0 ����
���о����塿MHC��major histocompatibility complex��������������Ҫ��֯�����Ը������ͳ��,����������3��,��Class-��Class-���Class-�������MHC����������ͬ����ֲ�ųⷴӦ,����Ҫ�����������������Ӧ�����ߵ��ڼ�ijЩ����״̬�IJ�����������ء���ͬ���ಸ�鶯���MHC�������нṹ�����ƴ��ڲ���[1,2,3,4]��˫��������Ӧ�Զ��ӻ�����ս������������������MHC���о�,˫������һ�־���ʵ�����������ģ��[5,6]����λ��ע��˫�����е�MHC����,��˫����MHC�Ľ�һ���о�,�Լ���˫���ջ��������Ƶ��о�����Ҫ�Ŀ�ѧ���塣��ǰ���о���չ������MHC���о�������������ݵ�1936��,����****���о�С������ϸ���ųⷴӦ���״η�����MHC����;1951��,�о��߷�����С���MHC-H2ϵͳ;1958��,DAUSSET�״η���������İ�ϸ����ԭ;1961��SCHIERMAN��NORDSKOGȷ���˼����е�MHCΪһ�ֺ�ϸ����ԭ,����������ΪB[2,7];����20����70��,���Ƿ������Ŵ�����H2ͬԴ��ϵͳͬ������������������[8,9,10];��������ȷ���������еļ����ﶼ����MHC����,�Ҹ���****���Ϸḻ�Ų�ͬ��������MHC������о���2012�꼪��ľͼ���ڵ��״����˫����ȫ����������ͼ���ƺͽ�����2016��ݿ˿�ѧ��ͨ��FISH������������MHC����λ����20��Ⱦɫ��̱��ϣ�20q12��[11]�������о�����㡿Ŀǰ˫���ղο����������װ��ע��ˮƽ������,��˫���ջ�������MHC����û�еõ���װ��ע��[12]����ͳ�Ļ���λ�����ձ���õ����ӽ�,�ཻ���Խ�,�ֱ���������Ľ����ʺ���Ծ���,Ȼ����Ⱦɫ����ȷ������������˳��,��Щ������������������,�ɱ�Ҳ�ܰ��������Ĺؼ����⡿���о�ͨ������Ŀǰ��֪��װˮƽ��õ�����MHC�������˫������Ե��ϵ�Ͻ���ţMHC����Ϊ��������,���ñȽϻ�����ѧ������̽�����ּ�����ͬԴ�Թ�ϵ,ȷ��λ��ע�͵�˫�����е�MHC����,Ϊ˫����MHC����Ľ�һ���о��춨���ۻ�����1 �����뷽��
������2017��6��9�������ɹ�ũҵ��ѧ�����ɹ�������������ҽѧ�ص�ʵ���ң�ũҵ�����R���ٴ����Ƽ����ص�ʵ���ң����С�1.1 ������Դ
˫����MHC�������дӼ���������˫���յIJο��������л��,�ο�������Camelus bactrianus (assembly Ca_bactrianus_MBC_1.0)��NCBI�����صõ�,����HLA��ţBoLA�������ж��Ǵ�ensemble��վ���صõ��������Ϣ���£���1����Table 1
��1
��1˫���ա����ࡢţ�������м�������Դ
Table 1Gene sequences and data sources of Bactrian camel, human, bovine
| ���� Name | ��Դ Source | ��ַ URL |
|---|---|---|
| ����˫���ղο������� Reference genome of domestic Bactrian camels | NCBI | ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/767/855/GCF_000767855.1_Ca_bactrianus_MBC_1.0/GCF_000767855.1_Ca_bactrianus_MBC_1.0_genomic.fna.gz |
| ����HLA�������� Human HLA coding sequence | ensemble | ftp://ftp.ensembl.org/pub/release-90/fasta/homo_sapiens/cdna/ |
| ����HLAȫ���������� Human HLA whole genome sequence | ensemble | ftp://ftp.ensembl.org/pub/release-90/fasta/homo_sapiens/dna/ |
| ����HLAע���ļ� Human HLA annotation file | ensemble | ftp://ftp.ensembl.org/pub/release-90/gtf/homo_sapiens |
| ţBoLA�������� Cattle BoLA coding sequence | ensemble | ftp://ftp.ensembl.org/pub/release-90/fasta/bos_taurus/cdna/ |
| ţBoLAȫ���������� Cattle BoLA whole genome sequence | ensemble | ftp://ftp.ensembl.org/pub/release-90/fasta/bos_taurus/dna/ |
| ţBoLAע���ļ� Cattle BoLA annotation file | ensemble | ftp://ftp.ensembl.org/pub/release-90/gtf/bos_taurus |
�´��ڴ�
1.2 ����
1.2.1 ����blast v2.3.0�������ֱ����1��ʾ��ַ��ȡ��HLA��BoLA�����������,�ֱ���˫����ת¼������blastn���бȶ�,ʶ������ƶȽϸߵ�scaffolds;��ͨ������HLA��BoLA�ȶ���scaffolds��λ��˳��,�Ӷ��Զ���scaffold����ƴ�ӡ�1.2.2 �ֱ���ȡHLA��BoLA������������˫������ƴ�ӵ�scaffolds���л����鹲���Է���,����lastz v.1.04.00������������ƴ�ӵ�scaffolds��HLA��BoLA���������е����Թ�ϵ,���ж�ɸѡ����scaffolds�Ƿ�ȷ��
1.2.3 ͨ������MHC�������ּ�����Թ�ϵ,����BLAT v.35������ȡ��MHC��˫���ջ�����Ļ�������,������Щ�������н��л���ע�͡�
1.2.4 ����MEGA 7.0.26�����Եõ���˫����MHC�������ϵͳ��������
2 ���
2.1 blastn���жԱȼ�ƴ��
�ֱ�������HLA��ţBoLA�������������˫����ת¼������blastn���жԱ�,���ʶ��������ƶȽϸߵ�3��scaffold,��NW_011511766.1��ȫ��4.1M����NW_011515227.1��ȫ��1.2M����NW_011514613.1��ȫ��15K��,��һ��ͨ������HLA��BoLA�ȶ���scaffolds��λ��˳��,����CBLA����͢������ֲ���NW_011515227.1��,��CBLA�������ֲ���NW_011511766.1��NW_011514613.1�϶�����3��scaffolds����ƴ��,�õ�˫����MHC��Pseudo chromosome,��ṹʾ��ͼ��ͼ1��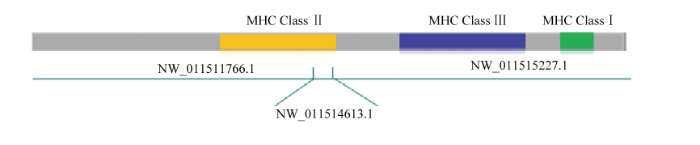 ��ʾԭͼ|����ԭͼZIP|����PPT
��ʾԭͼ|����ԭͼZIP|����PPTͼ1˫����MHC-Pseudo chromosome�ṹʾ��ͼ
-->Fig. 1Schematic diagram of structure for Bactrian camel MHC-Pseudo chromosome
-->
2.2 �����Է���
