0 引言
【研究意义】果树的矮化密植栽培因具有早果、高产、品质优良、易于管理、能够有效节约成本和增加利润等优点,已发展成为国内外极为重要的栽培模式。目前,实现矮密栽培的主要途径是利用矮化砧木和矮生品种,因此,发掘和利用优良的基因资源,进行矮化砧木和矮生品种的培育对果树生产具有重要意义。【前人研究进展】西洋梨矮生型实生变异品种‘Le Nain Vert’具有树体紧凑、矮小的特点,非常适合矮密栽培。研究发现,矮生梨在茎、叶的解剖结构上有区别于普通型梨的显著特征[1]。由于‘Le Nain Vert’的矮生性状受控于显性单基因[2],因此,该基因资源在梨矮生品种的培育方面具有重要应用价值。WANG等[3]将该基因命名为PcDw,并通过SSR标记将其定位于西洋梨基因组第16连锁群上。最近,李炜等[4]依据苹果与梨基因组间高度的保守性和共线性关系,以二者的参考基因组序列为基础,又筛选鉴定了一组与PcDw连锁的DNA分子标记,将该基因定位到了西洋梨的染色体片段scaffold00074上。利用简化基因组测序技术进行大规模高通量SNP分型是当前国际上动植物基因组学研究热点之一。美国Oregon大学发明的RAD(Restriction Association site DNA)技术是广泛应用的简化基因组测序技术[5]。2007年,MILLER等[6]发表了应用芯片杂交的RAD技术。2008年,BAIRD等[7]将RAD和NGS(Next-generation sequencing)结合,建立了RAD-seq技术。2012年,PETERSON等[8]对RAD技术进行改进,提出了ddRAD(double digest RAD)技术。针对RAD技术流程复杂的问题,ELSHIRE等[9]曾提出GBS(genotyping- by-sequencing)技术。随后,WANG等[10]推出基于llB型限制性内切酶的2b-RAD技术,该技术流程简便,重复性和标签代表性高,基于混合泊松分布模型的de novo SNP分型新算法(iML),可使假阳性率得到较大幅度降低[11]。目前,RAD测序技术在许多物种上得到了应用,例如大麦(Hordeum vulgare L. )[12]、黑麦草(Lolium perenne)[13]等,但在果树上还未见报道。【本研究切入点】为了最终达到分离鉴定PcDw的目的,还需要大量的遗传标记进一步对该基因进行更为精细的染色体定位。因此,在以往研究的基础上,继续高效开发更多与其紧密连锁的遗传标记是十分必要的。将2b-RAD测序技术与HRM分析技术相结合,进行果树重要农艺性状SNP标记的开发,是一个值得探索的途径。【拟解决的关键问题】随着西洋梨全基因组序列的公布[14],使大量获得SNP标记成为可能。本研究利用分离群体分组分析的原理[15],采用2b-RAD技术在矮生型/普通型梨对比基因池间进行比较分析,初步筛查可能与目标性状相关的SNP位点,然后利用高效的高分辨率熔解曲线(HRM)分析技术进行大规模的群体分析,以确认与梨矮生性状决定基因PcDw共分离的SNP标记,为进一步实现该基因的精细定位及分离鉴定提供重要依据。1 材料与方法
试验于2016年4—11月在青岛农业大学园艺学院植物遗传改良与育种重点实验室进行。1.1 基因组DNA 的提取与对比基因池的构建
以‘矮生梨’(Pyrus communis L.)ב茌梨’(P. bretschneideri Rehd.)及‘2-3’(P. communis L.×P. bretschneideri Rehd.)ב绿宝石’(P. Serotina Nakai.)2个F1杂交分离群体(即群体1和群体2)为试材。‘矮生梨’和‘2-3’为矮生型亲本;‘茌梨’和‘绿宝石’为普通型亲本。群体1共有215株,其中矮生型和普通型分别有115株和100株。群体2共有168株,其中矮生型和普通型各有89株和79株。2016年4月取梨树的幼叶,用天根植物基因组DNA提取试剂盒提取并纯化基因组DNA,用琼脂糖凝胶电泳和NanoDrop 2000超微量分光光度计(Thermo Fisher Scientific, USA)对DNA的质量和浓度进行检测,并将其浓度稀释到10 ng·L-1。从每个群体中分别抽取15个矮生型杂种和15个普通型杂种,将其DNA等量混合,形成矮生型基因池D1和D2以及普通型基因池S1和S2。
1.2 2b-RAD测序分析与SNP位点筛选
