0 引言
【研究意义】割手密(Saccharums pontaneum L.)又称甘蔗细茎野生种,是禾本科蜀黍族甘蔗亚族甘蔗属多年生草本植物[1],成熟茎微甜,因此又称“甜根子草”,具有发达的地下横走茎,宿根性好、分蘖能力强、固土力强,一般生长于田埂、坡地和干旱沙地中,是甘蔗品种改良最重要的野生亲本和甘蔗抗旱、耐瘠等重要基因源[2-3]。在现代甘蔗品种的基因组中,割手密基因组约占10%—25%[4-5],对甘蔗产量、抗性育种具有重要作用。由于近年来全球气候变化的影响,农作物干旱问题日益突出,对山坡地种植的甘蔗作物尤为明显[6]。割手密发达的地下横走茎,叶片窄小的形态特征,且适宜在热带、亚热带的沙壤环境中生存,对干旱具有极强的耐受性。因此,甘蔗育种工作者十分重视割手密的抗旱、强分蘖等优良特性对甘蔗遗传改良的作用。【前人研究进展】由于割手密资源分布范围广,遗传多样性丰富[7],在资源利用上具有一定的盲目性。苏火生等[8]、齐永文等[9]通过割手密初级核心种质库的构建,成功筛选出代表不同生态类型和基因型的核心种质,为割手密后续的研究利用提供了参考;同时国内外****对割手密不同无性系的抗旱水平进行了较多的研究,认为在旱坡地、干旱的沙壤土中生长的割手密具有较好的抗旱和耐旱能力[10-12];这类土壤环境下的土壤持水量差,短时间的高温天气即可导致土壤和植物缺水,因此割手密在生长旺盛时期的抗旱性可能具有短时效应;同时割手密在干旱胁迫下的电导率较低,细胞破坏程度小,抗旱能力强于甘蔗品种[13]。与其他作物的野生亲本相比,割手密抗旱相关基因的研究还很少见报道。姚艳丽等[14]通过同源基因比较的方法,克隆了割手密2个与抗旱相关的DREB转录因子,通过原核表达分析,证明这两个基因是功能基因;国外****PARK等[15]通过转录组学研究,证明割手密TUS05-05的SspNIP2在冷处理30 min后表达量明显增加,同时通过转SspNIP2植株的水分胁迫测试表明,转基因植株比正常植株的蒸腾速率低,证明了该基因与抗寒和抗旱性状具有一定的关系。【本研究切入点】植物的抗旱机理是一个非常复杂的过程,仅通过部分抗旱相关基因的克隆与功能研究,不能更好地诠释抗旱机理,割手密作为甘蔗遗传改良重要的野生亲本,其本身受到野外环境的胁迫,抗旱性与甘蔗相比具有一定的遗传优势。然而割手密不同干旱胁迫时期的转录组数据及其表达谱等方面的研究还未见报道,且与割手密抗旱相关功能基因的研究也非常有限;目前,中国甘蔗主产区主要分布在广西和云南两省,且主产区大部分为湿热气候生态型。因此,非常有必要利用该生态类型的割手密资源进行干旱胁迫下的转录组分析研究,它不仅可以探寻到适合该区域的抗旱类型,从育种的角度分析,还可以使选育出的品种具有本土生态基因型,更适合于当地种植。云南割手密82-114属于湿热气候生态型资源,株高、茎径等性状表现优异,是割手密种质中利用较为成功的种质;通过前期的抗旱性筛选评价,云南割手密82-114的叶片持绿性与其他割手密材料相比有较好的表现,水分浇灌后恢复快,耐短时高温及干旱环境,适合中国甘蔗种植区域的气候类型;且利用该种质杂交获得的F2种质中有5份材料抗旱性超过中国主栽品种ROC22,3份材料抗性中等[16],说明该种质在抗旱性育种方面具有一定的潜力。【拟解决的关键问题】本研究以内陆湿热生态型割手密云南82-114为试验材料,通过ІlluminaHiSeqTM4000高通量测序平台,比较该种质在拔节期干旱胁迫24、48和72 h的根部转录组水平差异,分析可能参与抗旱或耐旱调控的相关基因,并进一步揭示割手密受干旱胁迫的响应机理,为割手密在甘蔗抗旱育种研究中的应用奠定分子基础。1 材料与方法
1.1 试验材料的培育及胁迫处理
以来自于国家甘蔗种质资源圃内的湿热型割手密云南82-114为材料,于2015年4月10日种植于云南省农业科学院甘蔗研究所抗旱温室,土壤以沙和红土按1﹕1的比例混合(此土壤基质保水能力弱,易于水分胁迫),栽种于12个直径为40 cm且含有15 cm厚的珍珠岩桶中。基于前期预试验结果,植株在这种栽培条件下停水24 h后即可出现根部缺水症状。试验在割手密拔节期前按正常浇水处理,待拔节期开始第15天早晨对桶栽材料进行正常灌溉,下午14时取3桶对照(CK)材料的根系,此后其余材料停止浇水,并在24 h(植株表现为叶片萎蔫,早晨植株无吐水现象,标记为A)、48 h(植株表现为中度干旱,叶片萎蔫下垂,下部叶鞘脱水,标记为B)、72 h(植株表现为重度干旱,植株下部叶片、叶鞘枯黄,土壤极度干旱,标记为C)后分别取一次根系组织,每次取3桶,混合取样,并将根系样品放入液氮中迅速冷冻,置于-70℃超低温冰箱中保存备用。1.2 割手密根总RNA的提取及cDNA的合成
