
 , 张少飞3,4, 申传璞3
, 张少飞3,4, 申传璞3 1. 江西师范大学 生命科学学院,江西 南昌 330022;
2. 安徽医科大学 生命科学学院,安徽 合肥 230032;
3. 安徽医科大学 药学院,安徽 合肥 230032;
4. 淮北师范大学 生命科学学院,安徽 淮北 235000
收稿日期:2020-02-18;接收日期:2020-06-30;网络出版时间:2020-08-13
基金项目:国家自然科学基金(Nos. 41806158,41406195),江西省自然科学基金(No. 20202BAB203021)安徽省高校自然科学研究项目(No. KJ2017A177)资助
摘要:Pictet-Spengler (P-S)反应通常是指β-芳基乙胺在酸性条件下与醛或者酮发生缩合后环化形成四氢异喹啉或β-咔啉类生物碱结构的一类化学反应。而催化这类高度立体选择性和区域选择性反应的酶称为Pictet- Spenglerase (P-S酶)。P-S酶是一类重要的活性产物生物合成催化酶,广泛分布于吗啡、那可丁、奎宁、小檗碱、阿吗灵等临床药物以及一些先导化合物的生物合成途径中,应用开发前景广阔。鉴于此,文中对P-S酶的发现、功能鉴定、生物学特性以及催化应用等方面的研究进展进行了归纳总结,以期为P-S酶的后续发掘及系统研究提供良好的借鉴和参考。
关键词:Pictet-Spengler反应Pictet-Spengler酶生物催化生物碱生物合成
Research progress of Pictet-Spenglerases
Yunchang Xie1, Qi Chen2
, Shaofei Zhang3,4, Chuanpu Shen3 1. College of Life Sciences, Jiangxi Normal University, Nanchang 330022, Jiangxi, China;
2. School of Life Sciences, Anhui Medical University, Hefei 230032, Anhui, China;
3. School of Pharmacy, Anhui Medical University, Hefei 230032, Anhui, China;
4. School of Life Sciences, Huaibei Normal University, Huaibei 235000, Anhui, China
Received: February 18, 2020; Accepted: June 30, 2020; Published: August 13, 2020
Supported by: National Natural Science Foundation of China (Nos. 41806158, 41406195), Jiangxi Provincial Natural Science Foundation (No. 20202BAB203021), Provincial Natural Science Research Project of Anhui Colleges and Universities (No. KJ2017A177)
Corresponding author: Qi Chen. Tel: +86-551-65172130; E-mail: chenqi@ahmu.edu.cn.
Abstract: Pictet-Spenglerases (P-Sases) catalyze the Pictet-Spengler (P-S) reactions and exhibit high stereoselectivity and regioselectivity under mild conditions. The typical P-S reaction refers to the condensation and recyclization of β-arylethylamine with aldehyde or ketone under acidic conditions to form tetrahydroisoquinoline and β-carboline alkaloid derivatives. The related enzymatic products of P-Sases are the backbones of various bioactive compounds, including clinical drugs: morphine, noscapine, quinine, berberine, ajmaline, morphine. Furthermore, the activity of P-Sases in stereoselective and regioselective catalysis is also valuable for chemoenzymatic synthesis. Therefore, this review summarizes the research progress in the discovery, functional identification, biological characteristics and catalytic applications of P-Sases, which provide the useful theoretical reference in future P-Sases research and development.
