0 引言
【研究意义】牡丹(Paeonia suffruticosa Andr.)是中国著名的传统观赏花卉之一,同其他多年生木本植物一样,其在自然条件下必须经过冬天一段时间低温,才能解除休眠来年正常开花。足够低温累积是解除休眠的有效措施。低温解除牡丹休眠进程中,甲基化水平是下降的[1],暗示表观遗传修饰在其中发挥了重要作用。生产上,常用赤霉素辅助解除休眠,进行反季节花期调控生产。因此,明确赤霉素解除休眠是否与甲基化调控有关对于牡丹反季节生产具有重要意义。【前人研究进展】真核生物中,DNA胞嘧啶的甲基化与基因表达密切相关[2],5-胞嘧啶的甲基化是最普遍的DNA修饰。胞嘧啶甲基化在多种生物发育过程中发挥关键作用,包括基因沉默、转座子的失活和印记基因表达[3,4]。DNA甲基转移酶(Dnmt)家族是植物进行甲基化修饰的工具,它能催化S-adenosylmethionine(SAM)上的甲基转移到胞嘧啶嘧啶环的C-5位点,在表观遗传基因调控中起中心作用[5]。植物中的DNA甲基转移酶主要包括甲基转移酶MET1、染色质甲基转移酶CMT、结构域重排甲基转移酶DRM这3类[6]。拟南芥中,AtMET1主要维持重复序列和单拷贝DNA序列中CpG位点的DNA甲基化[7]。MET1能维持重复和单拷贝DNA序列中CG类型的甲基化,并影响其形态特征[8],DRM是植物中特有的一类甲基化酶,可以在异染色质发生DNA甲基化,其功能主要是维持CHG位点的DNA甲基化[2,9]。另外,植物DNA去甲基化酶ROS1(repressor of silencing 1)是一种DNA糖基化酶,它可以去除DNA5-甲基胞嘧啶上的甲基而发动DNA的去甲基化过程。目前,其功能和特性仅在拟南芥中进行了较为详细的研究[10]。【本研究切入点】关于赤霉素对甲基化影响的报道大都集中在模式植物上,在木本植物牡丹中的研究未见报道。【拟解决的关键问题】探究赤霉素促进休眠解除与DNA甲基化变化的潜在联系,为进一步研究赤霉素解除休眠的机制打下基础。1 材料与方法
1.1 试验材料
供试材料为4年生‘鲁菏红’(‘Luhehong’)健壮植株,由菏泽牡丹研究所提供。2016年10月下旬定植于盆中,11月12日移入温室,用500 mg·L-1的GA3处理花芽(棉花缠绕花芽,滴加GA3溶液1 mL),并在处理后0.5、1、3和5 d后取健壮顶芽,每组设3个重复,清水为对照;每处理15株,共90株,剥去鳞片后,液氮速冻后于-80℃保存备用。另取上述处理60株,移入温室后60 d调查植株生长情况。取初花期(大风铃期)牡丹花芽的各个组织(根、茎、叶、花瓣、萼片、苞片、雄蕊、心皮),每组取3个重复,用于组织表达分析。1.2 总RNA提取与cDNA合成
采用MiniBEST Plant RNA Extraction Kit(TaKaRa,大连)提取总RNA,-80℃保存。cDNA的合成按照PrimeScriptTM RT Reagent Kit with gDNA Eraser 试剂盒(TaKaRa,大连)说明书进行。RACE- Ready-cDNA第一链合成按照SMARTer? RACE 5′/3′ Kit说明书进行。1.3 RACE扩增和实时定量PCR
