为了获得PVDF的电活性相,许多不同的制备方法已经被提出,主要分为两类:特殊方法处理纯PVDF和添加填料诱导.由α-PVDF在温度80℃和拉伸比R=3~5经机械拉伸能够得到β相[10];熔融状态PVDF经高温高压淬火(280℃,≥500 MPa),或者经超高的冷却速度(>2 000 K·s-1)非等温结晶得到PVDF电活性相[11];高压条件下由溶液结晶得到电活性相或者高压条件下通过控制结晶温度(80℃最佳)直接在N,N-二甲基甲酰胺(DMF)或者二甲基亚砜(DMSO)等溶液中结晶得到电活性相[12];添加黏土、石墨纳米片或者水和盐等填料诱导[13, 14]以及与其他物质共聚[15]等方法都可以获得PVDF电活性相.机械拉伸法是最常用的方法但是会引入很多缺陷,造成拉伸后薄膜强度性能的下降.
本文通过最简单易行的溶液法制备PVDF电活性相薄膜,研究前驱体溶液的浓度、前驱体溶液搅拌时间和热压工艺对PVDF晶型形成的影响,通过红外光谱仪(Fourier Transform infrared spectroscopy,FTIR)和X射线衍射仪(X-Ray Diffraction,XRD)确定不同工艺条件制备样品的晶型,最终得到电活性相含量高的PVDF薄膜.
1 实 验1.1 样品制备1.1.1 原 料PVDF粉末,FR904型号,上海三爱富新材料有限公司生产;DMF,分析纯,北京化工厂生产.
1.1.2 前驱体溶液的配制量取一定体积的DMF溶液于烧杯中,加入溶液浓度1%~10%的PVDF粉末,60℃磁力搅拌1~3 h,使PVDF完全溶解,得到PVDF/DMF前驱体溶液.前驱体溶液配制的具体参数条件(溶液浓度和处理时间)列于表 1.
表 1 前驱体溶液制备的工艺条件 Table1 Procesing parameters of precursor solutions preparation
| 样品编号 | 溶液浓度/% | 处理时间/h |
| 1 | 7 | 1 |
| 2 | 7 | 2 |
| 3 | 7 | 3 |
| 4 | 1 | 2 |
| 5 | 10 | 2 |
表选项
1.1.3 基板的清洗实验所用的基板为20 cm×15 cm的厚玻璃板.基板表面洁净度对薄膜质量有重要影响.因此为了制得质量优良、不发生龟裂、剥离现象的薄膜,必须在制膜前做好厚玻璃板的清洗工作.先用1 g/L的NaOH稀溶液浸泡厚玻璃板24 h,除去表面的附着物;再用去离子水冲洗厚玻璃板,洗净表面的NaOH溶液,然后用一定量的乙醇溶液清洗玻璃板表面,将黏附在玻璃板表面的油脂等污染物除去;最后用去离子水冲洗厚玻璃板表面3遍,将清洗干净的厚玻璃板放入80℃的烘箱干燥备用.
1.1.4 薄膜制备采用涂膜机涂膜的方法制备薄膜.以清洗干净的厚玻璃板为基板,将其放置于涂膜机的加热板上,加热板温度设置为80℃,整个系统预热10 min.然后将前驱体溶液倾倒于厚玻璃板,涂膜得到厚度均匀的薄膜.为确保溶剂完全挥发,将样品放入80℃烘箱热处理24 h,最后得到薄膜样品.
1.1.5 熔融热压通过粉末压片机给样品提供单轴压力,将薄膜样品放在工作空间中央位置,薄膜在230℃热熔20 min,然后加压4 MPa热压20 min,最后保持压力情况下自然冷却至室温,得到热压后PVDF样品.
1.2 测 试用美国Nicolet公司的iN10MX型红外光谱仪(实验条件:ATR模式)和日本理学公司的D/max-2200/PC型X射线衍射仪(实验条件:Cu靶,40 KV,30 mA,扫描速度4(°)/min)表征分析样品的晶型结构.