将采取的样品用干冰盒包装好,送杭州联川生物技术有限公司提取RNA和测序。利用Nanodrop检测RNA质量,并经琼脂糖凝胶检测RNA完整性。样品总RNA提取后,经Oligo(dT)的磁珠富集纯化mRNA。经抽提的mRNA被随机打断成短片段后,以片段化的mRNA为模板,合成cDNA,并通过纯化、粘性末端修复为平末端,3′末端加碱基A并加接头,片段选择,最后进行PCR扩增,经文库质量检测合格后,安排上机测序。高通量测序用ІlluminaHiSeqTM 4000进行,整个流程由杭州联川生物技术有限公司完成。1.3 测序数据质量的评估和RAW数据的处理、Trinity组装
由于高通量测序错误率会随着测序序列的读长的增加而升高[16],需要对原始数据进行质量评估,分析不同长度片段测序的错误率。将由测序仪造成的错误率小于0.1%的碱基所占比例(Q30)来衡量测序的质量[17]。将测序仪测得的初始数据(raw reads)以FASTQ文件进行存储,并将Raw reads去除adapter,去除Raw reads中的杂质数据,得到clean reads,计算Raw Data和过滤后的Valid Data的比率。利用短reads组装软件Trinity进行序列的组装[18],将组装好的Contig进行聚类,用reads验证每个Component 的reads支持情况,并通过Butterfly合并在de Bruijn图中有连续节点的线性路径,剔除read支持少的路径。通过统计每个样品由raw reads 得到的测序碱基数、clean reads占Raw reads的比例、clean reads比对到contig的比例以及测序饱和度分析,来评估测序的质量[19]。1.4 割手密干旱胁迫下差异基因的筛选
利用组装好的基因作库,用序列相似性比对的方法求各基因在各样本中的表达丰度,基因的表达量利用RPKM[20](Reads Per Kilobase of exon model per Million mapped reads)值来衡量,RPKM值计算方法如下:RPKM=
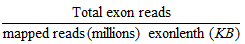
统计CK、A、B和C 4个样本的基因表达丰度,从整体水平观察样本间的表达情况和基因表达水平的离散程度;以P-value(假设认为2个不同样本间的所有基因都不存在差异的情况下出现的极端情况概率)<0.01为非常显著,利用错误发现率(false discovery rate,FDR)≤0.001为阈值[21](FDR值越低,表明基因表达差异越显著),筛选出样本间的差异表达基因(differentially expressed genes,DEGs),并从差异倍数(log2foldchange≥2)和显著性水平(P-value)进行评估,以未进行水分胁迫处理的CK为对照,对A、B、C 3个胁迫样本表达基因的上下调关系进行统计。
1.5 差异表达基因的功能注释与分析
Gene Ontology功能显著性富集分析首先把所有得到的差异表达基因向Gene Ontology数据库(http://www. geneontology.org)的各个term映射,计算每个term的基因数目,然后应用超几何检验,找出与整个基因组背景相比,在DEGs中显著富集的GO条目,其计算公式为:P=1–
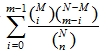
其中,N为所有基因总数;n为N中DEGs的数目;M为所有基因注释为某特定GO term的基因数目;m为注释为某特定GO term的DEGs数目。以P-value≤0.05为阈值,满足此条件的GO term定义为在DEGs中显著富集的GO term。通过GO功能显著性富集分析确定DEGs行使的主要生物学功能。同时将DEGs序列在KEGG数据库(kyoto encyclopedia of gene and genomes,京都基因与基因组百科全书)中进行注释[22],其显著性富集分析的P-value值计算和阈值同GO富集分析相同。通过Pathway显著性富集确定DEGs参与的最主要生化代谢途径和信号转导途径。
2 结果
2.1 转录组测序质量检测和RAW数据整理分析
