MoS2作为一种制氢的电催化剂,因其产量丰富、价格便宜而得到了广泛的关注.理论和实验[6, 7]证实,MoS2的电催化制氢的活性位点主要在其边缘位置[8, 9].因此,制备纳米级的MoS2,使其更多的边缘暴露在外面,是提高其电催化制氢活性的有效途径.但是纳米级的MoS2很容易团聚,而以某种导电材料为基底,就有可能合成出层状的、具有丰富边缘的MoS2电催化剂.如文献[8, 10, 11, 12, 13]中所报道的以Au[8, 12]、碳纳米管[13]等为基体,成功地制备出了具有高效催化活性的层状的MoS2复合电催化剂.
本实验中采用水热合成的方法,以二水合钼酸钠(Na2MoO4·2H2O)为前驱体,硫脲(NH2CSNH2)为硫源和还原剂,合成了层状的MoS2/Graphene电催化剂.MoS2通过化学耦合选择性地生长在石墨烯上,使得MoS2边缘拥有丰富的活性位点,石墨烯作为良好的导电基体也能大大加快电子由层状边缘的活性位点转移到溶液中的速度.因此,MoS2/Graphene表现出了良好的电催化制氢性能,其作为Pt族贵金属的替代品,具有广阔的应用前景.
1 实验部分1.1 化学试剂与材料Na2MoO4·2H2O(阿法埃莎,99.99%),NH2CSNH2(阿法埃莎,99%),石墨薄片(阿法埃莎,99.8%),高锰酸钾(KMnO4,北京化工厂,AR(分析纯)),浓硫酸(H2SO4,北京化工厂,AR).其他所有化学试剂均为分析纯,所有溶液均由高纯水配制.
1.2 氧化石墨烯与MoS2/Graphene层状电催化剂的制备采用改进的Hummers法[14, 15]来制备氧化石墨烯(Graphene Oxide,GO).具体合成步骤如下:将1 g石墨鳞片放在一定量的NaCl中一起研磨20 min,然后进行抽滤,用大量水反复冲洗石墨粉.将洗后的石墨放在烘箱中70℃干燥30 min.干燥的固体石墨粉随后被转移到容量瓶中,加入23 mL浓硫酸,室温下搅拌24 h.溶液水浴加热至40℃,分别加入100 g硝酸钠(NaNO3)和500 g高锰酸钾,磁力搅拌30 min,同时在反应的过程中隔一段时间加入一定量的水.30 min后,将烧瓶从水浴中取出,加入140 mL水和10 mL 30%的过氧化氢(H2O2),溶液室温下搅拌5 min.将悬浮液进行离心(7 500 r/min,15 min),得到的沉淀物进行充分洗涤,然后干燥.最终得到黑色的固体,即氧化石墨烯.氧化石墨烯的产率大约为40%.
采用水热合成的方法来制备MoS2/Graphene层状电催化剂[11].因为NH2CSNH2在合成过程中做硫源和还原剂,为了研究不同含量的NH2CSNH2对最终形成的MoS2/Graphene的结晶性的影响,本文采用了不同的摩尔浓度的NH2CSNH2来制备MoS2/Graphene复合催化剂,即Na2MoO4·2H2O的浓度为1 mmol,NH2CSNH2的浓度分别为5、6、7和8 mmol来水热合成MoS2/Graphene.以NH2CSNH2用量为6 mmol为例,合成方法如下:取0.013 5 g的氧化石墨烯加入到30 mL的二甲基甲酰胺(DMF)中,超声1 h,然后分别加入1 mmol的Na2MoO4·2H2O和6 mmol的NH2CSNH2,混合均匀,放入反应釜中220℃水热24 h.所得固体分别用无水乙醇和高纯水各洗3次,即得到纳米级的MoS2/Graphene电催化剂.在水热过程中NH2CSNH2在高温下分解放出的H2S将MoO-4和氧化石墨烯还原成MoS2和Graphene.纳米MoS2的合成过程同上,只是合成过程中不加入氧化石墨烯.
