MT, 2-MG, C5-alkene triols, and 3-MeTHF-3,4-diols | 16.0 | cis-pinonic acid, pinic acid, 3-HGA, HDMGA, MBTCA | 50.9 | ? | ? | (Zhang et al., 2019b) |
| Nansha (sub-urban) | Year 2015 | 25.6 | 67 | 14.4 | 38.3 | 1.8 | 8.3 | 3.7 | 2-MT, 2-MG, C5-alkene triols, and 3-MeTHF-3,4-diols | 17.0 | cis-pinonic acid, pinic acid, 3-HGA, HDMGA, MBTCA | 26.5 | ? | ? | (Zhang et al., 2019b) |
| Zhuhai (suburban) | Year 2015 | 24.2 | 74 | 7.3 | 57.4 | 1.4 | 8.5 | 3.3 | 2-MT, 2-MG, C5-alkene triols, and 3-MeTHF-3,4-diols | 10.8 | cis-pinonic acid, pinic acid, 3-HGA, HDMGA, MBTCA | 40.3 | ? | ? | (Zhang et al., 2019b) |
| Nanjing (urban) | Summer 2013 | 32.4 | 59.7 | 128 i | 39.3 h | ? | 17 | ? | 2-MT, 2-MG, and C5-alkene triols | 0.3 | ? | ? | 70 | 2.6 | (Zhang et al., 2017c) |
| Beijing (urban) | Summer 2017 | 16–38 | ? | ? | 225 i | ? | ? | ? | 2-MT, 2-MT OSs, 2-MG, 2-MG OSs, glycolic acid sulfate, hydroxyacetone sulfate, lactic acid sulfate, cyclic, and 9 NOSs | 107 | ? | ? | ? | ? | (Bryant et al., 2020) |
| Wanqingsha (forest) | Summer 2010 | 29.6 | 79.7 | 29.4 | 42.4 h | 2.8 | 9.1 | 3.1 | 2-MT sulfate ester, 2-MG sulfate ester | 0.68 | cis-pinonic acid, pinic acid, 3-HGA, HDMGA, MBTCA, NOSs (three isomers of MW 295) | 75.9 | ? | ? | (He et al., 2014) |
| Fall 2010 | 21.6 | 69.1 | 45.1 | 37.5 | 10.4 | 18.6 | 8.8 | 0.66 | 205.4 | ? | ? | ||||
| Wanqingsha (forest) | Summer 2008 | 29.0 | 66 | ? | ? | 5.3 | 23.0 | 4.9 | 3-MeTHF-3,4-diols, 2-MT, C5-alkene triols, 2-MT sulfate ester, 2-MG, 2-MG sulfate ester | 130.1 | ? | ? | 24.5 | 0.5 | (He et al., 2018) |
| Fall 2008 | 22.6 | 47 | ? | ? | 8.9 | 15.9 | 5.3 | 26.7 | ? | 11.8 | 2.8 | ||||
| Wuyi Mountain | Spring 2014 | 16 | 78 | 1.7 | 4.2 h | ? | ? | ? | 3-MeTHF-3,4-diols, C5-alkene triols, 2-MT, 2-MG | 6.6 | cis-pinic acid, cis-pinonic acid, 3-HGA, MBTCA | 26 | 9.7 | 0.2 | (Ren et al., 2019) |
| Summer 2014 | 23 | 79 | 0.9 | 1.7 h | ? | ? | ? | 21 | 36 | 7.4 | 0.1 | ||||
| Autumn 2014 | 17 | 75 | 3.1 | 4 | ? | ? | ? | 16 | 36 | 10.8 | 0.7 | ||||
| Winter 2014 | 6.4 | 64 | 6.7 | 6.2 | ? | ? | ? | 3 | 20 | 7.2 | 1.6 | ||||
| Qinghai Lake | Summer 2012 | 11 | 59 | — | ? | 0.4 | 2.2 | 0.4 | 3-MeTHF-3,4-diols, C5-alkene triols, 2-MT | 3.8 | cis-pinic acid, cis-pinonic acid, 3-HGA, MBTCA | 16 | ? | ? | (Ren et al., 2018) |
| Winter 2012 | ?9 | 26 | ? | ? | 0.8 | 2.2 | 0.1 | 0.6 | 1.3 | ? | ? | ||||
| ürümqi (urban) | Summer 2012 | 26 | 46 | ? | ? | 3.4 | 6.4 | 0.4 | 3-MeTHF-3,4-diols, C5-alkene triols, 2-MT | 10 | cis-pinic acid, cis-pinonic acid, 3-HGA, MBTCA | 44 | ? | ? | (Ren et al., 2018) |
| Winter 2012 | ?14 | 78 | ? | ? | 19 | 65 | 21 | 1.9 | 6.6 | ? | ? | ||||