通过对照样品CK和A、B、C胁迫组样品的转录组测序,分别获得50 408 133、46 705 498、43 087 971和49 777 187条Raw reads,按照测序仪造成的错误率小于0.1%的碱基来衡量测序质量,如图1所示,CK、A、B和C样品的质量检测,测序仪读取1—99个碱基质量的平均值均高于Q30,测得序列的GC含量占47%,其他样本的Q30和GC含量均符合上述质量要求,说明转录组测序质量较高,可以进行下一步的数据整理和分析。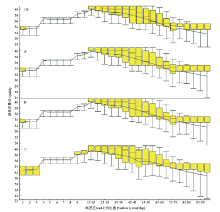 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图1CK、A、B和C样本测序错误率检测横坐标表示分布在read上碱基的位置,纵坐标代表碱基的质量
-->Fig. 1The sequencing error rate detection of CK, A, B and C samples X-axis represents the base position distributing in read, Y-axis represents the base quality
-->
经过原始Raw数据的整理和筛选分析,CK、A、B和C样本的有效数据量均有所下降,与Raw数据相比,筛选后的有效数据量分别占98%、98.5%、97.8%和98.7%(表1)。经过denovo的拼接结果显示,Transcript的总量为346 744,而unigene的总量为152 225,其中CK样本总量为134 724个,A样本总量为130 368个,B样本总量为133 564个,C样本总量为131 321个。转录本的长度分布主要集中在200 bp左右,并随着长度的增加片段数量迅速下降,在片段长度达到1 900 bp时降低到5 426个,此后大于2 000 bp的片段总计又增加到45 307个,与unigene长度的分布相比,整体分布较一致。但unigene的频数有所下降,说明存在一定量的可变剪接或单倍型序列。
Table 1
表1
表1数据产量统计
Table 1The statistics of data yield
| 样本 Sample | 初始数据 Raw data | 有效数据 Valid data | 有效率 Valid ratio(reads)(%) | ||
|---|---|---|---|---|---|
| 序列数 Read | 序列长度 Base | 处理后序列数 Clean reads | 处理后序列长度 Clean bases | ||
| CK | 50408133 | 5040813300 | 49399970 | 4939997000 | 98 |
| A | 46705498 | 4670549800 | 46004916 | 4600491600 | 98.5 |
| B | 43087971 | 4308797100 | 42140036 | 4214003600 | 97.8 |
| C | 49777187 | 4977718700 | 49130084 | 4913008400 | 98.7 |
新窗口打开
2.2 样本基因的注释和差异表达基因的筛选分析
将获得的unigene基因分别在Swiss-Prot、Nr、Pfam、KEGG和KOG中注释,结果云南割手密82-114在对照和不同胁迫处理组中共包含152 225个基因,其中70 355个基因得到注释,81 870个基因未得到注释(表2),在注释的基因中,来自Nr数据库注释的基因最多,达到70 186个。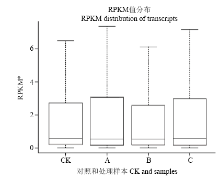 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图2样本表达水平RPKM盒形图*RPKM为每百万reads中来自于某基因每千碱基长度的reads数
-->Fig. 2The RPKM box map of expression levels for samples *RPKM is fragments per kb per million fragment
-->
Table 2
表2
表2割手密unigene基因的BLAST注释结果
Table 2The unigene annotation result of Saccharum spontaneum for BLAST
| 基因数目 Gene numbers | Swiss-Prot | Nr | Pfam | KEGG | KOG |
|---|---|---|---|---|---|