通过红外光谱图可以确定样品中β相的相对含量.为了确定样品中β相的含量,用762 cm-1和840 cm-1处分别代表α相和β相的吸收峰.通过文献[5]提出的方法可以确定每个样品中β相的含量.假设红外吸收满足Lambert-Beer定律,则对于厚度为L的薄膜样品,其吸收率为
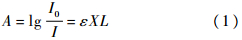
式中:I0和I分别为入射和透过光的光强;X为各晶相的结晶度;ε为各晶相的红外吸收系数,是单体密度(C=0.030 5 mol/cm3)和PVDF各晶相在特定吸收峰吸收系数的结合(α相和β相的吸收峰的吸收系数分别为:Kα=6.1×104cm2/mol和Kβ=7.7×104 cm2/mol).所以,对于1个只含有α和β相的样品,β相的相对含量为
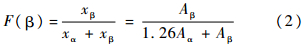
式中:Aα和Aβ分别为α和β相在762 cm-1和840 cm-1处的吸收峰强度.
2 结果与讨论2.1 前驱体溶液搅拌时间对PVDF结晶形态的影响原料PVDF和溶液浓度7%的溶液经不同搅拌时间得到PVDF样品的FTIR谱图如图 1所示.表 2为波数范围700~1 500 cm-1内PVDF不同晶型的FTIR特征吸收峰[4, 5, 16].图 1样品(a)和样品(b)均在762、796和976 cm-1处出现了所有代表α相的吸收峰,而在840和1 275 cm-1处代表β相的峰强非常弱,说明在原料PVDF和搅拌时间为1 h的样品中,α相占主导地位.随着搅拌时间的增长,代表β相的840和1 275 cm-1处吸收峰强度明显增强,而代表α相的特征吸收峰的强度明显下降,当搅拌时间为2 h时(样品(c)),在796和976 cm-1处α相特征峰完全消失,在762 cm-1处α相特征峰强度明显下降,只表现为一个小突起;当搅拌时间增加为3 h时,从图 1中样品(d)可以看出样品的762、796和976 cm-1处α特征峰都完全消失,此时样品中β相占主导地位.
 |
| (a)—原料PVDF; (b)—溶液浓度7%溶液搅拌1 h制备的PVDF; (c)—溶液浓度7%溶液搅拌2 h制备的PVDF; (d)—溶液浓度7%溶液搅拌3 h制备的PVDF; α—α相吸收峰;β—β相吸收峰.图 1 原料PVDF和溶液浓度7%的PVDF溶液经不同搅拌时间制备PVDF样品的FTIR谱图(波数范围700~1 500 cm-1)Fig. 1 FTIR spectra of raw PVDF and PVDF solution at mass fraction of 7% with different stirring time (Wavenumber range of 700-1 500 cm-1) |
| 图选项 |
表 2 PVDF主要晶型的FTIR吸收峰[4, 5, 16](波数范围:700~1 500 cm-1) Table2 FTIR absorption bands of major crystal phases of PVDF[4, 5, 16](Wavenumber range of 700-1 500 cm-1)
| 晶型 | 吸收峰位置/cm-1 |
| α-PVDF | 762,796,976 |
| β-PVDF | 840a,1 275b |
| γ-PVDF | 833a,1 233c |
表选项
高分子聚合物材料具有软物质特性,常常对外界弱作用做出强反应.从热力学观点来看,高分子链在没有外力作用时总是自发采取卷曲的形态,使构象熵趋于最大[17].对溶液施加磁力搅拌,溶液核心与周围液体交界处因速度梯度会形成强剪切,作用在聚合物分子链上时,剪切力所带来的能量打破了分子链的自发状态,能够起到扭转分子链的作用,并对分子链的无规热运动产生“导向”效果,从而造成分子构象的改变引起晶型变化.不同的剪切时间会影响高聚物的结晶行为[17].