| Xi'an (urban) | Summer 2012 | 24 | 78 | ? | ? | 8.8 | 15 | 4.3 | 3-MeTHF-3,4-diols, C5-alkene triols, 2-MT | 20 | cis-pinic acid, cis-pinonic acid, 3-HGA, MBTCA | 58 | ? | ? | (Ren et al., 2018) |
| Winter 2012 | 1 | 66 | ? | ? | 26 | 36 | 13 | 2.1 | 22 | ? | ? | ||||
| Shanghai (urban) | Summer 2012 | 28 | 78 | ? | ? | 4.2 | 7.2 | 1.3 | 3-MeTHF-3,4-diols, C5-alkene triols, 2-MT | 5.1 | cis-pinic acid, cis-pinonic acid, 3-HGA, MBTCA | 20 | ? | ? | (Ren et al., 2018) |
| Winter 2012 | 6 | 70 | ? | ? | 16 | 16 | 6.1 | 2.5 | 16 | ? | ? | ||||
| Chengdu (urban) | Summer 2012 | 25 | 81 | ? | ? | 6.5 | 14 | 3.4 | 3-MeTHF-3,4-diols, C5-alkene triols, 2-MT | 23 | cis-pinic acid, cis-pinonic acid, 3-HGA, MBTCA | 88 | ? | ? | (Ren et al., 2018) |
| Winter 2012 | 10 | 74 | ? | ? | 26 | 32 | 12 | 5.9 | 17 | ? | ? | ||||
| Guangzhou (urban) | Summer 2012 | 29 | 79 | ? | ? | 3.3 | 6.2 | 0.8 | 3-MeTHF-3,4-diols, C5-alkene triols, 2-MT | 10 | cis-pinic acid, cis-pinonic acid, 3-HGA, MBTCA | 43 | ? | ? | (Ren et al., 2018) |
| Winter 2012 | 17 | 73 | ? | ? | 12 | 15 | 5.1 | 6 | 46 | ? | ? | ||||
| Tibetan Plateau (Qinghai Lake) | Summer 2010 | 14.4 | 64.4 | ? | ? | 0.8 | 3.9 | 0.6 | C5-alkene triols, 2-MG, 2-MT | 2.5 | norpinic acid, pinonic acid, pinic acid, 3-HGA, MBTCA | 3.0 | 5.8 | ?1.2 | (Li et al., 2013) |
| Changbai Mountain | Summer 2007 | 25 | 59 | 5.35 | 1.3 | ? | ? | ? | 2-MT, 2-MG, C5-alkene triols | 53 | pinic acid, norpinic acid, 3-HGA, MBTCA | 31 | ? | ? | (Wang et al., 2008) |
| Chongming Island | Summer 2006 | 29 | 68 | 25.9 | 40.9 j | ? | ? | ? | 2-MT, 2-MG, C5-alkene triols | 4.8 | pinic acid, norpinic acid, 3-HGA, MBTCA | 1.8 | ? | ? | (Wang et al., 2008) |
| Notes: a The mean concentration of tracers; b Isoprene-derived SOA (SOAI) tracers: 2-MG (2-methylglyceric acid), 2-MT (2-methyltetrols that represent the sum of 2-methylthreitol and 2-methylerythritol), 3-MeTHF-3,4-diols (the sum of trans-3-methyltetrahydrofuran-3,4-diol and cis-3-methyltetrahydrofuran-3,4-diol), C5-alkene triols (the sum of cis-2-methyl-1,3,4-trihydoxy-1-butane, trans-2-methyl-1,3,4-trihydoxy-1-butane, and 3-methyl-2,3,4-trihydoxy-1-butane), OSs (organosulfates), NOSs (nitrooxy organosulfates); c The sum of SOAI tracers; d Monoterpene-derived SOA (SOAM) tracers: 3-HGA (3-hydroxyglutaric acid), HDMGA (3-Hydroxy-4,4-dimethylglutaric acid), MBTCA (3-methyl-1,2,3-butanetricarboxylic acid); e The sum of SOAM tracers; f Aerosol liquid water content; g AIM-derived in situ pH of the aqueous phase on aerosols; h The concentration of NO2; i The max concentration. | |||||||||||||||
Table3. Summary of gaseous and particulate species in different regions with anthropogenic–biogenic interactions in China.