Keywords: Pictet-Spengler reactionPictet-Spenglerasebiocatalysisalkaloidbiosynthesis
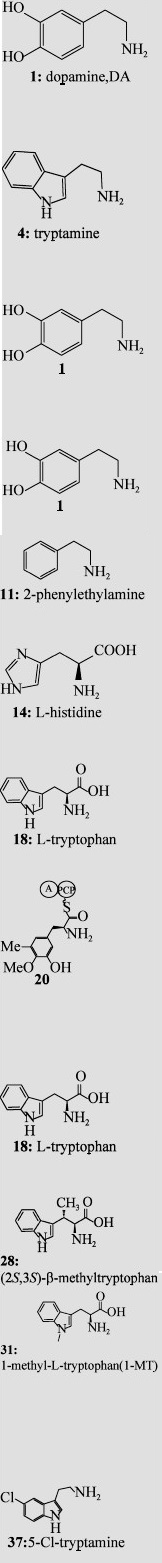
生物碱是一类自然界中(或人工合成的)常具有复杂环状结构的含氮碱性有机化合物,因其具有似碱特性,过去也称之为“赝碱”;其所含的氮原子往往包含于环状结构中,也被认为是一类含负氧化态氮原子的环状有机化合物[1-3]。生物碱类化合物结构多样、活性优良,是临床药物开发的重要来源,典型例子包括抗疟疾药物奎宁、抗菌药物小檗碱、缩宫麦素角胺、止痛药吗啡等[1-6]。四氢异喹啉(Tetrahydroisoquinoline,TIQ)和β-咔啉类生物碱(β-carboline alkaloid,βC)是具有含氮杂环结构的新颖生物碱类化合物,这类化合物表现出广泛的生物学活性而具有极高的研究开发价值。其骨架结构往往是由β-芳基乙胺在酸性条件下与醛或者酮发生缩合后环化形成(图 1)[7-12]。在常规的化学合成过程,这类成环反应往往需要一些特殊的催化条件,包括酸、碱、无机盐、微波等[8-10]。反应底物首先发生缩合生成烯胺,并进一步活化后与富电子的芳香环反应,生成新的碳碳单键而成氮杂环(图 1)[8-12]。而在生物合成途径中,这一高度立体选择性和区域选择性反应步骤则是由一类Pictet-Spenglerase (P-S酶)催化完成。P-S酶能够在温和条件下完成这一立体选择性的催化,从而生成氮杂环骨架[2, 9-10, 12]。生物体内的芳香胺、芳香族氨基酸或相应的取代物可在这些P-S酶的作用下与对应的醛或者酮缩合形成含氮杂环骨架结构,并经后续结构修饰而生成不同的生物碱[9-10, 12]。
 |
| 图 1 Pictet-Spengler反应机制示意图[2] Fig. 1 The general scheme for the Pictet-Spengler reaction[2]. |
| 图选项 |
P-S酶归属于国际酶学委员会(I.E.C)分类体系中的裂合酶大类(Lyases,EC 4)。迄今为止,多种来源于高等动、植物和微生物的P-S酶陆续被发现并鉴定(表 1)。例如来源于植物的去甲乌药碱合酶(Norcocalurine synthase,NCS)和异胡豆苷合酶(Strictosidine synthase,STR),它们催化苄基异喹啉生物碱(Benzylisoquinoline alkaloids,BIA)和萜类吲哚生物碱(Monoterpene indole alkaloids,MIA)生物合成途径中最为关键的一步反应[2, 10]。再比如来源于微生物的McbB、StnK2和SfmC[2, 10]。这些已鉴定的P-S酶是植物和微生物来源的MIA、BIA、β-咔啉以及TIQ等生物碱分子骨架合成的核心催化酶[1-2, 4-6, 8-12]。然而,目前已有报道的P-S酶的种类和数量有限,严重制约了这类酶进一步的研究与开发。针对当前存在的资源瓶颈,利用已有P-S酶研究工作作为参考,选择相应的P-S酶序列信息作为分子标记进行后续资源的深入发掘,不但有利于发掘更多新颖活性生物碱分子,同时为P-S酶及P-S反应在生物催化中的深入研究提供必要的理论参考和借鉴。鉴于此,文中对已有P-S酶在基因水平、酶学属性和结构生物学方面的研究进展进行了系统总结。
表 1 已有研究报道的Pictet-Spengler酶及相应的天然底物Table 1 The natural substrate scope of known Pictet-Spenglerases