| 152225 | 32478 | 70186 | 49160 | 66872 | 40746 |
| 21.34% | 46.11% | 32.29% | 43.93% | 26.77% |
新窗口打开
利用RPKM来衡量各样本间的基因表达丰度,将各样本中RPKM值用最大值、上四分位数、中值、下四分位数和最小值来统计,以RPKM盒型图来表示(图3),样本A和C的表达丰度高于CK和B样本,但B样本的中位数水平整体高于其他样本;从胁迫样本间基因表达的离散程度来看,A和C样本的离散程度较高,B样本的离散程度相对较低。
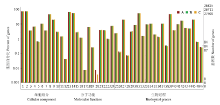 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图3A、B、C三个胁迫样本与CK样本差异表达基因的GO功能分类1:细胞;2:细胞部分;3:包膜;4:细胞外区域;5:细胞外区域部分;6:大分子复合物;7:膜封闭腔;8:细胞器;9:细胞器部分;10:共质体途径;11:抗氧化剂;12:辅助转运蛋白;13:结合;14: 催化剂;15:电子载体;16:酶调节因子;17:金属伴侣;18:分子传感器系统;19:营养库活性;20:蛋白标记;21:结构性分子;22:转录调节因子;23:翻译调节活性;24:运输;25:组织结构形成;26:生物附着;27:生物调节;28:细胞杀伤;29:细胞组分起源;30:细胞组分定位;31:细胞过程;32:致死;33:发育过程;34:定位建立;35:生长;36:免疫系统过程;37:定位;38:移位;39:代谢过程;40:多细胞/组织过程;41:多细胞有机过程;42:色素沉淀;43:生殖;44:生殖过程;45:响应刺激;46:节律过程;47:病毒复制
-->Fig. 3The GO functional classification of the differential expression gene from A, B, C samples and CK1: Cell; 2: Cell part; 3: Envelope; 4: Extracelluar region; 5: Extracellular region part; 6: Macromolecular complex; 7: Membrane-enclosed lumen; 8: Organelle;9: Organelle part; 10: Symplast; 11: Antioxidant; 12: Auxillary transport protein; 13: Binding; 14: Catalytic; 15: Electron carrier;16: Enzyme regulator; 17: Metallochaperone; 18: Molecular transducer; 19: Nutrient reservoir; 20: Protein tag; 21: Structural molecule; 22: Transcription regulator; 23: Translation regulator; 24: Transporter; 25: Anatomical structure formation; 26: Biological adhesion; 27: Biological regulation; 28: Cell killing; 29: Cellular component biogenesis; 30: Cellular component organization; 31: Cellular process; 32: Death; 33: Developmental process; 34: Establishiment of localization; 35: Growth; 36: Immune system process; 37: Localization; 38: Locomotion; 39: Metabolic process; 40: Multi/organism process; 41: Multicellular organismal process; 42: Pigmentation; 43: Reproduction; 44: Reproductive process; 45: Response to stimulus; 46: Rhythmic process; 47: Viral reproduction