根据已知序列设计特异性引物进行RACE扩增。反应条件:94℃ 30 s,72℃ 3 min,共5个循环;94℃ 30 s,70℃ 30 s,72℃ 3 min,共5个循环;94℃ 30 s,68℃ 30 s,72℃ 3 min,共25个循环。将反转录得到的cDNA稀释3倍作为实时定量PCR反应的模板,设定3个技术重复。Actin为内参,引物见表1,数据分析采用2-ΔΔCT法。Real-time PCR反应体系为20 μL,包括ddH2O 6.8 μL,SYBR Premix DimerEraser(2×)10.0 μL,PCR Forward Primer(10 μmol·L-1)0.6 μL,PCR Reverse Primer(10 μmol·L-1)0.6 μL,cDNA 2.0 μL。反应在Light Cycler 480 real-time PCR System上进行,反应条件参考司福会等[11]。
Table 1
表1
表1引物相关信息
Table 1Primers information used in this study
| 引物名称Primers name | 引物序列Primers sequence (5′-3′) | 用途Purpose |
|---|---|---|
| PsMET 5′ | GCCAGTACCCTTCACGTATCGATAATAAC | RACE |
| PsCMT 5′ | GATTCATTCCTGATTTGGGTTTCTGG | RACE |
| ActinF | GAGAGATTCCGTTGCCCTGA | qPCR |
| ActinR | CTCAGGAGGAGCAACCACC | qPCR |
| PsMBD5F | CGACAAGAACAATAACATAGGC | qPCR |
| PsMBD5R | GGTGGTCCCTTTGAGTTGTC | qPCR |
| PsMETF | GCTGCTTTGTACTCCAGGCCC | qPCR |
| PsMETR | CTGGTGGCATTCAAGAAGTCG | qPCR |
| PsCMTF | TTGAATCAGGGAGTGGTAATGTG | qPCR |
| PsCMTR | TTCTTCGCCGACCCTGTTG | qPCR |
| PsDRMF | GAGTTTAGAGTTCCCAAGCGAG | qPCR |
| PsDRMR | GCTCTCATCCTTTCCGTCATC | qPCR |
| PsROS1F | CAACAACTCCGACGCCAGATGCT | qPCR |
| PsROS1R | CTCTACTTCAGCATCTGGCGT | qPCR |
新窗口打开
1.4 生物信息学分析
测序结果利用DNAMAN6.0进行分析和拼接,利用Blast(http://blast.ncbi.nlm.nih.gov/Blast.cgi)进行蛋白质Blast对比并分析结构域。利用IBS 1.0.2绘制蛋白质结构域分布图。系统发育树采用MEGA 5.0中的邻接法(neighbor-joining,NJ)构建,置信度检验设置为Bootstrap=1 000。1.5 甲基化水平的测定
利用CTAB法提取植物总DNA[12],100℃加热2 min使约5 μg DNA变性,迅速置于冰上。甲基化测定用HASBUN等的方法[13],变性的DNA在37℃经核酸酶P1(Sigma)和碱性磷酸酶(Sigma)处理后,15 000×g离心20 min,上清液经0.025 μm滤纸(Millipore,Bedford,MA)过滤,上样Eclipse XDB-C18柱(5 μm,150×4.6 nm,Aglient)用于HPLC分析(Aglient 1100 Series),流动相为KH2PO3/ methanol(98﹕2,v/v),约20 min。标样胞嘧啶(C)和甲基胞嘧啶(mC)购自Sigma。使用下列公式计算甲基胞嘧啶百分比:mC(%)=(mC/(C+mC))×100。2 结果