利用2b-RAD技术构建梨4个基因池的标签测序文库,并在Hiseq2500 v2平台进行single-end测序(此项工作由上海欧易生物医学科技有限公司完成)。将原始reads进行质量过滤,剔除不含有BsaXⅠ酶切识别位点的序列,剔除低质量序列(即大于10个碱基的质量分数小于20的序列),剔除有10个以上连续相同碱基的序列。从梨参考基因组序列(https:// www.rosaceae.org/species/pyrus/pyrus_communis/genome_v1.0)中提取包含BsaXⅠ酶切位点的标签作为参考序列。将每个样本的高质量reads利用SOAP软件[16](参数设置为:-M4-v2-r0)定位到参考序列上。获得可用于分型的unique标签数目及深度。利用最大似然法(maximum likelihood,ML)进行SNP位点的分型(分型条件:只留取标签内最多有3个SNP的标签位点;分型标签深度设置为500以下。选取在样本间能分型的SNP位点,采用种群结构分析软件对样本间及组间差异SNP位点筛选数据进行分析。1.3 与PcDw连锁的SNP标记的鉴定
对在4个对比基因池上产生的SNP标记进行分析,筛选出矮生型基因池与普通型基因池间的多态性SNP标记,然后查找位于西洋梨染色体片段scaffold00074上的SNP位点,在其两侧合适位置设计引物(Primer 4.0 software),用HRM技术分析这些SNP标记在群体上的分离行为,以确定是否为与PcDw紧密连锁的遗传标记。1.4 高分辨率熔解曲线(HRM)分析
HRM分析在LightCycler®480Ⅱ荧光定量PCR仪(Roche)上进行。反应试剂来自LightCycler®480 High Resolution Melting Master 试剂盒。反应体系为10 μL,内含10 ng基因组DNA 1 μL,1×Master Mix 5 μL,2.0 mmol·L-1 MgCl2 1 μL,上、下游引物(0.2 μmol·L-1)各1 μL,ddH2O 1 μL。PCR扩增程序为95℃预变性10 min,然后按95℃变性10 min、55℃退火15 s、72℃延伸10 s的程序进行45个循环。在PCR循环结束后,立即对扩增产物进行HRM检测,程序为:95℃ 1 min、40℃ 1 min、65℃ 1 s;在65℃升温至95℃的过程中,以25次/℃的频率收集荧光信息,最后降温至40℃。HRM分析用LightCycler®480的Gene Scanning软件(1.5 version)进行。
2 结果
2.1 2b-RAD测序结果对4个样本(即4个近等基因池)的2b-RAD标签测序文库的测序结果共产生67 186 260条reads,平均每个样本测序reads数为16 796 565。将原始reads进行质量过滤后的统计结果表明,每个样本获得平均unique标签数目为86 810,平均测序深度为77×。测序深度能够达到准确分型的标准,可以进行下一步SNP标记分型分析。SOAP软件定位结果表明,4个测序文库中含有酶切位点的高质量reads占测序原始reads的70%以上(表1),表明梨文库的测序质量较好。
2.2 矮生型/普通型对比基因池间SNP位点筛选
选取在样本间能分型的SNP位点,分别在来自群体1和群体2的4个对比基因池间进行比较分析,共筛查出可在矮生型/普通型表型间进行分型的SNP标记1 317个,进一步的标签筛查结果表明,其中有8个SNP位于西洋梨染色体片段scaffold00074(即PcDw的定位染色体片段,长度441 741 bp)上,其染色体位置及样品分型结果见表2。Table 1
表1
表12b-RAD测序结果
Table 1The result of 2b-RAD sequencing
| 基因池 Gene bulks | 原始reads数 No. of raw reads | 高质量reads数 No. of filtered reads | 高质量reads百分率 Percentage of high quality reads | unique标签数 Unique tags | 标签深度 Tags depth |
|---|---|---|---|---|---|
| D1 | 16796565 | 12558380 | 74.80% | 88503 | 68 |