| Substrate (amine or amino acids) | Substrate (aldehyde or ketone) | P-S enzyme and its typical sources | Enzyme reaction Product(s) or Related chemical structure(s) |
 |  | NCS *# (Thalictrum flavum; Coptis japonica) | 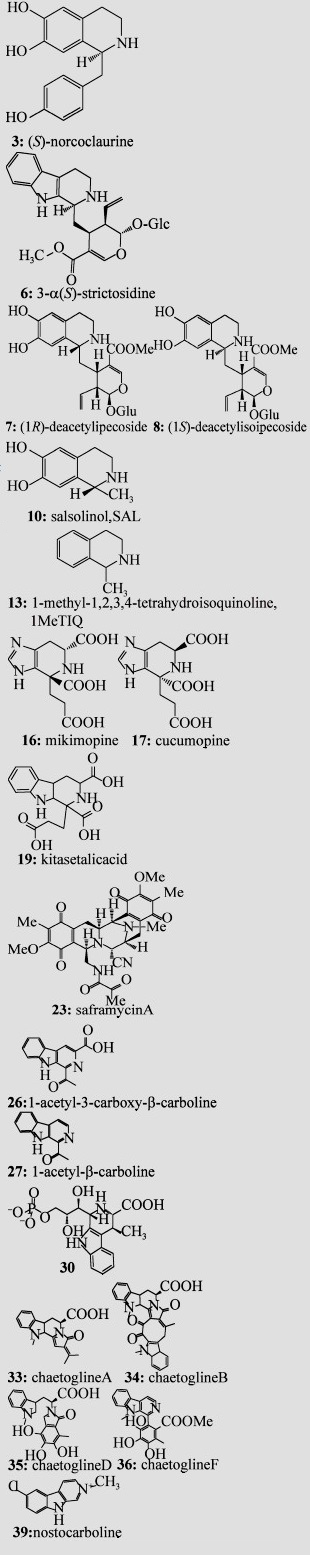 |
| STR *# (Rauvolfia serpentia; Catharanthus roseus; Ophiorrhiza pumila) | |||
| DIS/DIIS* (Alangium lamarckii Thw.) | |||
| SAL synthase (Parkinsonian patient brain; rat brain) | |||
| 1MeTIQ synthase (Animal brain) | |||
| MIS/CUS* (Agrobacterium rhizogenes derived from Ajuga reptans) | |||
| KslB (Kitasatospora setae NBRC 14216T) | |||
| SfmC (Streptomyces lavendulae) | |||
| McbB*# (Marinactinospora thermotolerans SCSIO 00652) | |||
| NscbB* (Nocardiopsis synnemataformans DSM 44143) | |||
| StnK2* (Streptomyces flocculus) | |||
| FPS (Chaetomium globosum 1C51) | |||
| Nostoc 78-12A (Freshwater cyanobacterium) | |||
| *Indicate enzyme activity was characterized; #Indicate three dimensional (3D) structures were elucidated. | |||
表选项
1 植物来源的Pictet-Spengler酶目前,植物中已经鉴定的P-S酶包括3类:NCS[2, 13-38]、STR[39-49]和Deacetylipecoside合酶[50-51]。前两种酶的研究较为透彻,已有的酶学和结构生物学方面的研究表明两者之间序列同源性极低,推测它们可能由不同祖先经趋同进化而来[10]。目前尚未获得Deacetylipecoside合酶的序列信息。这3种酶都是植物生物碱合成代谢途径中的关键酶,催化合成了大量的生物碱,其中包括多种具有抗肿瘤、抗感染活性的药用先导化合物,研究价值较大[2-10]。
1.1 去甲乌药碱合酶(NCS)NCS能够催化多巴胺(Dopamine,DA,1)和4-羟基苯乙醛(4-Hydroxyphenylacetaldehyde,4-HPAA,2)发生立体选择性P-S反应生成(S)-norcoclaurine(3),即2 500多种BIA生物合成途径的起始结构单元[2, 13-14]。而这些BIA活性分子广泛应用于临床治疗,其中包括止痛药吗啡(Morphine)、止咳药诺斯卡品(Noscapine)等[2]。