2.1 赤霉素处理解除牡丹花芽休眠的效应
GA3处理后,牡丹花芽迅速萌动,处理5 d时,大部分花芽即已萌动;至60 d时调查,萌动率为97.5%,成花率为92.5%。对照萌动较晚,20 d时才有少量花芽萌动,至60 d时萌动率仅为23.1%,未见花朵开放,仅有5.1%的成花率(图1)。可见,赤霉素处理可在一定程度上代替低温,促进休眠解除,显著提高萌动率和成花率。 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图1外施GA3的牡丹(移入温室60 d)
A:对照 Control;B:500 mg·L-1 GA3处理
-->Fig. 1The tree peony after applied GA3 (60 days after moved into greenhouse)
Treated with 500 mg·L-1 GA3
-->
2.2 赤霉素对甲基化水平的影响
利用HPLC分析了GA3处理后牡丹花芽的甲基化水平。由图2可以看出,GA3处理显著降低了花芽的甲基化水平,且随着施加GA3天数的增加,花芽甲基化水平呈下降趋势。从38.9%降至处理5 d时的28.7%,而对照甲基化水平维持在37.5%—41.5%,相对稳定。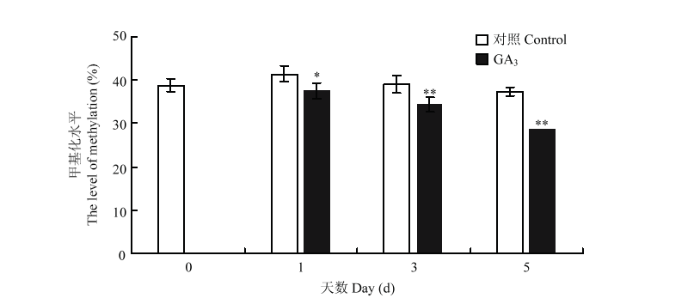 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图2GA3处理后牡丹花芽的甲基化水平
*P<0.05;** P<0.01。
-->Fig. 2Methylation level of tree peony buds after GA3 treatment
The same as
-->
2.3 甲基化相关酶基因的克隆
根据DNA甲基化相关酶基因保守序列,从转录组数据库中筛选出相关片段[12],其中DRM(Accession number:MH038045)和ROS1(Accession number:MH087469)已包含完整的ORF,而MET(ID:JI448809)和CMT(ID:JI448935)编码序列不完整,进行了RACE扩增。根据目的基因的序列设计了特异性引物(表1),经过RACE PCR扩增,得到了997 bp的PsMET 5′ RACE PCR产物和1 245 bp的PsCMT 5′ RACE PCR产物(图3)。测序后利用DNAMAN与已知cDNA进行序列拼接,最终获得1 506 bp的PsMET cDNA序列(Accession number:MH038046)、1 582 bp的PsCMT cDNA序列(Accession number:MH038044)。PsCMT开放阅读框的长度为1 118 bp,编码的氨基酸数目为372,蛋白质分子量为41.469 kD。PsMET开放阅读框的长度为1 056 bp,编码351个氨基酸,蛋白质分子量为39.853 kD。PsDRM开放阅读框的长度为2 175 bp,编码724个氨基酸,蛋白质分子量为81.477 kD。PsROS1含有6 636 bp的开放阅读框,编码2 211个氨基酸,分子量为246.675 kD。