摘要/Abstract
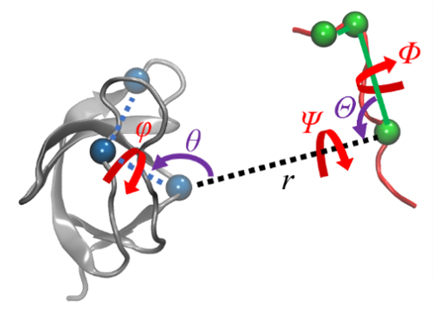
蛋白质-配体的结合过程伴随着复杂的结构变化, 在分子模拟可及的时间尺度内难以完全捕获, 这使得准确估计蛋白质-配体的结合自由能十分困难. 一种有效的解决途径是采用几何约束减小需要采样的构象空间, 再通过后处理方式扣除约束的影响. 本文综述了三种几何约束策略——漏斗状约束、球形约束和七自由度约束与自由能计算算法结合准确计算结合自由能的原理和进展, 重点概述理论严谨的七自由度约束的最新进展以及与Alchemistry或重要性采样方法的联用策略, 最后, 讨论了如何针对不同体系选择合适的计算策略以及蛋白质-配体准确结合自由能计算在药物设计等领域中的挑战和前景, 并提出了将上述方法进一步运用于研究更复杂的蛋白质-蛋白质问题的可能性.
关键词: 蛋白质-配体, 结合自由能, 分子模拟, 自由能微扰, 重要性采样
Binding free energy is the most crucial physical quantity for describing recognition-association of protein-ligand hybrids. Accurate estimation of protein-ligand binding free energies is of paramount importance in the field of drug design and biological engineering. However, the association process of protein-ligand hybrids is usually coupled with complex conformational changes of molecular objects, which is not amenable to the timescale of classical molecular simulations. This limitation makes it difficult to accurately estimate the protein-ligand binding free energies using classical free-energy calculation strategies. An effective solution is to apply geometric restraints to reduce the configurational space needed to be sampled, so as to boost up the convergence rate of simulations, and then calculate and deduct the contribution of these restraints to the binding free energy by post-processing. In this review, we firstly introduce the recent developments of three geometric restraints, namely, funnel, spherical, and seven-degree-of-freedom restraints, used in accurate binding free-energy calculations, with emphasis on the latest progress of the third one. Specifically, the theoretically rigorous seven-degree-of-freedom restraint describes translational, orientational, rotational, and conformational degrees of freedom by means of a center-of-mass distance, spherical angles, Euler angles and the root-mean-square deviation. Moreover, we demonstrate the theoretical backgrounds and methods of how to achieve accurate protein-ligand binding free-energy estimation by combination of geometric restraints and importance-sampling or alchemical algorithms. In the geometric routes, the degrees of freedom of the relative movement of the protein-ligand complex are addressed in a stepwise fashion by one-dimensional importance-sampling simulations. In the alchemical routes, a special thermodynamics cycle is designed, in which additional simulations are performed to address the contribution of the restraints. A general suggestion for how to choose a suitable strategy for a given molecular assembly based on our experience is provided. Last but not least, we discuss the applications and challenges of using accurate protein-ligand binding free-energy calculation methods in fields such as drug design, and present the possibility of extending these methods for investigating complex protein-protein interaction.
Key words: protein-ligand, binding free energy, molecular dynamics, free-energy perturbation, importance sampling
PDF全文下载地址:
点我下载PDF