-->
以FDR≤0.05和|log2 fold change|≥1来评估样本间的差异基因,与CK相比,A、B、C 3个样本在分别胁迫24、48和72 h后,上调和下调基因均明显增加,其中,C样本的上调基因为78 644个,是3个样本中上调表达基因数目最多的,其次为B样本,A样本最少;下调表达最多的样本为A,最少的样本为C(表3)。
Table 3
表3
表3差异基因的比较分析
Table 3The comparative analysis of differential genes
| 与CK比较Compare to CK | 上调 Up | 下调 Down | 显著上调 Remarkably up | 显著下调 Remarkably down |
|---|---|---|---|---|
| A | 74906 | 68761 | 3061 | 1302 |
| B | 76369 | 68182 | 2304 | 2841 |
| C | 78644 | 67654 | 3236 | 2525 |
新窗口打开
以各样本中前50个DEGs为筛选对象,FDR值=0为筛选条件,共筛选出79个DEGs,其中上调表达占39个,下调表达占40个。3个胁迫样本间的极显著上调和极显著下调表达基因数不同,其中A样本包含2个上调,10个下调基因;B样本包含23个上调和18下调基因;C样本包含13上调和12下调基因。从A、B和C 3个样本的前50个差异基因来看,大部分涉及转录激活、水分运输、DNA结合、ATP结合和蛋白酶结合相关基因,以及与细胞膜、跨膜运输、蔗糖代谢和防御反应相关的基因。从不同的文献报道来看,这些基因的激活和过量表达与干旱胁迫有着十分密切的关系[23]。
2.3 差异表达基因的Gene Ontology富集分析
Gene Ontology(简称GO)是一个国际标准化的基因功能分类体系,能够全面描绘生物体中基因和基因产物的属性。根据云南割手密82-114在干旱胁迫下筛选的不同DEGs,研究DEGs在GO中的分布状况,阐明试验中样本处理差异在基因功能上的体现。以CK作为对照,A、B、C 3个胁迫处理的样本经过GO功能显著性富集分析,在参与的生物过程、细胞组件和分子功能中,A和C样本均有47个小类别,而B样本则缺少一个代谢通路(图3)。与CK对比,在生物学途径中,A样本共有1 480个DEGs被注释,其中富集最多的是与DNA依赖型转录(transcription DNA-dependent)相关和转录调节子(regulation of transcription)相关的基因;B样本有2 499个DEGs被注释,其中富集最多的类别与A相同;C样本有3 165个DEGs被注释,其中富集最多的类别是与翻译(translation)相关的基因。在细胞组件中,A样本共有1 517个DEGs被注释,其中与内在的膜(integral to membrane)和核(nucleus)相关的基因富集最多;B样本共有2 629个DEGs被注释,富集最多的基因类别与A样本相同;C样本共有3 326个DEGs被注释,其中富集最多的基因类别同样与A相同。在分子功能中,A样本有1 760个DEGs被注释;B样本有3 093个DEGs被注释;C样本有3 825个DEGs被注释;而3个样本中富集最多的类别均是与ATP结合相关的基因。从图3中可以看出,A、B和C样本的差异基因数小于GO注释的差异基因数目,说明差异基因中存在多拷贝现象,符合异源多倍体植物含有多个单倍型基因的特点。2.4 差异表达基因的KEGG富集分析
通过KEGG富集分析,确定云南割手密82-114不同干旱胁迫时期,DEGs参与的最主要生化代谢途径和信号途径。找出与整个基因组背景相比,在DEGs中显著性富集的Pathway。结果表明,A样本有2 248个DEGs注释到KEGG数据库中107个分类代谢途径中,B样本有2 114个DEGs注释到KEGG数据库中130个分类代谢途径中,C样本有2 392个差异表达基因注释到KEGG通路中144个分类代谢途径中。以CK作为对照,经KEGG显著性富集分析,如表4所示,A样本上调的代谢通路有2个,下调的代谢通路有3个;B样本上调的代谢通路有1个,下调的代谢通路有6个;C样本下调的代谢通路有2个,上调的代谢通路有3个。Table 4
表4
表4KEGG代谢途径显著性富集的代谢通路
Table 4The significant enrichment in metabolic pathways from KEGG