在线分析牡丹Dnmts和ROS1的序列表明,PsCMT在152—269 aa间含有一个BAH结构域(The Bromo-adjacent homology domain),在311—375 aa间存在Cyt_C5_DNA_methylase结构域;PsMET在22—351 aa间存在一个SAM-dependent MTase C5-type结构域;在PsDRM序列上找到两个结构域,一个是位于232—276 aa间的UBA(Ubiquitin-associated domain)结构域,另一个是在393—723 aa的SAM-dependent MTase DRM-type结构域;PsROS1上存在DNA糖基化结构域、HhH-GPD 结构域、Perm-CXXC结构域和RRM-DME结构域(图3)。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图3DNA甲基化酶(A)和去甲基化酶(B)的结构域
-->Fig. 3The domain diagram of DNA methylase (A) and demethylase (B)
-->
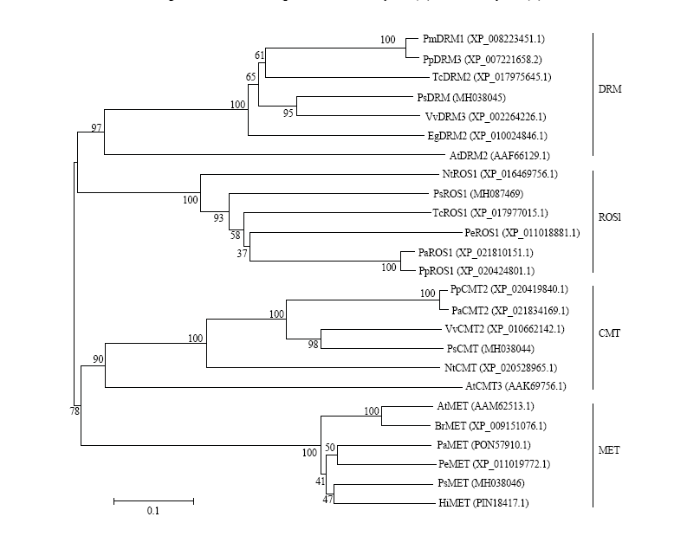
为了解牡丹PsDRM、PsMET、PsCMT、PsROS1蛋白与其他植物相应基因的进化关系,对牡丹中4种DNA甲基化相关酶进行系统发育分析(图4)。结果表明,PsCMT和PsDRM与葡萄的VvCMT2亲缘关系最近,与模式植物拟南芥亲缘关系较远;PsMET和PsROS1与大部分木本植物聚为一支,比如胡杨,与烟草等距离较远。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图4牡丹甲基化相关酶的系统发育分析
-->Fig. 4The phylogenetic analysis of methylation-related enzymes in tree peony
-->
2.4 牡丹甲基化相关酶基因的组织表达特性
在牡丹的初花期(大风铃期)取各个组织(根、茎、叶、苞片、萼片、花瓣、雄蕊、心皮),利用荧光定量PCR对DNA甲基化酶基因的表达模式进行了分析。结果表明,PsDRM、PsMET、PsCMT在各个组织中均能检测到表达,但表达量不同。PsMET和PsCMT在根、茎、雄蕊和苞片中表达量相对较高,在叶片和心皮中较低;而PsDRM在根中的表达量最高,在心皮中的表达量相对较高,其次为茎和萼片,在雄蕊中最低;PsROS1在心皮中表达量最高,其次是萼片,在叶片和花瓣中表达较少(图5)。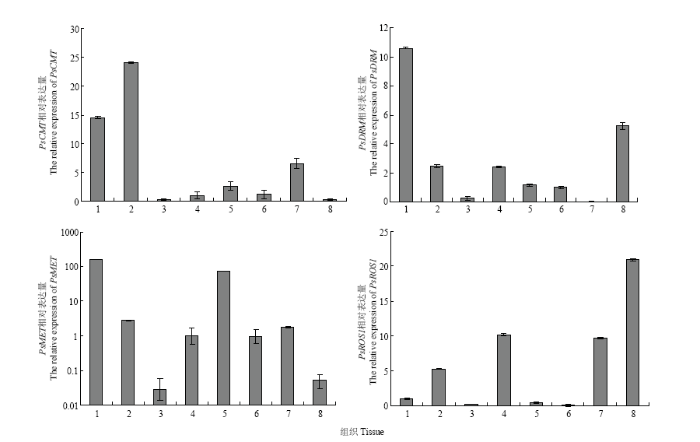 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图5甲基化相关酶基因的组织表达模式
1:根Root;2:茎Stem;3:叶Leaf;4:萼片Sepal;5:苞片Bract;6:花瓣Petal;7:雄蕊Stamen;8:心皮Carpel
-->Fig. 5The tissue expression pattern of methylation-related enzyme genes
-->
2.5 赤霉素对甲基化相关酶基因表达的影响