1.3 材料的表征与性能测试氧化石墨烯和MoS2/Graphene的形貌使用场发射显微镜(FE-SEM,Hitachi S-4800)进行表征.电化学测试均在0.5 mol/L的H2SO4中进行,LSV的扫速为1 mV/s.电化学工作站为辰华CHI 750C型,采用三电极体系,对电极和参比电极分别为饱和甘汞和大块Pt片,工作电极为MoS2/Graphene/FTO或MoS2/FTO.以MoS2/Graphene为例,工作电极的制备方法为:取10mg的MoS2/Graphene粉末溶于2 mL无水乙醇中,超声1 h,然后旋涂到掺杂氟的SnO2透明导电玻璃(FTO)电极上(每次10 μL,旋涂次数为8~16次).
2 结果讨论2.1 氧化石墨烯及MoS2/Graphene电催化剂的结构表征图 1为氧化石墨烯的紫外可见光谱图与扫描电镜(SEM)照片.图 1(a)为用改进的Hummers法制得的氧化石墨烯的紫外可见吸收光谱.氧化石墨烯有2个特征吸收峰,231 nm处的强吸收峰对应于C=C键的π→π*跃迁,而300 nm附近的弱吸收峰对应于C=O键的n→π*跃迁[16, 17].氧化石墨烯的SEM照片如图 1(b)所示.可以看见氧化石墨烯的表面不是很平整,其表面褶皱且其边缘具有明显的阶梯状形貌,从图可清晰观察到氧化石墨烯特有的二维层状结构.这确认了本实验中制备出了氧化石墨烯.
 |
| 图 1 氧化石墨烯的紫外可见光谱图与SEM照片Fig. 1 UV-vis spectra and SEM photograph of graphene oxide |
| 图选项 |
图 2为MoS2/Graphene复合催化剂的SEM照片.MoS2/Graphene电催化剂呈现出二维的火花状形态,从图 2(b)中也可以看到少量片状的石墨烯,证明用水热方法成功地将MoS2和石墨烯合成到了一起.且火花状的MoS2的边缘大量的暴露在了外面,这也有利于电催化制氢反应活性的提高[8].
 |
| 图 2 MoS2/Graphene复合催化剂的SEM照片Fig. 2 SEM photographs of MoS2/Graphene composite catalyst |
| 图选项 |
图 3为MoS2/Graphene复合催化剂的高分辨率的透射电镜(HRTEM)和透射电镜(TEM)照片.图 3(a)中0.62 nm的晶格条纹间距对应于MoS2的(002)晶面[10],0.34 nm的晶格条纹间距对应于Graphene的(002)晶面[18].从图 3(b)中可以清晰地看出层状的MoS2有序紧密地分散在Graphene上,而不是MoS2杂乱无序地堆叠在Graphene上.这说明水热合成过程中MoS2与Graphene之间有着强烈的耦合作用,因而使得MoS2可以选择性地生长在石墨烯上,从而充分暴露其边缘的活性位点.
 |
| 图 3 MoS2/Graphene复合催化剂的HRTEM和TEM照片Fig. 3 HRTEM and TEM photographs of MoS2/Graphene composite catalyst,respectively |
| 图选项 |
图 4为用不同摩尔含量的NH2CSNH2做硫源和还原剂,经水热反应最终形成的MoS2/Graphene电催化剂的X射线衍射(XRD)图谱.从中可以看出,不论NH2CSNH2的含量为多少,在2θ=26°处均可观察到石墨烯的特征峰(002)[19],由此证明了在水热过程中氧化石墨烯被还原成了石墨烯.由XRD图谱也可知所制备的MoS2为六方晶系(JCPDS No.65-1951).因此,XRD图谱清晰地证明了以Na2MoO4·2H2O为前驱体,NH2CSNH2为硫源和还原剂,用水热合成法可以成功地制备出MoS2/Graphene复合电催化剂.对比分别用5、6、7和8 mmol的NH2CSNH2制得样品的XRD发现,在NH2CSNH2用量为6 mmol时,样品的整体峰型更尖锐,其中在2θ=14.2°的(002)面的峰强也有明显的提高.表明在NH2CSNH2用量为6 mmol时,MoS2/Graphene的结晶度最好,且较强的(002)衍射峰表明得到的MoS2沿c轴方向的层状结构生长良好,这也与HRTEM照片中的结果吻合的很好.因此,在后续表征MoS2/Graphene催化剂的电催化制氢性能的实验中均用的是NH2CSNH2用量为6 mmol时所制备的电催化剂.