PVDF粉体投入DMF溶液磁力搅拌时,首先发生粉体颗粒的表面湿润过程,即DMF液体取代PVDF颗粒表面层的空气,并进入颗粒之间的间隙;接着是DMF液体进一步渗入,发生PVDF粉体的溶胀,随着搅拌的进行这种溶胀团聚体被流体所打散;最后PVDF完全溶解在DMF中,分子链脱离了晶格束缚,移动性增加,继续搅拌就会对分子链产生剪切作用力且高聚物材料具有强烈的对力作用时间的依赖性.所以搅拌时间为1 h时,样品中α相居多.当搅拌时间增加到2 h和3 h时,PVDF完全溶解,磁力搅拌系统所提供的能量能够满足扭转分子链所需要的能量,这就为全反式β相的形成提供了可能.
XRD是分析PVDF晶型结构的重要手段,原料PVDF和质量分数7%经不同搅拌时间得到PVDF样品的XRD谱图如图 2所示.多数情况下,原料PVDF(见图 2(a))的晶型以α相为主,在2θ为17.66°、18.30°、20.1°和26.56°存在全部α相的峰.在搅拌时间1 h(见图 2(b))时,17.66°处α相的(100)峰消失,18.30°处α相的(020)峰和26.56°处α相的(021)峰强明显受到抑制,此时样品中仍以α相为主.随着搅拌时间增加到2 h和3 h(见图 2(c)和图 2(d)),α相的18.6°和26.56°衍射峰消失,而β相的20.86°处(200)衍射峰成为主衍射峰,此时样品中主要含有β相.这与图 1中FTIR的结果是一致的.
 |
| (a)—原料PVDF; (b)—溶液浓度7%溶液搅拌1 h制备的PVDF; (c)—溶液浓度7%溶液搅拌2 h制备的PVDF; (d)—溶液浓度7%溶液搅拌3 h制备的PVDF.图 2 原料PVDF和溶液浓度7%的PVDF溶液经不同搅拌时间制备PVDF样品的XRD谱图Fig. 2 XRD patternsof raw PVDF and PVDF solution at concentration of 7% with different stirring time |
| 图选项 |
2.2 前驱体溶液浓度对PVDF结晶形态的影响对于高分子溶液,不同溶剂中高分子链之间的净相互作用力、链的形态、尺寸以及排除体积效应均是不同的,因此即使同一溶剂不同浓度,溶剂性能也可能会对高分子链的解缠绕程度和移动性产生影响,进而对所制得的高聚物的结晶构型产生影响.图 3所示为相同搅拌时间(2 h),不同前驱体溶液浓度对PVDF结晶形态影响的FTIR谱图.由图 3可知,前驱体溶液浓度为1%,代表α相的特征吸收峰762、796和976 cm-1都存在,随着溶液浓度的增大,在浓度为7%和10%时,762、796和976 cm-1处的α相特征吸收峰逐渐消失,而840和1 275 cm-1处β相的吸收峰强度逐渐增加.所以如图 3(a)样品主要含有α相,如图 3(b)和图 3(c)样品中大部分晶相为β相.
 |
| 图 3 相同搅拌时间(2 h),不同前驱体溶液浓度对PVDF结晶形态影响的FTIR谱图(波数范围:700~1 500 cm-1)Fig. 3 FTIR spectra of PVDF at the same stirring time (2 h) with different precursor solution concentrations (Wavenumber range of 700-1 500 cm-1) |
| 图选项 |
为了进一步证明晶型结构,本文做了XRD测试.将相同搅拌时间(2 h)下,不同前驱体溶液浓度对PVDF结晶形态影响的XRD谱图如图 4所示.当前驱体溶液浓度为10%时,仅存在20.86°处β相的(200)衍射峰,说明此时样品中大部分结晶相为β相.随着前驱体溶液浓度的降低,18.6°处代表α相的衍射峰逐渐增强,前驱体溶液浓度为1%时,α相的18.6°和26.56°处的衍射峰都存在.这与图 3中FTIR的分析结果是一致的.