植物中的甲基化水平是由甲基化酶和去甲基化酶共同维持的[14]。因此,对相关酶基因进行荧光定量PCR分析(图6)。结果表明,GA3处理的牡丹花芽与对照相比PsMET表达量差异不显著(P>0.05)。用GA3处理后,PsCMT的表达量在0.5 d内就有了大幅度的提高,这表明GA3显著地提高了PsCMT的表达水平,PsCMT可快速响应GA3。GA3处理0.5—3 d,PsDRM的表达明显减弱。与此相反,经GA3处理后,去甲基化酶PsROS1表达量显著提高,且有逐渐增加的趋势。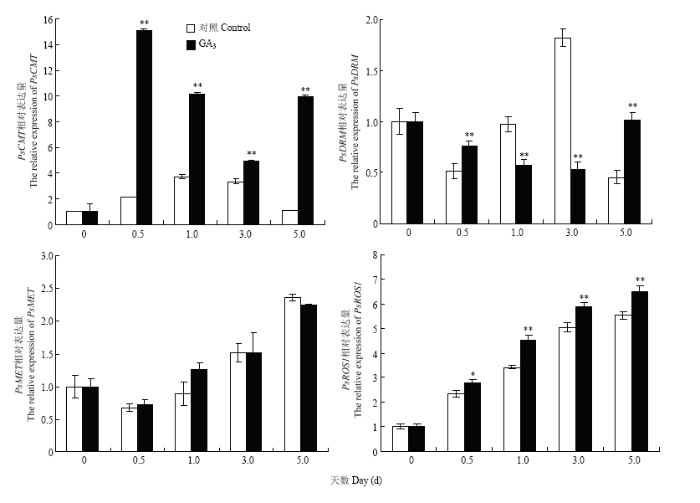 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图6GA3处理后甲基化相关酶基因的表达水平
-->Fig. 6The expression level of methylation-related enzyme genes after GA3 treatment
-->
3 讨论
DNA胞嘧啶甲基化、组蛋白修饰和RNA干扰(RNAi)是最常见的分子表观遗传学机制,在基因调控中发挥重要作用[14]。DNA胞嘧啶甲基化大约占总胞嘧啶的30%[15],越来越受到人们的重视。DNA甲基化水平主要是由DNA甲基转移酶和去甲基化酶创建和维持的。相关基因克隆和功能分析主要见于模式植物,但木本植物中相关基因克隆的报道较少。本研究从牡丹花芽中克隆了DNA甲基化酶基因PsCMT、PsMET、PsDRM和DNA去甲基化酶PsROS1,序列分析表明具有相应保守结构域,并在不同组织中检测到了它们的表达,暗示它们可能参与了牡丹细胞中DNA甲基化的调控。赤霉素是一种重要的植物激素,参与调控多种生长和发育过程[16]。目前已对赤霉素的生物合成和代谢途径有了比较明确的了解[17]。然而,赤霉素在表观遗传调控中发挥怎样的作用仍未可知。李梅兰等[18]研究表明,在春化诱导不结球白菜开花期间,DNA甲基化水平降低与芽尖分生组织中内源GA含量增加存在间接关系。本研究表明,赤霉素导致牡丹的甲基化水平下降。已有研究表明,休眠解除进程中伴随着DNA低甲基化[1,19]。因此,赤霉素处理导致DNA低甲基化,可能是其发挥作用促进休眠解除的一种重要方式。
另外,赤霉素处理可能通过调节DNA甲基化相关酶基因的表达而影响甲基化水平。从小麦幼苗和发芽胚的细胞核中纯化的DNMT的活性受到GA3抑制,但GA3添加到小麦胚的核提取物中似乎刺激了DNA甲基化酶活性[20]。然而,在水稻中,外施GA3不会引起OrDNMT表达的显著变化[21]。近期在烟草中的试验表明,NtMET1和NtCMT3响应GA3信号表达水平下降[14]。显示了不同生物中的Dnmt(MET、DRM和CMT)对GA响应是有差异的[19,20,21,22,23]。ROS1介导的主动去甲基化可以导致内外源基因的转录沉默[24],引起5S rDNA染色质的解聚及植物对生物胁迫和环境胁迫的响应[25,26,27]。本研究中,GA3处理导致PsCMT的表达量提高。CMT主要功能是维持CG位点甲基化,GA3处理促进了细胞分裂,需要CMT大量表达维持甲基化水平。DRM可导致CHG位点重头甲基化,GA3处理后PsDRM表达受到抑制,而PsROS1表达量显著提高,二者联合作用,导致总体甲基化水平的下降。
4 结论
利用转录组分析和RACE技术,得到了牡丹DNA甲基转移酶基因(PsCMT、PsMET和PsDRM)和DNA去甲基化酶基因PsROS1,具有相应保守结构域。外施GA3显著降低DNA甲基化水平,导致DNA低甲基化,进而促进了休眠解除,是赤霉素发挥作用的一种方式。DNA甲基化水平的降低可能与赤霉素诱导PsDRM表达水平下降和PsROS1表达量提高有关。The authors have declared that no competing interests exist.