 |
| A,B,C,D—水热合成过程中Na2MoO4·2H2O的浓度为1 mmol,NH2CSNH2的浓度分别为5、6、7和8 mmol时所制备得到的MoS2/Graphene.图 4 用不同摩尔含量的NH2CSNH2做硫源和还原剂,所制备得到的MoS2/Graphene复合催化剂的XRD谱图Fig. 4 XRD image of MoS2/Graphene composite catalysts which were prepared by different molar content of thiourea that served as source of sulfur and reducing agent |
| 图选项 |
2.2 MoS2/Graphene的电催化制氢性能表征本实验中将催化剂旋涂到FTO上,采用三电极体系来测定催化剂的电催化制氢性能.图 5为不同电催化剂(体相MoS2、纳米MoS2、MoS2/Graphene以及Pt)的电催化制氢性能的比较以及旋涂不同层数的MoS2/Graphene催化剂的电催化活性的比较.从图 5(a)中可以看到,体相MoS2几乎没有电催化制氢活性[20],而经水热合成的纳米MoS2其电催化制氢活性有了质的提高,其起峰电位提前到了0.15 V左右,在过电位更负时其电流快速地增加,在0.2 V的过电位下其电流密度达到了-2.1 mA/cm2.当MoS2与石墨烯复合后,MoS2/Graphene的起峰电位进一步提前到0.1 V,在0.2 V的过电位下其电流密度达到了-3.7 mA/cm2,几乎是纯MoS2的2倍.这是因为在电催化制氢反应中,MoS2的活性位点大多位于MoS2的边缘上[6, 8].体相中MoS2杂乱无序地堆叠在一起,活性位点少.而经水热反应合成的纳米MoS2具有层状结构,因此大大地增加了其边缘的活性位点,所以具有良好的电催化制氢活性.而MoS2与石墨烯复合后,可能是它们之间的良好电化学耦合作用提高了复合催化剂的电催化活性.当然,MoS2/Graphene的电催化制氢性能离Pt还是有一定的差距,但相比贵金属Pt,MoS2更为便宜,原料来源更为丰富,因此具有良好的发展与应用前景.
图 5(b)为旋涂不同层数的MoS2/Graphene催化剂的电催化活性的比较.可以看到,随着旋涂层数的增加,其电催化制氢活性先增加后减小.当旋涂层数为12层时,MoS2/Graphene电催化剂具有最佳的活性,其起峰电位为0.085 V,在0.2 V的过电位下其电流密度达到了-4.5 mA/cm2.MoS2/Graphene催化剂层数的增加对电催化制氢活性有两方面的影响:①MoS2/Graphene催化剂在FTO上的厚度越厚,其表面积会越大,活性位点相应的也就会增多,因此电流密度也会增大;②厚度的增加会造成催化剂和FTO基底之间的导电性变差,电子转移阻抗变大[21],这会降低催化剂的电催化活性.因此,从催化活性的角度考虑,12层是MoS2/Graphene电催化剂旋涂到FTO上的最佳层数,此时阴极电流密度最大.
 |
| 图 5 不同电催化剂(体相MoS2、纳米MoS2、MoS2/Graphene以及Pt)以及旋涂不同层数的MoS2/Graphene催化剂的极化曲线Fig. 5 Polarization curves obtained with different catalysts (bulk MoS2,nano MoS2,MoS2/Graphene and Pt) and polarization curves obtained with different layered MoS2/Graphene |
| 图选项 |
图 6为对MoS2/Graphene电催化剂的稳定性进行测试.电催化制氢的稳定性是衡量一个催化剂性能好坏最重要的指标之一.为此,本实验中对MoS2/Graphene复合电催化剂循环不间断地扫描1 000圈.可以看出,第1 000圈和最初的第1圈相比,起峰电位几乎没有变化,阴极的电流密度的损失也几乎可以忽略不计.因此,实验中所制备的MoS2/Graphene复合电催化剂具有非常好的稳定性[22].