首个鉴定的NCS是由Samanani等从一种唐松草Thalictrum flavum的细胞提取液中分离纯化获得的TfNCS。其天然蛋白的分子量约为28 kDa,由2个亚基组成;反应最适pH范围为6.5–7.0,最适温度范围为42–55 ℃,活性不受CaCl2和MgCl2的影响[14]。经大肠杆菌表达纯化,获得的重组酶分子量为23.3 kDa,等电点(Isoelectric point,pI)为5.5,酶反应最适pH值为7.0,最适反应温度为40 ℃,对4-HPAA底物的Km为700 μmol/L[15]。起初,基于核磁共振研究和TfNCS与对羟基苯甲醛的共结晶蛋白(PDB ID 2VQ5)结构分析,研究人员提出了羰基供体首先与活性中心结合的机理[16-17]。然而,这并不能解释TfNCS对醛底物的宽泛性,因为从机理上讲,需要活性位点残基使得DA去质子化,以便与亚胺离子中间体进行分子内环化。相比之下,DA首先与活性中心结合(即“DA”优先)的机理则更为合理,具体机制如下:首先,DA的3位羟基与Lys122结合,N原子与Glu110和Asp141结合;随后,羰基底物结合上来,由Tyr108、Glu110和Asp141催化形成亚胺;Glu110和Lys122依次诱发相应基团的去质子化、亲电加成形成终产物[16-18]。随后,Lichman通过对中间体类似物与TfNCS的共结晶(PDB ID 5N8Q,5NON)分析,发现DA能够结合到Lys122上,并证明了“DA”优先的机制合理性;此外晶体学分析发现羰基供体可结合于活性位点入口处(靠近溶剂),这种结合方式可有效拓展酶的底物利用范围,这也是TfNCS醛底物谱宽泛的直接原因[18-19]。Sheng和Himo通过量子化学理论计算发现,从能量消耗角度来说,虽然两种机制在酶-底物复合体形成时均可实现,但“DA”优先机制在能量上更为有利,且更加可行[19-20]。
Minami等证实来自于日本黄连Coptis japonica的CjNCS与TfNCS具有同样的功能,其天然酶和在大肠杆菌中表达得到的重组酶均能识别苯乙醛、4-HPAA和3, 4-二羟基苯乙醛,从而与DA反应[21]。随后的研究证实TfNCS和CjNCS能够识别多个醛基供体,从而酶促合成了一系列TIQ生物碱,彰显了它们作为生物催化剂的实力,但两者对底物胺的识别范围相对较为局限[22-23]。此外,TfNCS还能够识别α-取代醛,优先接受(R)-构型,其点突变株(Met97Val)能够以高非对映体比率(98︰2)制备(1S, 1’R)-TIQ产物[24]。值得一提的是,Lichman等研究发现TfNCS能够以一系列未活化的酮作为底物,在温和条件下形成1, 1’-取代的手性和螺环TIQ[25]。而来自于C. japonica的CjNCS2 (即CjPR10A)仅能识别活化的α-酮酸4-羟基苯丙酮酸和丙酮酸,但对醛的识别较为宽泛[21, 25]。因此,这是首次报道NCS能够催化未活化的酮的反应,促进了人们对NCS在体内和体外产生不同的TIQ家族化合物的催化能力的认识。此外,TfNCS和CjNCS也能识别一系列线性脂肪醛,应用“一锅法”化学-酶级联反应,生产了金莲花碱(Trolline)衍生物、(S)-BIA和(S)-四氢原小檗碱[26-28]。同时,研究人员还利用NCS及其他相关酶作为生物合成功能元件,在模式菌株大肠杆菌和酵母中构建了生产BIA和TIQ家族多个成员的细胞工厂[29-36],如阿片(Opioid)类生物碱蒂巴因(Thebaine)和氢可酮(Hydrocodone)[31]、抗肿瘤药物诺斯卡品[32-33]、(S)-网状番荔枝碱及其卤代物和磺酸化产物[33-36]。这些研究充分显示了NCS及其突变株在大量制备异喹啉生物碱家族成员中的应用,建立了相应的微生物生产平台,为未来更广泛的相关研究提供了方便,有望促进针对多种人类疾病的药物研发。
此外,还有一些来源于其他植物(如蓟罂粟Argemone mexicana、大红罂粟Papaver bracteatum、石生黄堇Corydalis saxicola)的NCS也能高效产生对映体过量的TIQ[37]。另有报道从水芙蓉Nelumbo nucifera的种子胚中分离到了(R)-norcoclaurine[38],表明NCS可能具备催化(R)-选择反应的能力或是存在对应的差向异构酶。