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
| [1] | . DNA 甲基化是表观遗传调控的重要组成部分, 在真核生物的基因表达调控中发挥了重要作用。本实验研究了感受0、6、12、18和24 d 4℃低温的牡丹(Paeonia suffruticosa)鲁荷红移入温室后的萌动和开花表现,并对其进行了甲基化敏感扩增多态性(methylation sensitive amplified polymorphism, MSAP)分析。结果表明,4℃低温处理18 d 可解除鲁荷红内休眠,继续增加低温则进入生态休眠阶段。MASP分析表明,牡丹内休眠期间花芽内DNA甲基化程度很高,甲基化位点占总位点比率的60%以上,以半甲基化为主,在低温处理进程中,甲基化水平总体呈下降趋势。与未经低温的对照相比,约50%的位点发生了甲基化模式变化,低温6、12、18和24 d去甲基化位点数和比例分别为97 (17.1%)、104 (18.6%)、148 (24.6%)和218 (36.1%),过甲基化的位点数分别为197 (34.7%)、137 (24.6%)、95 (15.8%)和79 (13.1%)。研究结果提示,DNA甲基化参与了牡丹内休眠的调控。 . DNA 甲基化是表观遗传调控的重要组成部分, 在真核生物的基因表达调控中发挥了重要作用。本实验研究了感受0、6、12、18和24 d 4℃低温的牡丹(Paeonia suffruticosa)鲁荷红移入温室后的萌动和开花表现,并对其进行了甲基化敏感扩增多态性(methylation sensitive amplified polymorphism, MSAP)分析。结果表明,4℃低温处理18 d 可解除鲁荷红内休眠,继续增加低温则进入生态休眠阶段。MASP分析表明,牡丹内休眠期间花芽内DNA甲基化程度很高,甲基化位点占总位点比率的60%以上,以半甲基化为主,在低温处理进程中,甲基化水平总体呈下降趋势。与未经低温的对照相比,约50%的位点发生了甲基化模式变化,低温6、12、18和24 d去甲基化位点数和比例分别为97 (17.1%)、104 (18.6%)、148 (24.6%)和218 (36.1%),过甲基化的位点数分别为197 (34.7%)、137 (24.6%)、95 (15.8%)和79 (13.1%)。研究结果提示,DNA甲基化参与了牡丹内休眠的调控。 |
| [2] | . |
| [3] | . |
| [4] | . |
| [5] | . Abstract The DNA methyltransferase (DNMT) family comprises a conserved set of DNA-modifying enzymes that have a central role in epigenetic gene regulation. Recent studies have shown that the functions of the canonical DNMT enzymes - DNMT1, DNMT3A and DNMT3B - go beyond their traditional roles of establishing and maintaining DNA methylation patterns. This Review analyses how molecular interactions and changes in gene copy numbers modulate the activity of DNMTs in diverse gene regulatory functions, including transcriptional silencing, transcriptional activation and post-transcriptional regulation by DNMT2-dependent tRNA methylation. This mechanistic diversity enables the DNMT family to function as a versatile toolkit for epigenetic regulation. |
| [6] | . |
| [7] | . |
| [8] | . |
| [9] | . |
| [10] | . Mutations in the Arabidopsis ROS1 locus cause transcriptional silencing of a transgene and a homologous endogenous gene. In the ros1 mutants, the promoter of the silenced loci is hypermethylated, which may be triggered by small RNAs produced from the transgene repeats. The transcriptional silencing in ros1 mutants can be released by the ddm1 mutation or the application of the DNA methylation inhibitor 5-aza-2 -deoxycytidine. ROS1 encodes an endonuclease III domain nuclear protein with bifunctional DNA glycosylase/lyase activity against methylated but not unmethylated DNA. The ros1 mutant shows enhanced sensitivity to genotoxic agents methyl methanesulfonate and hydrogen peroxide. We suggest that ROS1 is a DNA repair protein that represses homology-dependent transcriptional gene silencing by demethylating the target promoter DNA. |
| [11] | . . |
| [12] | . 78 A total of 23,652 assembled UniGenes were obtained with 15,284 annotated UniGenes with a minimal length of 300bp. 78 Many UniGenes were assigned to putative metabolic pathway. 78 484 putative transcription factors were obtained. 78 2253 potential Simple Sequence Repeats (SSR) loci were identified in the 454-ESTs. 78 73 pairs of primers displayed polymorphism between P. ostti and the two wild species (P. rockii and P. qiui) from 149 pairs designed according to our 454 EST collection. |