 |
| 图 6 MoS2/Graphene电催化剂的稳定性测试Fig. 6 Durability test for MoS2/Graphene electrocatalyst |
| 图选项 |
2.3 MoS2/Graphene电催化制氢的机理分析塔菲尔斜率是电催化材料的内在特性,它是由电催化制氢反应的限速步骤所决定.对塔菲尔斜率进行阐释和说明对理解电催化反应的内在机理非常重要.纳米MoS2、MoS2/Graphene以及Pt的塔菲尔曲线图如图 7所示.塔菲尔曲线的公式为
η=b lg j+a
式中:η为过电位;j为电流密度;b为塔菲尔斜率;a为j=1 mA·cm-2时的过电势(塔菲尔曲线与过电位轴的截矩).
 |
| 图 7 纳米MoS2、MoS2/Graphene以及Pt催化剂的塔菲尔曲线Fig. 7 Tafel plots for nano MoS2,MoS2/Graphene and Pt catalysts |
| 图选项 |
因此,由图 7可以得出纳米MoS2、MoS2/Graphene以及Pt的塔菲尔斜率分别为121、70和31 mV·dec-1.
在酸性溶液中,电催化制氢反应有3步可能的反应步骤[23]:
1) 质子放电(discharge reaction)步骤:
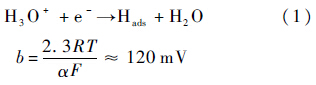
式中:R为理想气体常数;T为绝对温度;α≈0.5为对称系数;F为法拉第常数;Hads为吸附态氢离子.
2) 催化复合(combination reaction) 步骤:

3) 电化学脱附(electrochemical desorption reaction)步骤:

由此可知,电化学制氢反应中,由于Pt的塔菲尔斜率为31 mV·dec-1,因此在Pt表面发生的反应是步骤1)和步骤2),即快速的质子放电步骤1)和一个限速步骤即催化复合步骤2).然而,从发现MoS2对电化学制氢有良好的催化效果至今[24],MoS2电催化的反应机理仍未搞清楚.在本实验中,在纳米MoS2表面,由于其塔菲尔斜率为121 mV·dec-1,因此可以推测步骤1)可能为其限速步骤[25].MoS2/Graphene的塔菲尔斜率为70 mV·dec-1,文献[8, 26]中也有所报道.由Thomas的研究[27]可知,在电催化反应中较大的塔菲尔斜率(~60 mV·dec-1)意味着在化学吸附制氢过程中需要更多的活化能.因此,MoS2/Graphene电催化制氢的可能机理为:一个快速的质子放电步骤1),后续步骤为步骤2)或步骤3),即催化复合步骤2)或电化学脱附步骤3)均有可能为其限速步骤.单纯地从塔菲尔斜率上不能判断为哪一种机理.
图 8为不同电催化剂的电化学阻抗图谱.图 8(a)中旋涂层数为12层的MoS2/Graphene具有最小的电子转移阻抗,更小的电子转移阻抗意味着更快的电子转移速度[22],因此图 5(b)中旋涂层数为12层的MoS2/Graphene催化剂才会具有最好的电催化制氢活性.且不同旋涂层数电催化剂的电子转移阻抗变化的趋势和图 5(b)中催化剂的电催化制氢活性的变化是相一致的.图 8(b)中表明,MoS2/Graphene的电子转移阻抗Zf(Zf≈150 Ω)比纳米MoS2的(Zf≈5 kΩ)要小得多.从图 8(b)的局部放大图可以看出Graphene的电化学阻抗谱包含一个半圆和一个斜直线.低频区的斜直线可以看作Graphene的纯电容行为,而高频区的半圆则代表其电子转移阻抗[28, 29],约为5 Ω.因此石墨烯具有非常低的电子转移阻抗,所以其能作为良好的导电基体大大加快电子由层状边缘的活性位点转移到溶液中的速度.因而MoS2/Graphene具有比MoS2好得多的电催化制氢活性.
 |
| 图 8 在过电位η=0.12 V下,0.5 mol/L的H2SO4溶液中,不同旋涂层数的MoS2/Graphene的电化学阻抗图谱及纳米MoS2、MoS2/Graphene和Graphene的电化学阻抗图谱Fig. 8 AC impedance spectroscopy of different layered MoS2/Graphene and nano MoS2、MoS2/Graphene and Graphene catalysts in 0.5 mol/L H2SO4 at η=0.12 V |
| 图选项 |
图 9为MoS2和石墨烯经水热方法合成到一起的机理示意图.由HRTEM可以看出,MoS2是通过化学耦合和电子耦合选择性的生长在石墨烯基底上[10].MoS2/Graphene良好的电催化制氢性能正是来源于此.一方面,通过化学耦合作用,生长在基底上的MoS2有相对大的比表面积,这使得层状MoS2表面富有更多的边缘,因此大大增加了电催化制氢的活性位点.另一方面,石墨烯作为良好的电子导体,能够加快电子的转移速度,使电子更快地由层状边缘的活性位点转移到溶液中,从而提高电催化制氢的活性.