1.2 异胡豆苷合酶(STR)STR能够催化色胺(Tryptamine,4)和裂环马钱子碱(Secologanin,5)的P-S反应形成3-α(S)-异胡豆苷(6),其产物是超过2 000种植物来源MIA共同的生物合成前体[39]。MIA家族成员活性多样,临床应用广泛,典型例子包括抗感染药物喹啉、抗肿瘤药物喜树碱以及抗心律失常药物阿吗灵等[2]。STR家族成员中被研究得较为透彻的酶是来自于印度萝芙木Rauvolfia serpentia的RsSTR、来自于长春花Catharanthus roseus的CrSTR以及来自于短小蛇根草Ophiorrhiza pumila的OpSTR[39-41]。
Treimer等于1979年从C. roseus的悬浮培养物细胞中首次分离得到CrSTR,CrSTR比较稳定,分子量为34 kDa,酶反应最适pH值为6.8,裂环马钱子碱和色胺的Km值分别为3.4 mmol/L和2.3 mmol/L[42]。随后,McKnight等得到了其序列[43],de Waal等纯化得到了CrSTR的6种蛋白亚型(A1、A2、B、C1、C2、D),它们均为糖蛋白,Km值没有显著的差别,但在pI、分子量、离子交换的洗脱位置上差异较大[44]。2006年McCoy等研究表明CrSTR能识别18种色胺结构似物中的8种,能识别4种裂环马钱子碱结构类似物中的2种,但色胺催化效率最高[45]。2006年,Ma等研究发现RsSTR酶的晶体是一个新颖的六叶β-螺旋桨折叠结构蛋白,他们还得到了RsSTR与底物色胺和裂环马钱子碱(Secologanin)的共结晶(PDB ID 2FP8、2FP9、2FPB、2FPC)[40]。结构分析结合点突变实验证实RsSTR酶的关键活性位点是Glu309、Tyr151和His307。2008年,Maresh等通过动力学同位素效应和Ab initio理论计算等对CrSTR的催化机制进行研究,发现亚胺离子中间体的形成是酸催化,带正电荷中间体的重新芳构化是限速步骤,最终的去质子化过程是碱催化,整个反应过程并不生成螺环假吲哚中间体[46]。CrSTR的限速催化与P-S反应的酸碱催化机制相似,酶的存在保证底物在低浓度条件下发生反应,并产生单一立体对映体产物[46]。
Pressnitz等对CrSTR、OpSTR、RsSTR和RsSTR的突变株(Val208Ala)进行研究发现,它们在大肠杆菌中表达纯化后,能够识别一系列低分子量的脂肪醛,高产率生产C-1位置为(R)-构型的单一对映体(> 98%),并采用化学-酶级联反应法,得到了光学纯级的(R)-harmicine[47]。Wu等利用STR化学-酶法合成了多个结构新颖的生物碱[48];Cai等还将STR用于合成具有立体选择性的N-取代的异胡豆苷衍生物,具有拓扑异构酶Ⅰ抑制活性[49]。Eger等研究发现STR催化小分子醛类反应时会形成(R)-构型产物,他们结合X-单晶衍射、突变和MD方法,解析了OpSTR形成(R)-构型产物的基本原理:短链醛与裂环马钱子碱相比以反向方式结合,偏向于形成反向立体(R)-结构产物,这种通过对醛底物的不同结合方式而给出不同的产物绝对构型,表明相同的催化酶可以对一种底物具有两种不同的结合模式[41]。这些系统研究彰显了STR家族成员用于生物催化剂的较大潜力,可为酶的未来工程设计提供指导。Brown等还实现了Strictosidine在酵母中的从头合成,生产效价达0.5 mg/L,可为植物来源MIA的系统研究提供重要资源[39]。