 |
| 图 4 相同搅拌时间(2 h),不同前驱体溶液浓度对PVDF结晶形态影响的XRD谱图Fig. 4 XRD patterns of of PVDF at the same stirring time (2 h) with different precursor solution concentrations |
| 图选项 |
DMF是PVDF的良溶剂,PVDF在DMF中能够很好的溶解.溶液浓度较小时,高分子线团之间互相分离,随着溶液浓度增加,聚合物分子链之间的相互作用增加,高分子链相互接触、穿插、交叠,分子链的移动性变弱.搅拌时间同为2 h的PVDF溶液在厚玻璃板上成膜时,聚合物分子链的C-F键与厚玻璃板表面的羟基作用形成氢键,这些氢键的存在诱导近外层聚合物分子链上的F原子同侧排列,进而诱导近上层的聚合物分子链交替排列的有序结构[18, 19].溶液浓度低,分子链自由活动体积大;溶液浓度大,分子链之间缠结穿插严重,自由移动性变弱;所以这些浓度条件都不利于分子链有序结构的排列,只有浓度合适时,分子链有序结构的排列才更容易进行.根据结晶动力学可知,结晶过程包括成核和生长过程,成核首先发生在有序的高分子链束,然后在80℃晶粒结晶长大,这就为PVDF电活性相的形成提供了可能.
β相的含量随溶液搅拌时间和溶液浓度的变化如图 5所示.根据式(2)计算样品中β相的含量,图 5(a)为样品中β相含量随溶液搅拌时间的变化图,可以看出β相含量随搅拌时间先明显的增加,F(β)在搅拌时间2 h达到一个最大值81.3%,随后搅拌时间的增加F(β)有缓慢下降的趋势,F(β)的降低是由于随着时间的增长,溶剂的挥发造成溶液浓度的变大,从而造成分子链之间的缠结,影响β相的形成.图 5(b)是样品中β相含量随溶液浓度的变化图,可以看出F(β)随溶液浓度的增加迅速增加到一个最大值,而后缓慢下降.所以β相的含量在浓度7%,搅拌时间2 h时达到最大81.3%.
 |
| 图 5 β相的含量随溶液搅拌时间和溶液浓度的变化Fig. 5 Variations of β phase content with stirring imeand solution concentrations |
| 图选项 |
2.3 热压对PVDF结晶行为的影响图 6给出了溶液浓度7%搅拌时间3 h的PVDF样品在热压前后的FTIR谱图.由图 6可以看出热压前后代表β和γ相的吸收峰发生非常明显的变化.热压前只属于γ相的833和1 233 cm-1处吸收峰在热压后都移向了只代表β相的吸收峰840和1 278 cm-1处.
 |
| 图 6 溶液浓度7%,搅拌时间3h的PVDF样品在热压前后的FTIR谱图(波数范围:700~1 500 cm-1)Fig. 6 FTIR spectra of PVDF at concentration of 7% and with stirring time of 3 h (Wavenumber range of 700-1 500 cm-1) |
| 图选项 |
图 7给出了溶液浓度7%、搅拌时间3 h的PVDF样品在热压前后的XRD谱图.可以清晰地看出,20°附近的峰在热压后由20.2°向后移向了20.8°,前者对应γ相的(110)面,后者为β相的(200)面.热压后沿(200)面定向排布的β相增加,这与图 6 FTIR得出的结论是一致的.
 |
| 图 7 溶液浓度7%,搅拌时间3 h的PVDF样品在热压前后的XRD谱图Fig. 7 XRD patterns of PVDF at concentration of 7% and with stirring time of 3 h |
| 图选项 |
β相和γ相同属正交晶系,β相的晶格常数为a=0.858 nm,b=0.491 nm,c=0.256 nm,γ相的晶格常数为a=0.866 nm,b=0.493 nm,c=0.258 nm.由晶格常数可知β相的晶格常数比γ相的晶格常数小,这就使得β相晶格中具有平面锯齿状的聚合物分子链的堆积更加致密,所以β相的晶格密度也是所有晶型中最大的[20].高压下结晶产物的密度比常压下结晶产物的密度高,PVDF在相对较高的温度和较强的压力作用下,高分子链会紧密堆积,形成更加致密型的β相,所以本文在热压条件下观察到γ→β.