| 样本 Sample | 胁迫时间 Stress time (h) | 功能定义 Function definition | KEGG显著富集的代谢通路 The metabolic pathway of significantly enriched for KEGG | 表达水平 Regulation |
|---|---|---|---|---|
| A | 24 | 细胞外信号调节激酶 Extracellular signal-regulated kinase | MAP激酶信号途径 MAPK signaling pathway-fly | ↑ |
| 磷脂酶 D Phospholipase D | 醚脂类代谢途径Ether lipid metabolism | ↑ | ||
| β-氨基己糖苷酶 β-hexosaminidase | 鞘糖脂生物合成途径-环球系列 Glycosphingolipid biosynthesis - globo series | ↓ | ||
| β-氨基己糖苷酶 β-hexosaminidase | 糖胺聚糖降解途径 Glycosaminoglycan degradation | ↓ | ||
| β-氨基己糖苷酶 β-hexosaminidase | 鞘糖脂生物合成途径-神经节系列 Glycosphingolipid biosynthesis-ganglio series | ↓ | ||
| B | 48 | ATP结合盒B亚族(MDR/TAP) -1 ATP-binding cassette, subfamily B (MDR/TAP), member 1 | ABC转运蛋白 ABC transporters | ↑ |
| β-氨基己糖苷酶β-hexosaminidase | 鞘糖脂生物合成途径-环球系列 Glycosphingolipid biosynthesis - globo series | ↓ | ||
| β-半乳糖苷酶 β-galactosidase | 多聚糖降解途径 Other glycan degradation | ↓ | ||
| β-氨基己糖苷酶 β-hexosaminidase | 鞘糖脂生物合成途径-神经节系列 Glycosphingolipid biosynthesis-ganglio series | ↓ | ||
| β-氨基己糖苷酶 β-hexosaminidase | 糖胺聚糖降解途径 Glycosaminoglycan degradation | ↓ | ||
| 脂氧合酶 Lipoxygenase | 亚油酸新陈代谢途径Linoleic acid metabolism | ↓ | ||
| β-半乳糖苷酶 β-galactosidase | 鞘脂类代谢途径 Sphingolipid metabolism | ↓ | ||
| C | 72 | 细胞外信号调节激酶 Extracellular signal-regulated kinase | MAP激酶信号途径 MAPK signaling pathway-fly | ↑ |
| 乙酰乳酸合成酶I/II/III大亚基Acetolactate synthase I/II/III large subunit /支链氨基酸转氨酶 Branched-chain amino acid aminotransferase | 泛酸盐和辅酶A生物合成 Pantothenate and CoA biosynthesis | ↑ | ||
| 乙酰乳酸合成酶I/II/III大亚基 Acetolactate synthase I/II/III large subunit | C5二元酸分支代谢途径 C5-Branched dibasic acid metabolism | ↑ | ||
| 甘氨酸羟甲基转移酶 Glycine hydroxymethyl-transferase | 叶酸-碳代谢通路 One carbon pool by folate | ↓ | ||
| ATP结合盒B亚族(MDR/TAP) -1 ATP-binding cassette, subfamily B (MDR/TAP), member 1 | ABC转运蛋白 ABC transporters | ↓ |
新窗口打开