1.3 Deacetylipecoside合酶(DIS)和Deacetylisoipecoside合酶(DIIS)DIS与DIIS是在中性条件下,催化DA和裂环马钱子碱分别合成差向异构体(1R)-deacety- lipecoside (7)和(1S)-deacetylisoipecoside (8)的酶。这两种酶最早由De-Eknamkul等从热带植物印度八角枫Alangium lamarckii Thw.的叶子中提取得到,其产物为Deacetylipecoside和Deacetylisoipecoside,是在该植物中催化合成的两种异构体,分别是大量生物碱和含氮糖苷的前体[50]。DIS比较稳定且能通过柱层析法纯化得到,而DIIS不稳定且不易纯化。2000年De-Eknamkuld等分离纯化得到DIS,纯化倍数为570倍,回收率为6%,DIS为多肽单体,分子量为30 kDa,底物专一性较高,对DA和裂环马钱子碱的Km值分别为0.7 mmol/L和0.9 mmol/L[51]。DIS活性不被底物DA抑制,但能被一些TIQ类生物碱抑制,具体抑制机制尚不清楚。这两种酶的氨基酸序列目前尚未确定,但有研究显示,它们在分子大小、最适温度、pH和底物专一性等方面都与上述的STR类似[51]。
2 动物来源的Pictet-Spengler酶到目前为止,已经从动物体内分离得到了两类能合成异喹啉生物碱的酶,即去甲猪毛菜碱(Salsolinol,SAL)合酶和1-甲基-1, 2, 3, 4-四氢异喹啉(1-Methyl-1, 2, 3, 4-tetrahydroisoquinoline, 1MeTIQ)合酶,这两类酶的发现、鉴定以及相应产物的形成和活性鉴定,对人们了解相关的帕金森氏病(Parkinson’s disease,PD)的发病机制以及疾病预防具有重要意义。
2.1 SAL合酶SAL合酶能够催化DA (1)和乙醛(9)生成1-甲基-6, 7-二羟基-1, 2, 3, 4-四氢异喹啉(又称去甲猪毛菜碱,Salsolinol,SAL,10),是脑内合成SAL及其衍生物的关键酶,与帕金森氏病发病机制密切相关[52],SAL目前已在人体来源的尿液、脑脊液和脑组织中被检测到[53]。SAL是一种内源性儿茶酚异喹啉类物质,可以经由甲基化或氧化产生一系列具有神经毒性的衍生物,这些内源性神经毒素会引起多巴胺能神经元的死亡,从而导致帕金森氏病的发生。SAL有R和S两种构型,其C-1位是不对称中心,DA和乙醛发生P-S反应时会形成外消旋体,但在人脑中只检测到了(R)-SAL,并且SAL不能通过血脑屏障,间接说明脑内检测到的(R)-SAL极有可能是酶促催化形成[52, 54]。
1996年Naoi等从帕金森氏病患者脑灰质区首次分离得到SAL合酶,它能够催化DA和乙醛立体选择异构形成(R)-SAL,此酶专一性识别DA,能够识别甲醛和丙酮酸,但不能识别N-甲基多巴胺、肾上腺素、去甲肾上腺素和左旋多巴,其分子量约为34.3 kDa[52, 55]。Chen等发现SAL合酶也存在于斯普拉-道来氏大鼠脑中,继续研究发现鼠脑中的SAL合酶最适温度是37 ℃,耐热性较好,对强酸敏感,最适pH值为7.4,pI为6.6 [56-58]。SAL合酶含有77个氨基酸,大小约为10 kDa,该酶与泛素蛋白的氨基酸序列仅有4个氨基酸的差异[58]。因此,他们通过对泛素基因的4个位点进行定点突变获得了SAL合酶基因,并将其连接到载体pET30a-GST上,表达并纯化了GST融合蛋白,为进一步研究SAL合酶结构、酶学性质及其生物功能奠定基础,也为阐明内源性神经毒素儿茶酚异喹啉类物质诱发帕金森氏病的发病机制提供佐证[58-59]。