Biogenic organosulfates in ambient particles, which are formed through the cross-reaction between BVOCs and anthropogenic pollutants, are important markers of anthropogenic–biogenic interactions. Quantification of organosulfates in fine particle samples collected in the central Pearl River Delta in 2010 showed nearly three times higher pinene-derived nitrooxyorganosulfates (MW = 295) in fall than in summer, probably due to the higher levels of sulfates and NOx in fall (He et al., 2014). 2-Methyltetrol sulfate ester produced via isoprene-derived IEPOX oxidation under low-NOx conditions showed low concentrations. The high NOx mixing ratio (daily 65 ppb and hourly 163 ppb) here could be the reason why IEPOX formation was suppressed. Other observations in this region also showed the Ox and sulfate dependence of isoprene-SOA tracers (He et al., 2018; Zhang et al., 2019b). Simultaneously, SOA tracers originating from β-caryophyllene and high-generation monoterpene oxidation were positively correlated with Ox and sulfate (Zhang et al., 2019b). Interestingly, the reduction of 50% Ox in this region was estimated to be more efficient in reducing biogenic SOA than that of sulfate. In eastern China, combining field measurements and model analysis, the depression of IEPOX SOA by high-NOx levels was confirmed as the reactive uptake of IEPOX and the ratio of IEPOX to isoprene high-NOx SOA precursors were lower than those observed in regions with abundant biogenic emissions, high particle acidity and low-NOx concentrations (Zhang et al., 2017c). Biogenic SOA formation in summer 2012 over China was simulated using the Community Multiscale Air Quality (CMAQ) model, which considers the reactive uptake of isoprene-derived intermediates, multigenerational oxidation and detailed monoterpene SOA production (Qin et al., 2018a). Isoprene SOA tracers showed high concentrations in southwestern China owing to the abundant IEPOX and high particle surface area provided by sulfate. Similar positive correlations between biogenic SOA tracers and sulfate were also observed in urban ürümqi, Qinghai Lake and urban Xi’an, Beijing, Nanjing, Pearl River Delta, and -Wanqingsha (He et al., 2014, 2018; Zhang et al., 2017c; Ren et al., 2018; Zhang et al., 2019b; Bryant et al., 2020). The isoprene SOA formation pathway in some areas of the Yangtze River Delta Region and North China Plain was influenced by NOx emissions, as a high ratio of 2-methylglyceric acid and 2-methyltetrols (0.06–0.1 by the model and 0.58–0.78 in observations) showed in these regions (Qin et al., 2018a). We note that, although the simulated total biogenic SOA in summer 2012 in China accurately tracked the observed data (normalized mean bias of 1% and r2 of 0.59), CMAQ did not simulate the ratios of 2-methylglyceric acid and 2-methyltetrols well. The uncertainties in the fate of IEPOX, 2-methylglyceric acid reaction parameters and C5-alkene triols formation pathways could be possible reasons for the discrepancies between modeled and observed results. The linear correlations between SOA tracers of isoprene, monoterpenes and sesquiterpenes and anthropogenic pollutants, such as SO2 and NOx, were also observed at Wuyi Mountain and Changbai Mountain in southeastern and northeastern China, respectively, suggesting that SO2 and NOx can enhance biogenic SOA production in the remote mountain area through acid-catalyzed heterogeneous chemistry (Wang et al., 2008; Ren et al., 2019). For the more polluted urban Beijing, which is characterized by both local isoprene and anthropogenic pollutants, anthropogenic-influenced biogenic SOA formation in summer 2017 was also observed (Bryant et al., 2020). Isoprene-derived particulate organosulfates and nitrooxy-organosulfates, the formation of which is related to NOx and particulate SO42? levels, accounted for 0.62% and could be as high as ~3% on certain days.