 |
| 图 9 MoS2/Graphene复合催化剂里的电子转移机理图Fig. 9 Mechanism schematic diagram of the charge transfer in MoS2/Graphene composite catalysts |
| 图选项 |
3 结 论1) 采用水热合成的方法,以Na2MoO4·2H2O为前驱体,NH2CSNH2为硫源和还原剂,合成了层状的MoS2/Graphene电催化剂.MoS2通过化学耦合选择性地生长在石墨烯上,使得MoS2边缘拥有丰富的活性位点,石墨烯作为良好的导电基体也能大大加快电子由层状边缘的活性位点转移到溶液中的速度.
2) MoS2/Graphene表现出了良好的电催化制氢性能,其起峰电位为0.085 V,在0.2 V的过电位下其电流密度达到了-4.5 mA/cm2,塔菲尔曲线的斜率约为70 mV·dec-1.
3) MoS2/Graphene复合催化剂因其高的电催化制氢效率以及低的成本,很有希望成为Pt族金属的替代品,具有广阔的应用前景.
参考文献
| [1] | Turner J A.Sustainable hydrogen production[J].Science, 2004, 305(5686):972-974. |
| Click to display the text | |
| [2] | Dresselhaus M, Thomas I.Alternative energy technologies[J].Nature, 2001, 414(6861):332-337. |
| Click to display the text | |
| [3] | Barreto L, Makihira A, Riahi K.The hydrogen economy in the 21st century:A sustainable development scenario[J].International Journal of Hydrogen Energy, 2003, 28(3):267-284. |
| Click to display the text | |
| [4] | le Goff A, Artero V, Jousselme B, et al.From hydrogenases to noble metal-free catalytic nanomaterials for H2 production and uptake[J].Science, 2009, 326(5958):1384-1387. |
| Click to display the text | |
| [5] | Trasatti S.Electrocatalysis of hydrogen evolution:Progress in cathode activation[J].Advances in Electrochemical Science and Engineering, 1992, 2:1-85. |
| Click to display the text | |
| [6] | Laursen A B, Kegnæs S, Dahl S, et al.Molybdenum sulfides-efficient and viable materials for electro-and photoelectrocatalytic hydrogen evolution[J].Energy & Environmental Science, 2012, 5(2):5577-5591. |
| Click to display the text | |
| [7] | Merki D, Hu X.Recent developments of molybdenum and tungsten sulfides as hydrogen evolution catalysts[J].Energy & Environmental Science, 2011, 4(10):3878-3888. |
| Click to display the text | |
| [8] | Jaramillo T F, Jørgensen K P, Bonde J, et al.Identification of active edge sites for electrochemical H2 evolution from MoS2 nanocatalysts[J].Science, 2007, 317(5834):100-102. |
| Click to display the text | |
| [9] | Hinnemann B, Moses P G, Bonde J, et al.Biomimetic hydrogen evolution:MoS2 nanoparticles as catalyst for hydrogen evolution[J].Journal of the American Chemical Society, 2005, 127(15):5308-5309. |
| Click to display the text | |
| [10] | Li Y, Wang H, Xie L, et al.MoS2 nanoparticles grown on graphene:An advanced catalyst for hydrogen evolution reaction[J].Journal of the American Chemical Society, 2011, 133:7296-7299. |
| Click to display the text | |
| [11] | Chang K, Chen W.In situ synthesis of MoS2/graphene nanosheet composites with extraordinarily high electrochemical performance for lithium ion batteries[J].Chemical Communications, 2011, 47(14):4252-4254. |
| Click to display the text | |
| [12] | Wang T, Liu L, Zhu Z, et al.Enhanced electrocatalytic activity for hydrogen evolution reaction from self-assembled monodispersed molybdenum sulfide nanoparticles on an Au electrode[J].Energy & Environmental Science, 2013, 6(2):625-633. |
| Click to display the text | |
| [13] | Wang X, Yan Y, Ge X, et al.Facile synthesis of low crystallineMoS2 nanosheet-coated CNTs for enhanced hydrogen evolution reaction[J].Nanoscale, 2013, 5(17):7768-7771. |
| Click to display the text | |
| [14] | Dikin D A, Stankovich S, Zimney E J, et al.Preparation and characterization of graphene oxide paper[J].Nature, 2007, 448(7152):457-460. |