在上调的代谢途径中,MAP激酶(促分裂原活化蛋白激酶)信号途径在A样本和C样本中同时出现,该途径可将不同的细胞膜感受器与细胞应答联系起来,响应各种生物和非生物胁迫,在植物激素信号传递、细胞发育和分裂过程中起着重要的作用[24]。近年来,随着功能获得型突变体的获得,MAP激酶级联途径在干旱胁迫下信号传导中的功能和作用得到了广泛的验证。利用获得的MAP激酶序列进行蛋白相似性比对和进化分析,所获得的MAP激酶与水稻的OsMAPK5有84%的同源性,与栗(Setariaitalical)的MAPK5有88%的同源性,相似性最高,在蛋白进化分析中距离最近;而在水稻中的研究结果表明,水稻OsMAPK5的过表达对非生物胁迫的抵抗力增加,对非生物胁迫具有正调控作用[25],在盐、干旱、37℃处理都能诱导水稻OsMAPK5表达[26],与割手密云南82-114所获得的上调控MAP代谢途径相一致,说明A和C样本所获得的MAP激酶有可能参与非生物胁迫的正调控。
同时在KEGG富集中,3份胁迫样本中仅有A样本含有醚脂类代谢途径,且表现为上调,参与该途径调控的磷脂酶D(PLD)不仅是生物膜的骨架成分,而且能够通过水解后产生的产物来参与多种生理过程以及环境刺激引起的细胞反应,在PLD活性受反义抑制后,拟南芥叶片对ABA的敏感性下降,因此会阻碍气孔关闭,导致水分缺失,这种现象在拟南芥PLD缺失突变株[27]和更苏(Craterostigma plantagineum)植物的PLD活性检测[28]中已经得到证实;本研究在A样本中筛选到的差异表达基因PLD与玉米PLD1有94%的同源性,且在干旱胁迫中处于上调表达,表明PLD在割手密云南82-114干旱胁迫进程中同样发挥着重要调控作用。
在A与B样本中同时存在下调的代谢途径有鞘糖脂生物合成途径、糖胺聚糖降解途径,这些途径都涉及β-氨基己糖苷酶(β-HEX),且都是起到下调作用;鞘脂是细胞质膜和叶泡膜结构的重要成分,在植物膜稳定性、细胞信号和逆境反应中发挥重要的作用[29-32],如图4所示,鞘糖脂生物合成的2个途径都受到β-氨基己糖苷酶和α-半乳糖苷酶的调节,且在A和B样本中都处于下调表达,在拟南芥的β-氨基己糖苷酶的研究中,已经发现多个亚型,其中HEXO2和HEXO3蛋白主要位于质膜上[33],说明水分胁迫后鞘脂在膜的稳定性和细胞信号传导的作用受到影响。在图4中,β-氨基己糖苷酶下调导致催化形成的神经酰胺类物质减少,而神经酰胺具有启动细胞的能力,促进细胞的新陈代谢,促使角质蛋白有规律再生的功能,说明受干旱胁迫的A和B样本在根系细胞的新陈代谢、能力的恢复中产生了负面影响,增加上述基因的表达活性可对延缓细胞衰老,增加根系再生具有重要作用。
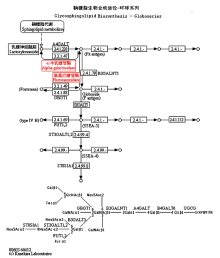 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图4鞘糖脂生物合成途径-环球系列红色字体为差异表达基因
-->Fig. 4Glycosphingolipid biosynthesis-globo seriesThe red fonts for the differentially expressed genes
-->
3 讨论
本研究应用RNA-seq技术对云南割手密82-114进行干旱胁迫前后的转录组比较分析,经GO和KEGG的显著性富集分析,筛选的差异基因类型与前人在甘蔗[34]、小麦[35]、楸树[36]等分析的结果基本一致,主要表现为植物膜结合和信号传导相关的基因最多。由于甘蔗和割手密的基因组还没完成测序,本研究从不同胁迫时间段对割手密进行转录组测序分析,探讨割手密随着干旱胁迫时间的变化,其差异表达基因、代谢通路是否有一定的差异,寻找与干旱胁迫相关的抗性基因;同时3个胁迫组分别测序,对增加数据分析的可靠性具有一定好处。通过GO富集分析,3个胁迫样本在参与的生物学途径并不完全相同,其中A和B样本富集的DNA依赖型转录相关和转录调节子相关的基因,从材料的胁迫程度来看,属于对干旱胁迫的生理应答反应,但C样本富集最多的类别则是与翻译相关的基因,这可能与部分细胞膜、各细胞器受到破坏有一定的关系。但这些变化是否随着干旱胁迫时间的延长代谢通路发生改变,目前还并不清楚。值得肯定的是,3个胁迫样本在GO注释的分子功能和细胞组件中富集最多的类别是相同的,通过3个胁迫样本的交集分析,如图5所示,在上调差异表达基因中,A、B、C 3个样本共有的差异表达基因有70个,主要包含氧化压力应答、碳水化合物代谢过程、电子载体活性、脂类转运、细胞壁组织和膜等相关的基因;在下调的差异表达基因中,A、B、C 3个样本共有的差异表达基因仅有11个,其中以基因序列comp117244_c0_seq7参与的代谢路径最多,主要包括过氧化氢应答、高温应答、病毒应答、高光强度应答、液泡膜、ATP结合、细胞壁和泛素蛋白连接酶结合相关代谢过程;其次为comp114714_c0_seq3,GO功能注释显示,在拟南芥中参与盐胁迫应答、蛋白结合、光形态发生、细胞核和锌离子结合相关的代谢过程。