| D2 | 16796565 | 15009108 | 89.40% | 84954 | 83 |
| S1 | 16796565 | 14336929 | 85.40% | 90151 | 76 |
| S2 | 16796565 | 14442251 | 86.00% | 83635 | 81 |
新窗口打开
Table 2
表2
表2定位到西洋梨染色体片段scaffold00074上的8个SNP标记及其分型信息
Table 2Eight SNP markers mapped on chromosome scaffold00074 of P. communis and their genotypes of each sample
| SNP位点 SNP locus | 2b-RAD标签参考序列 2b-RAD tags reference sequence | 染色体起始位置 Starting locus in the chromosome | 样本分型 Sample genotypes | |||
|---|---|---|---|---|---|---|
| D1 | D2 | S1 | S2 | |||
| SNP1 | TAACTTTTAACATGGGCTCCTGTTTGT | 18140 | AG | AG | GG | GG |
| SNP2 | GGCGGAAGGAGAAACGGTTTCTTGATA | 70046 | CT | CT | CC | CC |
| SNP3 | CCGTAACGGAGACAGGGTACCAAATAG | 99471 | AC | AC | AA | AA |
| SNP4 | GGCGAGTGGAGACCGTGTGTAAACAAA | 196511 | CG | CG | CC | CC |
| SNP5 | CGAACGACCACGCGCTCTCCATGTCAC | 267146 | AG | AG | GG | GG |
| SNP6 | GCTCAGCCTACTGAGCCTCCTGAGCAA | 333036 | AG | AG | GG | GG |
| SNP7 | GTTAAGGGGAGGTGGTGTGGGGGAATG | 353951 | AG | AG | GG | GG |
| SNP8 | TTCCTTGGGAGAGGGTGTTTATATAGT | 429465 | CT | CT | TT | TT |
新窗口打开
2.3 与PcDw位点共分离的SNP标记鉴定
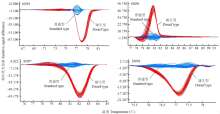
针对以上8个定位于scaffold00074上的SNP位点,分别设计8对PCR引物(表3),并分别在群体1和群体2上进行HRM分析,最终在群体1上检测到SNP1和SNP7两个与PcDw 位点共分离的SNP标记,在群体2上检测到SNP1、SNP6、SNP7和SNP8 4个可与PcDw 位点共分离的SNP标记。根据其扩增子的熔解曲线形状差异,可有效地区分矮生型和普通型表型(图1、图2)。在群体1的215个杂种后代和群体2的168个杂种后代中,未发现有标记与性状的重组类型。Table 3
表3
表38个SNP位点群体分型检测所用的HRM 引物
Table 3HRM primers designed for 8 SNPs genotyping in the whole populations
| SNP 位点 SNP loci | 正向引物序列 Forward primer sequence (5′-3′) | 反向引物序列 Reverse primer sequence (5′-3′) |
|---|---|---|
| SNP1 | GGGATTCTCTTCGGATCCTTTCT | TCTGCGCCGTTGAAAATTGA |
| SNP2 | GTGAGTTTGGCGGAAGGAGA | TGGCACAATTGAGCGGTACA |
| SNP3 | TGTTGGTGGCCATGAAGAGG | CCACTATTTGGTACCCTGTCTCC |
| SNP4 | GGATTCTCTCGATGCGCTTA | TTACACACGGTCTCCACTCG |
| SNP5 | TGGGAGGAGGGATCTTGCAT | AAAGGGGTGACATGGAGAGC |