For China as a whole, SOA formation in 2013 was modeled by incorporating updated two-product SOA yields and SOA formation from the reactive uptake of isoprene-derived IEPOX and methacrylic acid epoxide into the updated 3D air quality model (Hu et al., 2017). The enhancement effect of anthropogenic emissions on biogenic SOA was evidenced because the SOA concentration was less than 1 μg m?3 when solely considering biogenic emissions (Hu et al., 2017). Similar anthropogenic–biogenic interactions were found in a more recent study (Wu et al., 2020). With the modeled anthropogenic and biogenic emissions in China in 2016, the CMAQ model that includes updated POA aging, SOA properties and IEPOX organosulfates formation rate constants showed that removing all anthropogenic emissions while keeping biogenic emissions unchanged led to a 60% reduction of SOA formation. These studies suggest that, athough the emission of BVOCs is uncontrollable, biogenic SOA reduction can be achieved through controlling anthropogenic emissions. It should be noted that the modeled SOA concentrations have not been compared with the direct SOA measurements owing to data limitations. Many other studies show that current models usually underestimate or predict the SOA concentration with large uncertainties because of the missed SOA precursors, formation mechanism, components and complex atmospheric conditions (Shrivastava et al., 2017; Liu et al., 2018; Slade et al., 2019). With more detailed measurements of the particle composition and biogenic SOA tracer performed in many areas over China (Table 3) and the increased knowledge of the SOA formation mechanism by laboratory studies, models could be better constrained by the observed data and model performance could be better evaluated.
Currently available laboratory and field observations have made great progress in the scientific understanding of this kind of interaction. This paper reviews the effects of NOx, anthropogenic aerosols, SO2 and NH3/amines on biogenic SOA formation from BVOC photooxidation and ozonolysis, from the perspective of gas- and particle-phase reactions. The NOx level is effective in determining the RO2· fate by competing with HO2· in daytime oxidation and changing the atmospheric oxidation capacity by forming NO3· that acts as another sink for BVOCs besides O3 at night. These NOx-involved BVOC oxidation processes induce changes in the distribution of product volatility and thus SOA composition and yields. However, whether high- or low-NOx levels favor SOA formation depends on the hydrocarbon precursor itself, indicating that the spatial and temporal distribution of different BVOCs need to be carefully considered when evaluating NOx effects on biogenic SOA formation. The definition of high- or-low NOx levels for a specific BVOC, such as isoprene, is also unclear and a detailed NOx-involved mechanism warrants further attention.
POA from anthropogenic activity could alter the gas–particle partitioning of SOA-forming products if a homogeneous mixing phase occurs. Inorganic sulfates promote SOA formation through particle-phase reactions, which would simultaneously be associated with the particle acidity and water content. While strong correlation between IEPOX SOA and sulfates is frequently observed, the combined effects of these factors under certain circumstances should be checked in detail by further laboratory experiments.
SO2 enhances SOA formation dominantly by forming H2SO4 that triggers NPF and acid-catalyzed particle-phase reactions. SO2-introduced reduction of the oxidation capacity, such as ·OH and sCIs levels in the reaction system, would somewhat counterbalance the enhancement effect. New mechanisms of the direct interaction between SO2 and peroxides and other potential mechanism are possible but need further examination.