PVDF结晶形态的变化与各晶型的分子构象有关,在PVDF分子链中,由于C—F和C—H键的极性差异,分子链间的这两个键之间存在很强的相互作用力.大多认为α构型的空间群为P21/c,分子链组成是以21螺旋或者反式链的形式存在.文献[21]显示用统计的方法推导得出α-PVDF是一种“上-下”无序的排列结构,每一条链沿晶格c轴方向有50%可能为上-下取向排列[21].这种统计分子链排列的模型,可以很好地解释磁力搅拌条件下由α-PVDF直接制备γ-PVDF的现象.
3 结 论1) 通过非常简单易操作的溶液法制备了电活性相含量高的PVDF薄膜.
2) 前驱体溶液浓度和搅拌时间会影响PVDF的晶型结构:前驱体搅拌时间2~3 h,溶液浓度7%~10%更利于电活性相的形成;前驱体溶液浓度7%、搅拌时间2 h的条件下得到最大β相含量81.3%.
3) 熔融热压同样会影响PVDF的晶型结构,PVDF薄膜经过230℃、4 MPa熔融热压,发生了γ→β相变,在压力作用下形成了体积更小更致密的β相.
参考文献
| [1] | He X,Yao K.Crystallization mechanism and piezoelectric properties of solution-derived ferroelectric poly (vinylidene fluoride) thin films[J].Applied Physics Letters,2006,89(11):112909. |
| Click to display the text | |
| [2] | Zhu L.Exploring strategies for high dielectric constant and low loss polymer dielectrics[J].The Journal of Physical Chemistry Letters,2014,5(21):3677-3687. |
| Click to display the text | |
| [3] | Gomes J,Nunes J S,Sencadas V,et al.Influence of the β-phase content and degree of crystallinity on the piezo- and ferroelectric properties of poly (vinylidene fluoride)[J].Smart Materials and Structures,2010,19(6):065010. |
| Click to display the text | |
| [4] | Martins P,Lopes A,Lanceros-Mendez S.Electroactive phases of poly (vinylidene fluoride):Determination,processing and applications[J].Progress in Polymer Science,2014,39(4):683-706. |
| Click to display the text | |
| [5] | Salimi A,Yousefi A.Analysis method:FTIR studies of β-phase crystal formation in stretched PVDF films[J].Polymer Testing,2003,22(6):699-704. |
| Click to display the text | |
| [6] | Sencadas V,Gregorio R,Lanceros-Méndez S.α to β phase transformation and microestructural changes of pvdf films induced by uniaxial stretch[J].Journal of Macromolecular Science,Part B,2009,48(3):514-525. |
| Click to display the text | |
| [7] | da Silva A B,Wisniewski C,Esteves J V A,et al.Effect of drawing on the dielectric properties and polarization of pressed solution cast β-PVDF films[J].Journal of Materials Science,2010,45(15):4206-4215. |
| Click to display the text | |
| [8] | Hattori T,Kanaoka M,Ohigashi H.Improved piezoelectricity in thick lamellar β-form crystals of poly (vinylidene fluoride) crystallized under high pressure[J].Journal of Applied Physics,1996,79(4):2016-2022. |
| Click to display the text | |
| [9] | 傅万里,杜丕一,翁文剑,等.聚偏氟乙烯压电薄膜的制备及结构[J].材料研究学报,2009,19(3):243-248.Fu W L,Du P Y,Weng W J,et al.Preparation and structure of PVDF piezoelectric film[J].Chinese Journal of Materials Research,2009,19(3):243-248(in Chinese). |
| Cited By in Cnki (22) | |
| [10] | Salimi A,Yousefi A.Conformational changes and phase transformation mechanisms in pvdf solution-cast films[J].Journal of Polymer Science Part B:Polymer Physics,2004,42(18):3487-3495. |