| [13] | . Quantification of deoxynucleosides using micellar high-performance capillary electrophoresis (HPCE) is an efficient, fast and inexpensive evaluation method of genomic DNA methylation. This approach has been demonstrated to be more sensitive and specific than other methods for the quantification of DNA methylation content. However, effective detection and quantification of 5-methyl-2 -deoxycytidine depend of the sample characteristics. Previous works have revealed that in most woody species, the quality and quantity of RNA-free DNA extracted that is suitable for analysis by means of HPCE varies among species of the same gender, among tissues taken from the same tree, and vary in the same tissue depending on the different seasons of the year. The aim of this work is to establish a quantification method of genomic DNA methylation that lends itself to use in different Castanea sativa Mill. materials, and in other angiosperm and gymnosperm woody species. Using a DNA extraction kit based in silica membrane has increased the resolutive capacity of the method. Under these conditions, it can be analyzed different organs or tissues of angiosperms and gymnosperms, regardless of their state of development. We emphasized the importance of samples free of nucleosides, although, in the contrary case, the method ensures the effective separation of deoxynucleosides and identification of 5-methyl-2 -deoxycytidine. |
| [14] | . |
| [15] | . Abstract A plant cytosine methyltransferase cDNA was isolated using degenerate oligonucleotides, based on homology between prokaryote and mouse methyltransferases, and PCR to amplify a short fragment of a methyltransferase gene. A fragment of the predicted size was amplified from genomic DNA from Arabidopsis thaliana. Overlapping cDNA clones, some with homology to the PCR amplified fragment, were identified and sequenced. The assembled nucleic acid sequence is 4720 bp and encodes a protein of 1534 amino acids which has significant homology to prokaryote and mammalian cytosine methyltransferases. Like mammalian methylases, this enzyme has a C terminal methyltransferase domain linked to a second larger domain. The Arabidopsis methylase has eight of the ten conserved sequence motifs found in prokaryote cytosine-5 methyltransferases and shows 50% homology to the murine enzyme in the methyltransferase domain. The amino terminal domain is only 24% homologous to the murine enzyme and lacks the zinc binding region that has been found in methyltransferases from both mouse and man. In contrast to mouse where a single methyltransferase gene has been identified, a small multigene family with homology to the region amplified in PCR has been identified in Arabidopsis thaliana. |
| [16] | . |
| [17] | . |
| [18] | . 以普通白菜‘油冬儿’为试材, 研究了低温对白菜开花的诱导效应, 并分析了萌动种子和幼<BR>苗春化诱导后DNA 甲基化水平、赤霉素(GA) 含量和蛋白质种类的变化。结果表明, 不论萌动种子还是幼苗, 4 ℃低温处理30 d 均可完成春化。春化处理后, 两处理植株茎尖组织DNA 甲基化水平都较对照下降;但GA 含量明显上升, 为对照的2~3 倍; 低温诱导植株产生了一种分子量为58 kD 的特异蛋白质, 同时也使一种蛋白质消失。说明低温可能通过降低DNA 甲基化水平或增加GA 含量而诱导植物开花。<BR> . 以普通白菜‘油冬儿’为试材, 研究了低温对白菜开花的诱导效应, 并分析了萌动种子和幼<BR>苗春化诱导后DNA 甲基化水平、赤霉素(GA) 含量和蛋白质种类的变化。结果表明, 不论萌动种子还是幼苗, 4 ℃低温处理30 d 均可完成春化。春化处理后, 两处理植株茎尖组织DNA 甲基化水平都较对照下降;但GA 含量明显上升, 为对照的2~3 倍; 低温诱导植株产生了一种分子量为58 kD 的特异蛋白质, 同时也使一种蛋白质消失。说明低温可能通过降低DNA 甲基化水平或增加GA 含量而诱导植物开花。<BR> |