2.2.1 ����HLA��˫����Pseudo chromosome�����Է��� ��ȡ����MHC����HLA���Ļ�����DNA����,��˫���ջ�������й����Է������ڻ����鹲���Է�����һ�������Ի����Ӧһ����,���ܼ��Ĺ����Ի���������ӳ�Ƭ��,������������ʽ������ͼ�С�����˫����MHC����������HLA�������Թ�ϵͼ��ͼ2�������Կ���,�кܶ�˫����MHC����Ƭ�κ�����HLA����Ƭ���ǹ����Թ�ϵ,�����ڽϳ�������Ƭ��,������֧���˱��о�����λ����˫����MHC�����ȷ��,�Ӷ�˵��˫���յ�MHC���������Ҫ�ֲ��ڱ���ƴ�ӵ�scaffolds�ϡ�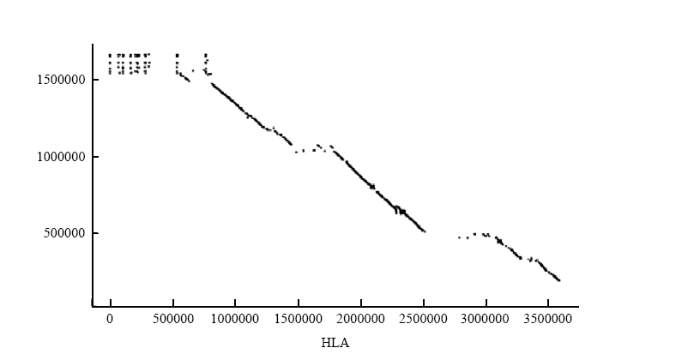 ��ʾԭͼ|����ԭͼZIP|����PPT
��ʾԭͼ|����ԭͼZIP|����PPTͼ2����HLA������˫����MHC����Ĺ����Թ�ϵ�Ƚ�
����Ϊ��HLA�Ļ�������,����Ϊ˫����MHC�Ļ�������
-->Fig. 2Comparison of collinearity between human HLA gene and Bactrian camel MHC gene
The horizontal axis is the gene sequence of human HLA, and the vertical axis is the gene sequence of Bactrian camel MHC
-->
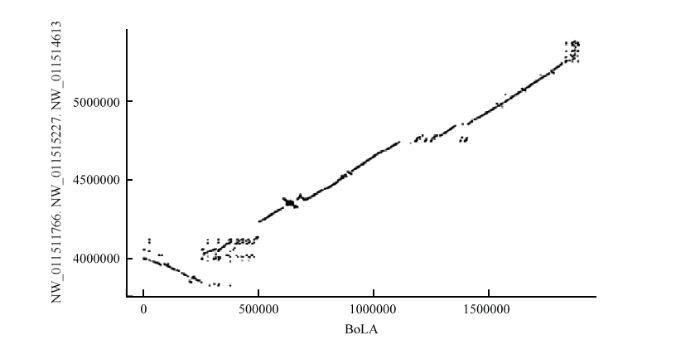
2.2.2 ţBoLA��˫����pseudo chromosome �����Է��� ͬ��,��ȡ��˫������Ե��ϵ�����ţ��MHC����BoLA��[13]�Ļ�����DNA����,��˫���ջ�������й����Է���,�����ʾţBoLA������˫���յ�NW_011511766.1��ȫ��4.1M����NW_011515227.1��ȫ��1.2M����NW_011514613.1��ȫ��15K�����ƶ����,�Ң���͢������ֲ���NW_011515227.1��,���������ֲ���NW_011511766.1��NW_011514613.1��,��һ��������֪���������Ҫ�ֲ���NW_011511766.1 ��3.5��4.1M��λ�á��ڻ����鹲���Է�����һ�������Ի����Ӧһ����,�ܼ��Ĺ����Ի���������ӳ�Ƭ��,������������ʽ������ͼ�С�����˫����MHC������ţBoLA�������Թ�ϵͼ��ͼ3������������ı���,�кܶ�˫����MHC����Ƭ�κ�ţBoLA����Ƭ���ǹ����Թ�ϵ,�����ڽϳ�������Ƭ��,������֧���˱��о�����λ����˫����MHC�����ȷ��,����˵��˫���յ�MHC���������Ҫ�ֲ���ƴ�ӵ�scaffolds��,�ý������1��һ�¡����ھֲ�˫���յ�Ƭ�γ��ָ������Ƭ,�������������4 000 000ʱţBoLA��˫����pseudo chromosome ������������ֵ�������
 ��ʾԭͼ|����ԭͼZIP|����PPT
��ʾԭͼ|����ԭͼZIP|����PPTͼ3ţBoLA������˫����MHC����Ĺ����Թ�ϵ�Ƚ�
����ΪţBoLA�Ļ�������,����Ϊ˫����MHC�Ļ�������
-->Fig. 3Comparison of collinearity between bovine BoLA gene and Bactrian camel MHC gene
The horizontal axis is the gene sequence of bovine BoLA, and the vertical axis is the gene sequence of Bactrian camel MHC
-->
2.3 BLAT����
�����ڹ���������Ļ�����������ȡ����,��ȶԵ�˫�����ϵ�CBLA����ı�����������BLAT v.35���з���,�Ӷ���˫����CBLA���붨λ�ڻ������ϡ��ȶԷ��������ʾ,��˫���ջ������й�ʶ���24����ţBoLA����߶����ƵĻ���,���ڱ��������з���˫���յ�CBLA����24������,���Т������1��,����10��, �������13������2�������ɴ˿ɻ�����/ţ/˫���� MHC�������ͳ�Ʊ�����3����Table 2
��2
��2BLAT�����ҵ���CBLA����
Table 2The CBLA gene found by BLAT analysis
| ������Class | �� | �� | �� |
|---|---|---|---|
| ������� Genetic code | XM 010961543.1 | XM 010949122.1 XM 010958395.1 XM 010949120.1 XM 010949126.1 XM 010949114.1 XM 010949113.1 XM 010949105.1 XM 010949127.1 XM 010949125.1 XM 010949153.1 | XM 010961490.1 XM 010961476.1 XM 010961495.1 XM 010961533.1 XM 010961451.1 XM 010961470.1 XM 010961594.1 XM 010961447.1 XM 010961538.1 XM 010961482.1 XM 010961484.1 XM 010961536.1 XM 010961596.1 |
�´��ڴ�
Table 3
��3
��3��/ţ/˫���� MHC�������ͳ�Ʊ�
Table 3Statistics table of MHC gene number in Human / bovine / Bactrian camels
| ������Class | ���� MHC����HLA | ţMHC����BoLA | ˫����MHC����CBLA |
|---|---|---|---|
| �� | 36/106 | 1 | 1 |
| �� | 33/59 | 11 | 10 |
| �� | 59/59 | 13 | 13 |
�´��ڴ�
2.4 ����ע��
���մ�ensemble�ϻ�ȡ������HLA��ţBoLA��֪MHC����ע����Ϣ���ҵ�����24���������ע��,�ɻ���˫����CBLA����ע����Ϣ������4����Table 4
��4
��4˫����CBLA����ע����Ϣ
Table 4CBLA gene annotation information of Bactrian camel
| ������� Gene id | ���� Length | ������ Exon | λ�� Scaffold | ��ʼ�� Start | ��ֹ�� End | ע����Ϣ Description |
|---|---|---|---|---|---|---|
| XM 010949105.1 | 2866 | 5 | NW 011511766.1 | 3837314 | 3842070 | PREDICTED:CBLA-DOA |
| XM 010949113.1 | 1189 | 5 | NW 011511766.1 | 3874933 | 3878475 | PREDICTED:CBLA-DMA |
| XM 010949114.1 | 1391 | 6 | NW 011511766.1 | 3885523 | 3891578 | PREDICTED:CBLA-DMB |
| XM 010949120.1 | 1381 | 6 | NW 011511766.1 | 3965547 | 3972059 | PREDICTED:CBLA-DOB |
| XM 010949122.1 | 813 | 5 | NW 011511766.1 | 4106164 | 4114129 | PREDICTED:CBLA-DRB2 |
| XM 010949125.1 | 1177 | 5 | NW 011511766.1 | 4047230 | 4051352 | PREDICTED:CBLA-DQA |
| XM 010949126.1 | 1033 | 4 | NW 011511766.1 | 4031979 | 4039091 | PREDICTED:CBLA-DQB |
| XM 010949127.1 | 1307 | 5 | NW 011511766.1 | 4120230 | 4124972 | PREDICTED:CBLA-DRA |
| XM 010949153.1 | 759 | 4 | NW 011511766.1 | 3992073 | 3995513 | PREDICTED:CBLA-DYA |