| Click to display the text | |
| [15] | Liang Y, Li Y G, Wang H, et al.Co3O4 nanocrystals on graphene as a synergistic catalyst for oxygen reduction reaction[J].Nature Materials, 2011, 10(10):780-786. |
| Click to display the text | |
| [16] | Marcano D C, Kosynkin D V, Berlin J M, et al.Improved synthesis of graphene oxide[J].ACS Nano, 2010, 4(8):4806-4814. |
| Click to display the text | |
| [17] | Li D, Muller M B, Gilje S, et al.Processable aqueous dispersions of graphene nanosheets[J].Nat Nanotechnol, 2008, 3(2):101-105. |
| Click to display the text | |
| [18] | Xiang Q, Yu J, Jaroniec M.Synergetic effect of MoS2 andgraphene as cocatalysts for enhanced photocatalytic H2 production activity of TiO2 nanoparticles[J].Journal of the American Chemical Society, 2012, 134(15):6575-6578. |
| Click to display the text | |
| [19] | Liu C J, Tai S Y, Chou S W, et al.Facile synthesis of MoS2/graphene nanocomposite with high catalytic activity towardtriiodide reduction in dye-sensitized solar cells[J].Journal of Materials Chemistry, 2012, 22(39):21057-21064. |
| Click to display the text | |
| [20] | Liu Y D, Ren L, Qi X, et al.Preparation, characterization and photoelectrochemical property of ultrathin MoS2 nanosheets via hydrothermal intercalation and exfoliation route[J].Journal of Alloys and Compounds, 2013, 571:37-42. |
| Click to display the text | |
| [21] | Guo X, Diao P, Xu D, et al.CuO/Pd composite photocathodes for photoelectrochemical hydrogen evolution reaction[J].International Journal of Hydrogen Energy, 2014, 39(15):7686-7696. |
| Click to display the text | |
| [22] | Liao L, Zhu J, Bian X, et al.MoS2 formed on mesoporous graphene as a highly active catalyst for hydrogen evolution[J].Advanced Functional Materials, 2013, 23(42):5326-5333. |
| Click to display the text | |
| [23] | Conway B, Tilak B.Interfacial processes involving electrocatalytic evolution and oxidation of H2, and the role of chemisorbed H[J].Electrochimica Acta, 2002, 47(22):3571-3594. |
| Click to display the text | |
| [24] | Tributsch H, Bennett J.Electrochemistry and photochemistry of MoS2 layer crystals.[J].Journal of Electroanalytical Chemistry and Interfacial Electrochemistry, 1977, 81(1):97-111. |
| Click to display the text | |
| [25] | Bonde J, Moses P G, Jaramillo T F, et al.Hydrogen evolution onnano-particulate transition metal sulfides[J].Faraday Discussions, 2009, 140(1):219-231. |
| Click to display the text | |
| [26] | Ji S, Yang Z, Zhang C, et al.Exfoliated MoS2 nanosheets as efficient catalysts for electrochemical hydrogen evolution[J].Electrochimica Acta, 2013, 109:269-275. |
| Click to display the text | |
| [27] | Thomas J.Kinetics of electrolytic hydrogen evolution and the adsorption of hydrogen by metals[J].Journal of Chemical Society, Faraday Transactions, 1961, 57:1603-1611. |
| Click to display the text | |
| [28] | Casero E, Parra-Alfambra A, Petit-Domínguez M, et al.Differentiation between graphene oxide and reduced graphene by electrochemical impedance spectroscopy (EIS)[J].Electrochemistry Communications, 2012, 20:63-66. |
| Click to display the text | |
| [29] | Tian H, Wang L, Qin X, et al.Influence of hydrophilic properties on capacitive behavior of functionalized graphene[J].Ionics, 2014, 20(8):1055-1061. |
| Click to display the text |