| [19] | . Cytosine DNA methylation is a conserved epigenetic regulatory mechanism in both plants and animals. DNA methyltransferases (DNA MTases) not only initiate (de novo) but also maintain the process of DNA methylation. Here, we characterized the genome-wide expression profiles of 10 cytosine DNA MTase genes belonging to 4 subfamilies, MET1, CMT, DNMT2, and DRM, in rice. Tissue-specific gene expression analysis showed that all family members varied widely in their expression and specificities and might be involved in some basic metabolic pathways. Similarly, the expression of all rice cytosine DNA MTase genes was not regulated by plant hormones except OsDRM1a and OsDRM1b, which were downregulated by jasmonic acid. The transcription level of 10 genes in rice shoots and roots was also measured under salt and osmotic stress. Meanwhile, quantitative polymerase chain reaction data of the japonica and indica rice cultivars revealed that there is large variation in the expression activities of all genes. The results provide a foundation to further explore the roles of DNA MTases and the epigenetic regulation of abiotic stress responses in rice. |
| [20] | . Abstract Cytosine DNA methyltransferases (MTases) were isolated from nuclei of wheat seedlings and germinating embryos. The MTases isolated from both sources were able to perform de novo and maintenance DNA methylations. The most purified MTase fraction showed the presence of one main 67-kDa protein (embryos) and of a 85-kDa protein (in seedlings) in SDS-PAGE. Some plant growth regulators (gibberellic acid A3, 6-benzylaminopurine and fusicoccin) elevate by 30-65% the extent of in vitro DNA methylation by nuclear extracts with a maximal effect at 10(-6) M phytohormone concentration. The same phytohormones do not increase the extent of in vitro DNA methylation by purified wheat MTase; rather they inhibit it at concentrations of 10(-4)-10(-5) M. Thus, DNA methylation in the plant nucleus is controlled by phytohormones. The phytohormone effect may be mediated by other proteins in nuclear extracts. |
| [21] | . A cDNA encoding a DNA methyltransferase, with a predicted polypeptide of 1556 amino acid residues containing all motifs conserved in this enzyme family, was isolated from tobacco plants, and the corresponding gene was designated as NtMET1. RNA blot analysis indicated NtMET1 transcripts to accumulate in dividing tissues of tobacco plants, and they could be detected during the S phase in synchronized dividing BY2 cells. In situ hybridization revealed the transcripts to be localized exclusively in actively proliferating tissues around axillary apical meristem. In order to ascertain physiological roles, transgenic tobacco plants that had the antisense construct were made and examined for phenotypes. Methylation levels of genomic DNA from transgenic plants significantly decreased in comparison with wild-type levels, and distinct phenotypic changes including small leaves, short internodes and abnormal flower morphology were noted. Microscopic observation revealed that leaf structure differed between transgenic and wild-type plants. These results suggest that NtMET1 functions during DNA replication, and that DNA methylation plays an important role in plant morphogenesis. |
| [22] | . |
| [23] | . |
| [24] | . |
| [25] | . |
| [26] | . |
| [27] | . |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}