| Click to display the text | |
| [11] | Matsushige K,Nagata K,Takemura T.Direct observation of crystal transformation process of poly (vinylidene fluoride) under high pressure by PSPC X-ray system[J].Japanese Journal of Applied Physics,1978,17(3):467. |
| Click to display the text | |
| [12] | Gregorio R Jr,Ueno E M.Effect of crystalline phase, orientation and temperature on the dielectric properties of poly (vinylidene fluoride) (PVDF)[J].Journal of Materials Science,1999,34(18):4489-4500. |
| Click to display the text | |
| [13] | Liang C L,Xie Q,Bao R Y,et al.Induced formation of polar phases in poly (vinylidene fluoride) by cetyl trimethyl ammonium bromide[J].Journal of Materials Science,2014,49(12):4171-4179. |
| Click to display the text | |
| [14] | Martins P,Costa C M,Benelmekki M,et al.On the origin of the electroactive poly (vinylidene fluoride)[small beta]-phase nucleation by ferrite nanoparticles via surface electrostatic interactions[J].CrystEngComm,2012,14(8):2807-2811. |
| Click to display the text | |
| [15] | Mohamadi S,Sharifi-Sanjani N.Investigation of the crystalline structure of PVDF in PVDF/PMMA/graphene polymer blend nanocomposites[J].Polymer Composites,2011,32(9):1451-1460. |
| Click to display the text | |
| [16] | Bormashenko Y,Pogreb R,Stanevsky O,et al.Vibrational spectrum of PVDF and its interpretation[J].Polymer Testing,2004,23(7):791-796. |
| Click to display the text | |
| [17] | 何曼君,张红东,陈维孝.高分子物理[M].上海:复旦大学出版社,2007:39-91.He M J,Zhang H D, Chen W X.Polymer physics[M].Shanghai:Fudan University Press,2007:39-91(in Chinese). |
| [18] | 惠迎雪,刘卫国,牛小玲,等.β相聚偏二氟乙烯PVDF薄膜的制备和热光效应研究[J].红外与激光工程,2006(S5):143-149.Hui Y X,Liu W G,Niu X L,et al.Preparation and thermo-optic effect of oriented β PVDF films[J].Infrared and Laser Engineering,2006(S5):143-149(in Chinese). |
| Cited By in Cnki (4) | |
| [19] | 惠迎雪,樊慧庆,刘卫国,等.PVDF铁电聚合物薄膜的溶液法制备[J].材料科学与工程学报,2008,26(3):331-334.Hui Y X,Fan H Q,Liu W G,et al.Ferroelectric polymeric PVDF films preparaed by solution casting method[J].Journal of Materials Science & Engineering,2008,26(3):331-334(in Chinese). |
| Cited By in Cnki (13) | |
| [20] | Wang Z Y,Fan H Q,Su K H,et al.Structure and piezoelectric properties of poly (vinylidene fluoride) studied by density functional theory[J].Polymer,2006,47(23):7988-7996. |
| Click to display the text | |
| [21] | 李吉超,王春雷,钟维烈.聚偏氟乙烯全反式分子链振动模式的研究[J].物理化学学报,2004,19(11):1010-1014.Li J C,Wang C L,Zhong W L.Vibrational modes analysis of poly (vinylidene fluoride) all-trans molecular chain[J].Acta Physico-Chimica Sinica,2003,19(11):1010-1014(in Chinese). |
| Cited By in Cnki (3) |