| XM 010958395.1 | 1315 | 7 | NW 011514613.1 | 5057 | 14519 | PREDICTED:CBLA-DRB3 |
| XM 010961447.1 | 1407 | 9 | NW 011515227.1 | 42577 | 46241 | PREDICTED:PBX2 |
| XM 010961451.1 | 1602 | 8 | NW 011515227.1 | 67307 | 73614 | PREDICTED:PPT2 |
| XM 010961470.1 | 1452 | 8 | NW 011515227.1 | 175625 | 182554 | PREDICTED:STK19 |
| XM 010961476.1 | 2773 | 18 | NW 011515227.1 | 207277 | 218549 | PREDICTED:C2 |
| XM 010961482.1 | 2376 | 1 | NW 011515227.1 | 303003 | 305378 | PREDICTED:HSPA1A |
| XM 010961484.1 | 2500 | 2 | NW 011515227.1 | 305769 | 309435 | PREDICTED:HSPA1L |
| XM 010961490.1 | 2657 | 25 | NW 011515227.1 | 340559 | 355003 | PREDICTED:MSH5 |
| XM 010961495.1 | 1158 | 6 | NW 011515227.1 | 357933 | 363242 | PREDICTED:CLIC1 |
| XM 010961533.1 | 668 | 6 | NW 011515227.1 | 462534 | 464279 | PREDICTED:AIF1 |
| XM 010961536.1 | 688 | 4 | NW 011515227.1 | 488003 | 489391 | PREDICTED:LTA |
| XM 010961538.1 | 1318 | 5 | NW 011515227.1 | 499275 | 507275 | PREDICTED:NFKBIL1 |
| XM 010961543.1 | 1513 | 9 | NW 011515227.1 | 1073777 | 1105682 | PREDICTED:CBLA-A |
| XM 010961594.1 | 6598 | 30 | NW 011515227.1 | 17926 | 38510 | PREDICTED:NOTCH4 |
| XM 010961596.1 | 12304 | 45 | NW 011515227.1 | 105330 | 155862 | PREDICTED:TNXB |
�´��ڴ�
2.5 ˫����MHC�������ϵͳ������
�Եõ���˫����MHC�������ϵͳ������,�����ʾע�͵�CBLA Class-�����Class-���������ͬһ��֧��ͼ4��,˵��CBLA Class-�����Class-������������ϵ����� ��ʾԭͼ|����ԭͼZIP|����PPT
��ʾԭͼ|����ԭͼZIP|����PPTͼ4˫����CBLA����ϵͳ������
��ɫ�߱�ʾCBLA-�������,��ɫ�߱�ʾCBLA-�����,��ɫ�߱�ʾCBLA-�������
-->Fig. 4Phylogenetic tree of Bactrian camel CBLA gene
The blue line indicates the CBLA class- I gene, the red line indicates the CBLA-II gene, and the black line indicates the CBLA class-�� gene
-->
3 ����
�����������˺ܶ�MHC����Ķ�λ����,���õķ�������ϸ���ӽ�������¡Ƕ�巨��ԭλ�ӽ���ӫ��ԭλ�ӽ�����FISH������������������Martin Plasil��2016��ͨ��ӫ��ԭλ�ӽ����������յ�12��MHC����λ����20��Ⱦɫ��̱ۣ�20q12���ϵľ���λ��,������������ͼ�ס����о�ͨ������Ŀǰ��֪��װˮƽ��õ�����HLA�������˫������Ե��ϵ�Ͻ���ţBoLA����[14]��Ϊ������������λ��ע��˫���յ�MHC����ͨ���������ο����ֵıȽϻ�����ѧ����,������˫���ջ������������ҵ���24��MHC�������Т������1��,����10��, �������13����,�����Ƴ���˫����MHC��Pseudo chromosome���������ǰ��MHC����Ķ�λ����,�÷���������㡢���Ѹ�١��ɱ�����,�����ƹ�Ӧ������������MHC����Ķ�λ��ע�͡���������������ͬһ�����ͷֻ������IJ�ͬ���ּ��������ͼ����˳��ı�����,�����ּ��һ�ֹ�ϵ,�����˻����ͬԴ���Լ����������˳���о�ͨ��ʹ��lastz�����ֱ������HLA��ţBoLA������ƴ�ӳ�����˫����CBLA�������н��й����Է�������,����ֻ�����кܸߵĹ�����,˵����3�ֶ���MHC������ͬԴ�Ժܸ�[15]���Ա�ͼ2��3,���Կ���ţBoLA����������ƴ�ӳ�����˫����CBLA�������о��и��õĹ�����,��ţ��˫���յĵ�MHC����ͬԴ�Ը��ߡ�˵�������������,ţ��˫������Ե��ϵ��������һ�����WU��[16]���ý���һ��,�Ӷ�֤�����������÷����Ľ����ȷ�ġ��ٶ�3�����ֽ���BLAT����,������MHC�������ϵĵ�λ�������ࡢ�ṹ��λ��Ҳ��������,��˿����϶�MHC����Ϊֱ��ͬԴ�����һ��[17,18]��������MHC�������ֹ��ܻ�����о�,ͨ��ѡȡ������Ե���������Ϊ���ս��бȽϻ�����ѧ�о�,���ü������ǿ������Ϣ������������ڸ����ֵ�ֱ��ͬԴ����,��ѡ�������Ҫ����ѧ���ܻ��ֵ�ĺ�ѡ��ǻ�����������о�,����Ѹ��ʵ�ֶ��������Ƚ���Ϣ�����ּ�ת��,��������µĹ��ܻ���Ͷ�λ��Ϣ,�Ӷ��ӿ���������ֻ�����ѧ�о�[19]��
Ŀǰ,������֪��HLA-���������106��,����36���������,����Ϊ�ٻ���;HLA-�������59��,���б������33��,����Ϊ�ٻ���;HLA-�������59��,��Ϊ�������[20,21]��������HLA�����������,˫��������ע�͵���MHC�����������ϴ�[22,23],�Ʋ�ԭ����������¼��㣺1��˫���ղο����������װˮƽ�ϲ�;2��˫����MHC������еͶ������ص�[24,25];3��������˫������Ե��ϵ��Զ,���������ֽ��������з����ı�[26,27,28]��������Ե��ϵ�Ͻ���ţBoLA���,˫����MHC������������һ�£���3��������,����3���ܹ����Թ۲쵽ţ��˫���յ�MHC�������ĸ���������������MHC�������ĸ���[29,30,31]�����,�Ƚ��о�˫����������MHC�����ڽ��������з����IJ��콫��һ����Ҫ���о�����,����ڽ�һ����ʾ˫����MHC�����þ�����Ҫ����,Ҳ���ǽ���ص��о����·���,ͬʱ,Ҳ����ţ��˫���յĻ������о����ƶ����á�
�ڶ�ţBoLA��˫����pseudo chromosome ���й����Է������ֵ����������4 000 000ʱţBoLA��˫����pseudo chromosome ������������ֵ�������,˵��˫����CBLA���ֻ�����ֵ��á�����2016��PLASILͨ��FISH�������Ƶ�����ͼ�������[11]������Ʋ�ò��ֻ�����ֵ��õ�ԭ������������ֽ��������з������øı�,���ֵ��øı�������MHC����Ķ�̬�ԡ�������һ����ķ��������Լ����������һ�����о���
4 ����
���о����ñȽϻ�����ѧ����,����Ŀǰ��֪��װˮƽ��õ�����HLA�������˫������Ե��ϵ�Ͻ���ţBoLA������Ϊ��������,�ɹ�������һ�����͵����ڶ�λ��ע��˫���յ�MHC����ķ�����ʹ�ø÷����ҵ��˴�����˫���ջ������е�24��MHC����,���Т��������1��,���������10��,���������13�����ֱ�ֲ���NW_011511766.1��ȫ��4.1M����NW_011515227.1��ȫ��1.2M����NW_011514613.1��ȫ��15K��3��scaffolds��,�Ң���͢������ֲ���NW_011515227.1��,���������ֲ���NW_011511766.1��NW_011514613.1��,��һ��������֪���������Ҫ�ֲ���NW_011511766.1 ��3.5��4.1M��λ��,�ɻ���˫����MHC��Pseudo chromosome��Լ1.8M,����˫������24��MHC�����������Ϣע��,Ϊ˫����MHC�Ľ�һ���о��춨�����ۻ�����The authors have declared that no competing interests exist.
�ο����� ԭ��˳��
������ȵ���
������������
�����ڿ�Ӱ������
| [1] | . . |
| [2] | . ��Ҫ��֯�����Ը�����(Major Histocompatibility Complex,MHC)�Ǽ��������������߹���������ص�һ���Ӵ�Ļ������,�ڵ�������ϵͳʶ������������֮��������Ҫ�����á���������ڶ��������Ŵ������ȷ�������˹㷺���о�,���Ĵ����ࡢ�����ࡢ����Ͳ����༸������ֱ����MHC������о���չ�����Ĵ�MHC����Ľṹ����ⷽ�������ܵȷ����������صIJ����� . ��Ҫ��֯�����Ը�����(Major Histocompatibility Complex,MHC)�Ǽ��������������߹���������ص�һ���Ӵ�Ļ������,�ڵ�������ϵͳʶ������������֮��������Ҫ�����á���������ڶ��������Ŵ������ȷ�������˹㷺���о�,���Ĵ����ࡢ�����ࡢ����Ͳ����༸������ֱ����MHC������о���չ�����Ĵ�MHC����Ľṹ����ⷽ�������ܵȷ����������صIJ����� |
| [3] | . |
| [4] | . The MHC, the most polymorphic and gene dense region in the vertebrate genome, contains many loci essential to immunity. In mammals, this region spans approximately 4 Mb. Studies of avian species have found the MHC to be greatly reduced in size and gene content with an overall locus organization differing from that of mammals. The chicken MHC has been mapped to two distinct regions (MHC-B and -Y) of a single chromosome. MHC-B haplotypes possess tightly linked genes encoding the classical MHC molecules and few other disease resistance genes. Furthermore, chicken haplotypes possess a dominantly expressed class I and class II B locus that have a significant effect on the progression or regression of pathogenic disease. In this study, we present the MHC-B region of the turkey (Meleagris gallopavo) as a similarly constricted locus, with 34 genes identified within a 0.2-Mb region in near-perfect synteny with that of the chicken MHC-B. Notable differences between the two species are three BG and class II B loci in the turkey compared with one BG and two class II B loci in the chicken MHC-B. The relative size and high level of similarity of the turkey MHC in relation to that of the chicken suggest that similar associations with disease susceptibility and resistance may also be found in turkey. |
| [5] | . |
| [6] | . Bactrian camels serve as an important means of transportation in the cold desert regions of China and Mongolia. Here we present a 2.0165Gb draft genome sequence from both a wild and a domestic bactrian camel. We estimate the camel genome to be 2.3865Gb, containing 20,821 protein-coding genes. Our phylogenomics analysis reveals that camels shared common ancestors with other even-toed ungulates about 55-60 million years ago. Rapidly evolving genes in the camel lineage are significantly enriched in metabolic pathways, and these changes may underlie the insulin resistance typically observed in these animals. We estimate the genome-wide heterozygosity rates in both wild and domestic camels to be 1.0 �� 10(-3). However, genomic regions with significantly lower heterozygosity are found in the domestic camel, and olfactory receptors are enriched in these regions. Our comparative genomics analyses may also shed light on the genetic basis of the camel's remarkable salt tolerance and unusual immune system. |
| [7] | . |