The acid–base reaction between NH3 and organic acid in the gas phase is the main way for the interference of NH3 on SOA generation. Whether NH3 enhances particle formation depends on organic acids formed from BVOC oxidation and thus the parent BVOCs and oxidants themselves. The particle-phase reaction between NH3 and carbonyls under acid conditions is efficient in forming NOC and thus enhancing the light absorption of SOA particles. The reactions of amines with epoxides/carbonyls/organic acids derived from biogenic sources may also modify biogenic SOA composition and properties, but this needs further exploration.
Despite the abovementioned advances having shed light on the importance of anthropogenic–biogenic interactions, the exploration of this topic is far from complete. More research efforts are recommended to be engaged toward the following directions:
(1) Generally, the concentrations of parent hydrocarbons and anthropogenic pollutants in laboratory experiments are much higher than the ambient levels, which might cause a deviation in some critical conditions, such as the change of the competitive advantages of different reaction paths. Therefore, the concentrations of the substance in the laboratory experiments need to be closer to the real atmospheric level while keeping other conditions, like RH and particle acidity, more atmospherically relevant.
(2) Besides the role of single pollutants in the formation of biogenic SOA, the combined effects of multiple anthropogenic pollutants, such as the simultaneous presence of NOx and SO2, SO2 and NH3/amines, in BVOC photooxidation and ozonolysis are scarcely investigated. Giving chemical insight into whether obvious synergistic, antagonistic actions, or irrelevance between different pollutants exist makes sense because these interactions could greatly affect the SOA yield.
(3) SOA tracers, such as organosulfates and organic nitrates from typical BVOCs are important compounds found in laboratory and field SOA samples. These compounds not only qualitatively represent anthropogenic biogenic–interactions, but their atmospheric concentrations could also be the basis for the quantification of controllable biogenic SOA. While using surrogate compounds for the quantification of these SOA tracers induces uncertainty, authentic quantitative standards can further assist in the accurate quantification and comprehensive interpretation of the mechanism for the interaction between BVOCs and anthropogenic pollutants. Yet, these authentic quantitative standards that can be used for the determination of SOA from various monoterpenes and sesquiterpenes, are often unavailable and require further development.
(4) The particle-phase state (liquid, semisolid, solid) of SOA is crucial for the partitioning of semi-volatile compounds, particle-phase reactions and, most importantly, climate. SOA morphology is related to the components and their hygroscopicity; for example, the presence of oligomers and high molecular weight compounds favors the amorphous solid state of SOA particles while hydrophilic products lead to a liquid state of the particles. Anthropogenic pollutants, such as NOx and SO2, and particle acidity, could potentially change biogenic SOA formation pathways and composition, and thus possibly the phase state of SOA particles. However, this is still poorly understood. Future research needs to expand on the exploration of anthropogenic effects on the morphology of SOA to better address the morphology-associated heterogeneous chemistry, optical properties, air quality and climate.
(5) Vast areas of the globe, like China, still experience both large BVOCs and anthropogenic pollutant emissions. To explore the extent to which biogenic SOA could be mitigated by controlling anthropogenic pollutants, collecting more field evidence regarding the correlation between region-specific types and amounts of BVOCs and human-induced pollutants is a demanding task. Furthermore, while climate change and land-use change tend to increase global BVOC emissions, emissions of anthropogenic pollutants are also changing. For example, NOx and SO2 emissions are expected to continually decrease in North America and Europe but increase in Asia. Besides the interaction mechanism among different BVOC precursors and pollutants, changes in the temporal and spatial distribution of both BVOCs and anthropogenic pollutants should be the basis for regional and, ultimately, global control of SOA formation.
Overall, shedding light on anthropogenic–biogenic interactions is necessary for better evaluation of the contribution of biogenic SOA to the total SOA budget, formulating more effective pollution control measures and reducing uncertainties in the current understanding of air pollution and climate change.
Acknowledgements. This work was supported by National Natural Science Foundation of China (Grant No. 91644214), Youth Innovation Program of Universities in Shandong Province (Grant No. 2019KJD007), and Fundamental Research Fund of Shandong University (Grant No. 2020QNQT012).