而这些富集的差异基因与上述KEGG富集分析结果和甘蔗干旱胁迫下转录组变化的结果是一致的[24]。说明割手密与甘蔗在干旱胁迫后的调控机制方面存在一定的共性。从3个样本在GO富集分析结果来看,A、B、C 3个样本差异基因数量都是不同的,割手密随着干旱胁迫时间的变化,其差异表达基因和代谢通路同样发生变化,在不同的时间段启动的干旱胁迫应答机制不同,说明在研究植物干旱胁迫转录组分析时,不同胁迫时间上调和下调的表达基因不同,响应机理也不完全相同,C样本由于处于极度的干旱水平,部分叶片已经枯死,因此,随着细胞内毒素物质的增多,对细胞的代谢产生严重影响,很多基因的表达处于一种紊乱的状态,抗旱功能基因的差异表达难以准确判断。通过3个胁迫样本的交集分析,70个上调基因和11个下调基因很可能对割手密抗旱性起着重要的调节作用,其所参与的代谢路径值得进一步的研究。
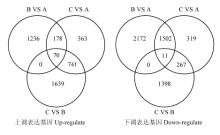 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图5A、B、C 3个胁迫样本的交集分析
-->Fig. 5The intersection analysis of A, B, and C samples under drought stress
-->
在差异表达基因所参与的代谢通路中,除了A、B、C 3个样本都有交叉的代谢通路外, B和C样本中也富集了一些不同代谢通路的差异基因,如B样本中独有的脂氧合酶(lipoxygenase),C样本中的甘氨酸羟甲基转移酶(Glycine hydroxymethyl-transferase)和乙酰乳酸合成酶(Acetolactate synthase)。B样本富集的脂氧合酶(LOX)是植物十八碳酸代谢途径中的关键酶,该酶对植物的生长发育及其对环境胁迫的反应具有重要的影响,可降解细胞膜,与植物的衰老功能相联系[37];在拟南芥[38]和水稻[39]水分胁迫的试验中,定量表达分析证明了水分胁迫样本LOX表达活性上升,且水稻的LOX3启动子中也证明存在一些逆境诱导元件。但本研究中割手密水分胁迫处理48 h后LOX处于下调表达,与已报道的结果不相符合,需要进一步研究验证。在C样本中富集的甘氨酸羟甲基转移酶在干旱胁迫中的报道并不多,但在水稻干旱胁迫的蛋白组学研究中筛选到该蛋白,在植物干旱胁迫后的具体功能尚不清楚。
ABC转运蛋白广泛存在于植物的细胞质膜、叶泡、线粒体和过氧化物酶中,其蛋白数目已超过100多种[40],随着转运底物的差异在植物生长素的外源毒素的解毒、气孔调节等生理活动中发挥作用,是植物器官生长、环境胁迫等重要的转运蛋白。经过序列的比对分析,该基因编码的蛋白属于ABC转运蛋白B亚族MDR/TAP-1类蛋白成员,在拟南芥中该基因与除草剂交叉抗性有关,参与拟南芥体内有毒物质的外排过程和次生代谢物质的跨膜运输[41]。本研究在KEGG富集的通路中,B和C样本都含有ABC转运蛋白,不同的是,B样本在胁迫48 h后处于上调表达,C样本在胁迫72 h后处于下调表达,说明不同胁迫时间该蛋白的表达水平不同,B样本的干旱胁迫较C样本轻,可能处于对干旱反应的生理调节阶段,对植株干旱处于正向调节,而C样本的ABC转运蛋白处于下调水平,可能细胞的代谢处于一种极度紊乱水平,影响了该基因的正常表达,使该基因不能发挥正常的功能,但这些猜测还需要后续的进一步研究验证。
4 结论
通过转录组分析,获得云南割手密82-114在干旱胁迫下的基因表达谱数据,阐明了参与干旱调控的信号传递、脂类代谢、多糖代谢、水分运输、DNA结合等相关基因的表达差异;通过富集的差异基因同源比对分析,证明了ATP结合盒B亚族(MDR/TAP) -1、脂氧合酶、β-氨基己糖苷酶、磷脂酶D和细胞外信号调节激酶等都与干旱胁迫存在一定的相关性,并在其他作物上得到证实;获得在整个干旱胁迫时期稳定上调表达基因70个,稳定下调表达基因11个,阐明了这些基因在各自的代谢通路中被强烈诱导或抑制表达,显示与干旱胁迫存在密切关系。The authors have declared that no competing interests exist.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}