| [8] | . Major histocompatibility complex ��MHC�� is widely found in vertebrate. Because of its high polymorphism, MHC genes have become the focus of attentionhop in relation to genetics and breeding and disease-resistance etc. This article make a retrospective conclusion on the major histocompatibility complex genes polymorphism and the relationship between this gene and disease, and other aspects on the domestic and foreign researchers�� papers in recent decades.Some of these contents as a reference for the next correlation experiments. . Major histocompatibility complex ��MHC�� is widely found in vertebrate. Because of its high polymorphism, MHC genes have become the focus of attentionhop in relation to genetics and breeding and disease-resistance etc. This article make a retrospective conclusion on the major histocompatibility complex genes polymorphism and the relationship between this gene and disease, and other aspects on the domestic and foreign researchers�� papers in recent decades.Some of these contents as a reference for the next correlation experiments. |
| [9] | . Comments on the major histocompatibility complex in genetic field. Development of immunological means to protect mice against the growth of transplanted syngeneic leukemias; Test of segregans for the presence of antigen II; Outcome of the linkage test. |
| [10] | . Abstract In the past few years the DNA sequence database for molecules of the MHC (major histocompatibility complex) has expanded greatly, yielding a more complete picture of the long-term rates and patterns of evolution of the MHC in vertebrates. Sharing of MHC allelic lineages between long-diverged species (trans-species evolution) has been detected virtually wherever it is sought, but new analyses of linked neutral regions and the complexities of sequence convergence and microrecombination in the peptide binding region challenge traditional phylogenetic analyses. Methods for estimating the intensity of selection on MHC genes suggest that viability is important, but recent studies in natural populations of mammals give inconsistent results concerning mate choice. The complex and interacting roles of microrecombination, parasite-mediated selection and mating preferences for maintaining the extraordinary levels of MHC polymorphism observed are still difficult to evaluate. |
| [11] | . The Major Histocompatibility Complex (MHC) is a genomic region containing genes with crucial roles in immune responses. MHC class I and class II genes encode antigen-presenting molecules expressed on the cell surface. To counteract the high variability of pathogens, the MHC evolved into a region of considerable heterogeneity in its organization, number and extent of polymorphism. Studies of MHCs in different model species contribute to our understanding of mechanisms of immunity, diseases and their evolution. Camels are economically important domestic animals and interesting biomodels. Three species of Old World camels have been recognized: the dromedary (Camelus dromedarius), Bactrian camel (Camelus bactrianus) and the wild camel (Camelus ferus). Despite their importance, little is known about the MHC genomic region, its organization and diversity in camels. The objectives of this study were to identify, map and characterize the MHC region of Old World camelids, with special attention to genetic variation at selected class MHC II loci. Physical mapping located the MHC region to the chromosome 20 in Camelus dromedarius. Cytogenetic and comparative analyses of whole genome sequences showed that the order of the three major sub-regions is entromere - Class II Class III Class I .DRA, DRB, DQAandDQBexon 2 sequences encoding the antigen binding site of the corresponding class II antigen presenting molecules showed high degree of sequence similarity and extensive allele sharing across the three species. Unexpectedly low extent of polymorphism with low numbers of alleles and haplotypes was observed in all species, despite different geographic origins of the camels analyzed. TheDRAlocus was found to be polymorphic, with three alleles shared by all three species.DRAandDQAsequences retrieved from ancient DNA samples ofCamelus dromedariussuggested that additional polymorphism might exist. This study provided evidence that camels possess an MHC comparable to other mammalian species in terms of its genomic localization, organization and sequence similarity. We described ancient variation at theDRAlocus, monomorphic in most species. The extent of molecular diversity of MHC class II genes seems to be substantially lower in Old World camels than in other mammalian species. The online version of this article (doi:10.1186/s12864-016-2500-1) contains supplementary material, which is available to authorized users. |