| SNP6 | TCGACTGTCCCACCCATTAA | ACAGACCGTTGGATTTTGATCC |
| SNP7 | TGGCGATTCTTTGTGGATTGG | TGCCCTATTCTCCATCCGAT |
| SNP8 | TCTCGACAACGGGCATGTTA | GTCCAGATTCGGCGTACAGA |
新窗口打开
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图 1SNP1和SNP7在‘矮生梨’ב茌梨’群体中的HRM分析结果
-->Fig. 1Results of HRM analysis of SNP1 and SNP7 in the population produced by ‘Aishengli’בChili’
-->
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图 2SNP1、SNP6、SNP7和SNP8在‘2-3’ב绿宝石’群体中的HRM 分析结果
-->Fig. 2Results of HRM analysis of SNP1, SNP6, SNP7 and SNP8 in the population produced by ‘2-3’ בLvbaoshi’
-->
3 讨论
单核苷酸多态性(SNP)是基因组中最常见的遗传多态性。SNP标记已在果树的遗传图谱构建、基因标记与定位等研究中发挥了重要作用[17-20]。与RFLP、AFLP、SSR等传统基因分型技术相比,SNP分子具有分布广泛、代表性强、稳定性好、易于分析等优势。SNP分子标记的获得往往依赖于大规模、高通量的基因组数据分析[21]。对果树来说,其复杂的基因组是造成测序分析成本高的主要原因。简化基因组测序技术2b-RAD是改进的RAD技术,使测序更加简易、精确,已在高密度遗传图谱的构建、群体的遗传学研究、群体进化分析、标记开发等领域得到了应用[22-28]。高分辨率熔解曲线分析是一种高效的遗传变异检测方法,被广泛的应用于突变扫描、基因分型和标记开发等方面。目前,HRM技术已发展成为SNP标记分型的重要手段[29-31]。李炜等[4]曾依据苹果与梨基因组间高度的保守性和共线性关系,以苹果基因组序列为参考设计了大量引物,通过HRM分析仅筛查到1个与梨矮生性状决定基因PcDw紧密连锁的SNP标记。为了提高SNP位点的检测效率,本研究利用2b-RAD测序技术通过对4个对比基因池间共有标签的序列分析,筛查到了大量的可能与目标性状相关的SNP位点,对于初步筛查到的SNP标记,再利用高通量的HRM分析技术进行群体上的大规模验证,以确定其是否为与目标性状紧密连锁的遗传标记。这样,可避免了在群体上继续用2b-RAD技术进行验证分析造成的高成本问题。本研究基于这一策略,鉴定获得了一组与梨矮生性状决定基因PcDw位点连锁的新标记。可见,将2b-RAD测序技术与HRM分析技术相结合,进行果树重要农艺性状分子标记的开发,是一种行之有效的方法。
根据不同亲本构建的杂交群体的遗传背景是有差异的,因此,依据不同群体开发的遗传标记是不可能完全相同的。本研究结果显示,在群体1上,鉴定出了2个与PcDw连锁的SNP标记;在群体2上,鉴定出了4个与PcDw连锁的SNP标记。另外,对在不同群体上鉴定到的相同的SNP标记来说,其HRM分析熔解曲线形状也可能会出现差异。出现这一现象的原因,也与群体的遗传基础差别有关。本研究从2个不同的杂种群体上共检测到了4个新的与梨PcDw紧密连锁的DNA分子标记。这些标记,不仅可以作为目标基因定位的参考依据,也可成为目标性状标记辅助选择的有力工具。
4 结论
应用2b-RAD技术对2对矮生型/普通型对比基因池进行基因组测序分析,在对比基因池间筛选出单核苷酸多态性(SNP)位点1 317个,其中有8个SNP位点位于PcDw的定位染色体scaffold0007上。用HRM分析技术对这8个SNP位点进行群体上的进一步检测,共鉴定出4个与PcDw位点共分离的SNP标记,其中SNP1和SNP7为2对群体共有的SNP标记。这一研究结果对PcDw的精细定位及其候选基因的筛查具有重要参考。The authors have declared that no competing interests exist.

{kind=link}
{kind=link}
{kind=link}
{kind=link}