| [12] | . Abstract Cytogenetic chromosome maps offer molecular tools for genome analysis and clinical cytogenetics and are of particular importance for species with difficult karyotypes, such as camelids (2n = 74). Building on the available human-camel zoo-fluorescence in situ hybridization (FISH) data, we developed the first cytogenetic map for the alpaca (Lama pacos, LPA) genome by isolating and identifying 151 alpaca bacterial artificial chromosome (BAC) clones corresponding to 44 specific genes. The genes were mapped by FISH to 31 alpaca autosomes and the sex chromosomes; 11 chromosomes had 2 markers, which were ordered by dual-color FISH. The STS gene mapped to Xpter/Ypter, demarcating the pseudoautosomal region, whereas no markers were assigned to chromosomes 14, 21, 22, 28, and 36. The chromosome-specific markers were applied in clinical cytogenetics to identify LPA20, the major histocompatibility complex (MHC)-carrying chromosome, as a part of an autosomal translocation in a sterile male llama (Lama glama, LGL; 2n = 73,XY). FISH with LPAX BACs and LPA36 paints, as well as comparative genomic hybridization, were also used to investigate the origin of the minute chromosome, an abnormally small LPA36 in infertile female alpacas. This collection of cytogenetically mapped markers represents a new tool for camelid clinical cytogenetics and has applications for the improvement of the alpaca genome map and sequence assembly. The American Genetic Association. 2012. All rights reserved. For permissions, please email: journals.permissions@oup.com. |
| [13] | . |
| [14] | . |
| [15] | . ����Ӳ�����������һ��ļ�����Ǿ���MHC�����ĵ͵ȼ������MHCI����ṹ��ߵȼ�������ȣ����ڽϴ���죬����Ѿ���Ϊ�Ƚϻ�����ѧ�о��Ľ��㡣��Ӳ������MHC�������о�ʼ��1990�꣬Hashimoto��ͨ���Ƚ��ˡ�����MHC�����3�ṹ��͢���½ṹ�������У���Ƹ߶ȼ�����������õ����㲿��MHC��������С���һ�о��ɹ��ҿ�������MHC�о�����Ļ�� . ����Ӳ�����������һ��ļ�����Ǿ���MHC�����ĵ͵ȼ������MHCI����ṹ��ߵȼ�������ȣ����ڽϴ���죬����Ѿ���Ϊ�Ƚϻ�����ѧ�о��Ľ��㡣��Ӳ������MHC�������о�ʼ��1990�꣬Hashimoto��ͨ���Ƚ��ˡ�����MHC�����3�ṹ��͢���½ṹ�������У���Ƹ߶ȼ�����������õ����㲿��MHC��������С���һ�о��ɹ��ҿ�������MHC�о�����Ļ�� |
| [16] | . Abstract Bactrian camel (Camelus bactrianus), dromedary (Camelus dromedarius) and alpaca (Vicugna pacos) are economically important livestock. Although the Bactrian camel and dromedary are large, typically arid-desert-adapted mammals, alpacas are adapted to plateaus. Here we present high-quality genome sequences of these three species. Our analysis reveals the demographic history of these species since the Tortonian Stage of the Miocene and uncovers a striking correlation between large fluctuations in population size and geological time boundaries. Comparative genomic analysis reveals complex features related to desert adaptations, including fat and water metabolism, stress responses to heat, aridity, intense ultraviolet radiation and choking dust. Transcriptomic analysis of Bactrian camels further reveals unique osmoregulation, osmoprotection and compensatory mechanisms for water reservation underpinned by high blood glucose levels. We hypothesize that these physiological mechanisms represent kidney evolutionary adaptations to the desert environment. This study advances our understanding of camelid evolution and the adaptation of camels to arid-desert environments. |
| [17] | . |
| [18] | . pAbstract/p pBackground/p pThe major histocompatibility complex (MHC) is a group of genes with a variety of roles in the innate and adaptive immune responses. MHC genes form a genetically linked cluster in eutherian mammals, an organization that is thought to confer functional and evolutionary advantages to the immune system. The tammar wallaby it(Macropus eugenii/it), an Australian marsupial, provides a unique model for understanding MHC gene evolution, as many of its antigen presenting genes are not linked to the MHC, but are scattered around the genome./p pResults/p pHere we describe the core tammar wallaby MHC region on chromosome 2q by ordering and sequencing 33 BAC clones, covering over 4.5 MB and containing 129 genes. When compared to the MHC region of the South American opossum, eutherian mammals and non-mammals, the wallaby MHC has a novel gene organization. The wallaby has undergone an expansion of MHC class II genes, which are separated into two clusters by the class III genes. The antigen processing genes have undergone duplication, resulting in two copies of TAP1 and three copies of TAP2. Notably, Kangaroo Endogenous Retroviral Elements are present within the region and may have contributed to the genomic instability./p pConclusions/p pThe wallaby MHC has been extensively remodeled since the American and Australian marsupials last shared a common ancestor. The instability is characterized by the movement of antigen presenting genes away from the core MHC, most likely via the presence and activity of retroviral elements. We propose that the movement of class II genes away from the ancestral class II region has allowed this gene family to expand and diversify in the wallaby. The duplication of TAP genes in the wallaby MHC makes this species a unique model organism for studying the relationship between MHC gene organization and function./p |
| [19] | . . |
| [20] | . |
| [21] | . The genomic organisation of the Major Histocompatibility Complex (MHC) varies greatly between different vertebrates. In mammals, the classical MHC consists of a large number of linked genes (e.g. greater than 200 in humans) with predominantly immune function. In some birds, it consists of only a small number of linked MHC core genes (e.g. smaller than 20 in chickens) forming a minimal essential MHC and, in fish, the MHC consists of a so far unknown number of genes including non-linked MHC core genes. Here we report a survey of MHC genes and their paralogues in the zebrafish genome. Using sequence similarity searches against the zebrafish draft genome assembly (Zv4, September 2004), 149 putative MHC gene loci and their paralogues have been identified. Of these, 41 map to chromosome 19 while the remaining loci are spread across essentially all chromosomes. Despite the fragmentation, a set of MHC core genes involved in peptide transport, loading and presentation are still found in a single linkage group. The results extend the linkage information of MHC core genes on zebrafish chromosome 19 and show the distribution of the remaining MHC genes and their paralogues to be genome-wide. Although based on a draft genome assembly, this survey demonstrates an essentially fragmented MHC in zebrafish. |
| [22] | . |
| [23] | . |
| [24] | .[D]. |
| [25] | . <FONT face=Verdana>����Ӧ�ñȽϻ�����ѧ��������Ϣѧ�����Ƚ�������ţ��¹��MHC-DQA1�������У����Ըû�����Ŵ������Լ�����ϵͳ���������˷��������������38���������м�107����̬λ�㣬������38�������͡����ּ估������MHC��DQA1���ڽϷḻ���Ŵ������ԡ���<BR> </FONT> . <FONT face=Verdana>����Ӧ�ñȽϻ�����ѧ��������Ϣѧ�����Ƚ�������ţ��¹��MHC-DQA1�������У����Ըû�����Ŵ������Լ�����ϵͳ���������˷��������������38���������м�107����̬λ�㣬������38�������͡����ּ估������MHC��DQA1���ڽϷḻ���Ŵ������ԡ���<BR> </FONT> |
| [26] | . |
| [27] | . The northern pig-tailed macaque(Macaca leonina) has been identified as an independent species from the pig-tailed macaque group.The species is a promising animal model in HIV/AIDS pathogenesis and vaccine studies due to susceptibility to HIV-1.However,the major histocompatibility complex(MHC) genetics in northern pig-tailed macaques remains poorly understood.We have previously studied the MHC class �� genes in northern pig-tailed macaques and identified 39 novel alleles.Here,we describe the MHC class �� alleles in all six classical loci(DPA,DPB,DQA,DQB,DRA,and DRB) from northern pig-tailed macaques using a sequence-based typing method for the first time.A total of 60 MHCII alleles were identified of which 27 were shared by other macaque species.Additionally,northern pig-tailed macaques expressed a single DRA and multiple DRB genes similar to the expression in humans and other macaque species.Polymorphism and positive selection were detected,and phylogenetic analysis suggested the presence of a common ancestor in human and northern pig-tailed macaque MHC class �� allelic lineages at the DQA,DQB and DRB loci.The characterization of full-length MHC class �� alleles in this study significantly improves understanding of the immunogenetics of northern pig-tailed macaques and provides the groundwork for future animal model studies. |
| [28] | . Twenty new world camelidae primer pairs were selected for cross-species amplification of loci in the old world camelids, the dromedary (Camelus dromedarius) and the bacterian (camelus bactrianus). A panel of DNA samples from 34 unrelated dromedaries (Kenya) and from 34 unrelated bactrians (32 domestic bactrians and two wild bactrians, China) were amplified. DNA samples from two llamas (Lama glama) and two alpacas (Lama pacos) were used as positive controls. PCR conditions; amplification and polymorphism, and microsatellite sequences were assessed and results presented. |
| [29] | . Both archaeological data and the presence of few mitochondrial DNA lineages suggest that most widespread domestic mammals (cattle, sheep, goats, pigs and dogs) derive from only a handful of domestication events. However, each of these species shows a high level of diversity at the nuclear genes of the major histocompatibility complex (MHC). Through simulations incorporating various degrees of population subdivision, growth rate and selection, we demonstrate that the numerous MHC DRB alleles that are present in modern domestic mammals implies that substantial backcrossing with wild ancestors, either accidental or intentional, has been important in shaping the genetic diversity of our domesticates. These results support the view that, contrary to common assumption, domestic and wild lineages might not have been clearly separated throughout their history. |
| [30] | . |
| [31] | . The single�\humped dromedary (Camelus dromedarius) is the most numerous and widespread of domestic camel species and is a significant source of meat, milk, wool, transportation and sport for millions of people. Dromedaries are particularly well adapted to hot, desert conditions and harbour a variety of biological and physiological characteristics with evolutionary, economic and medical importance. To understand the genetic basis of these traits, an extensive resource of genomic variation is required. In this study, we assembled at 65�� coverage, a 2.0602Gb draft genome of a female dromedary whose ancestry can be traced to an isolated population from the Canary Islands. We annotated 2102167 protein�\coding genes and estimated ~33.7% of the genome to be repetitive. A comparison with the recently published draft genome of an Arabian dromedary resulted in 1.9102Gb of aligned sequence with a divergence of 0.095%. An evaluation of our genome with the reference revealed that our assembly contains more error�\free bases (91.2%) and fewer scaffolding errors. We identified ~1.4 million single�\nucleotide polymorphisms with a mean density of 0.7102��0210613per base. An analysis of demographic history indicated that changes in effective population size corresponded with recent glacial epochs. Ourde novoassembly provides a useful resource of genomic variation for future studies of the camel's adaptations to arid environments and economically important traits. Furthermore, these results suggest that draft genome assemblies constructed with only two differently sized sequencing libraries can be comparable to those sequenced using additional library sizes, highlighting that additional resources might be better placed in technologies alternative to short�\read sequencing to physically anchor scaffolds to genome maps. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}