2.2 1MeTIQ合酶1MeTIQ合酶是一种催化2-苯基乙胺(11)与丙酮酸(12)生成1MeTIQ (13)的酶。1MeTIQ和TIQ广泛存在于人和各种其他动物的脑中,在PD患者中,1MeTIQ的检测水平比正常人脑低,且随着年龄的增加呈减少趋势[10, 60],因此,1MeTIQ可能是一种内源性的帕金森氏病预防因子。Yamakawa等研究发现1MeTIQ合酶是一种位于线粒体囊泡上的膜蛋白。该酶活性会被TIQ衍生物抑制,而不会被SAL或Norlaudanosoline抑制[10, 61]。Yamakawa等还发现该酶在大脑和丘脑有较高的活性,而1MeTIQ在纹状体和黑质区的分布要比脑中其他区域多[10, 61]。同时,Abis等发现,1MeTIQ合酶的活性在老年鼠脑内明显下降,1MeTIQ能改善由1-甲基-4-苯基-1, 2, 3, 6-四氢吡啶及一些内源性神经毒素引起的帕金森氏病症状[10, 62]。因此,1MeTIQ可能是一种能预防帕金森氏病的物质,而1MeTIQ合酶的缺失与否及功能水平则可能在先天性帕金森氏病致病机理中起到非常重要的作用。
3 微生物来源的Pictet-Spengler酶目前对微生物来源的P-S酶研究较为深入的是McbB、NscbB和StnK2。
3.1 Mikimopine合酶(MIS)和Cucumopine合酶(CUS)Mikimopine (16)和Cucumopine (17)是由L-组氨酸(14)与α-酮戊二酸(15)在NADPH的参与下经过P-S缩合成环形成,两者为镜像对称结构,均属于冠瘿碱[63-65]。冠瘿碱是由农杆菌感染引起的植物冠瘿瘤细胞的特殊基因产物,其合成是由于农杆菌Ti (或Ri)质粒的T-DNA (转移DNA)在植物细胞中发生了水平基因转移、整合与表达。冠瘿碱由植物细胞产生,能够为农杆菌的生长提供碳源和氮源[66]。2001年,Suzuki等从来源于匍匐筋骨草Ajuga reptans的毛状根中的农杆菌Agrobacterium rhizogenes出发,从其Ri质粒(pRi1724)的T-DNA区得到了mikimopine合酶编码基因mis,同时以来源于pRi2659的cucumopine编码基因cus为对照,将二者在大肠杆菌中表达纯化后检测到了相应蛋白的酶活[63-65]。分析表明mis和cus在核酸水平上并无序列同源性,但在氨基酸水平上部分同源,且转(mis)基因的烟草植株能够产生冠瘿瘤[66]。比较特殊的是该酶的其中一个底物为α-酮酸,而其他的P-S酶通常以醛作为天然底物。
3.2 KslB在菌株世里北里孢菌Kitasatospora setae NBRC 14216T中蕴含着一个P-S酶(KslB,KSE_70640),目前已获得该酶的基因序列,对其功能的鉴定依赖于异源表达和对应产物的鉴定,尚缺少蛋白纯化和体外生化实验以及蛋白结构等研究[67-70]。
前期工作中,Aroonsri等从K. setae NBRC 14216T分离到一个结构新颖的β-咔啉生物碱Kitasetaline[67];随后,Aroonsri等证实KsbC能够激活巴弗洛霉素(Bafilomycin)的产生,同时抑制Kitasetaline的产生,是Kitasetaline的负调控因子[68];同时,他们在利用异源表达的方式挖掘该菌株产次级代谢产物的过程中,从异源表达重组菌株中分离得到了两个β-咔啉生物碱类化合物JBIR-133和JBIR-134,与Kitasetaline有着相同的骨架,由此证实该段DNA序列中涵盖Kitasetaline的生物合成基因,其中含kslA (kse_70650)、kslB (kse_70640)、kslC (kse_70630)的基因区段最有可能是Kitasetaline的生物合成基因簇,而这3个功能基因编码蛋白的预测功能分别为FAD依赖的氧化还原酶、黄瓜碱合酶和细胞色素P450单加氧酶[68]。
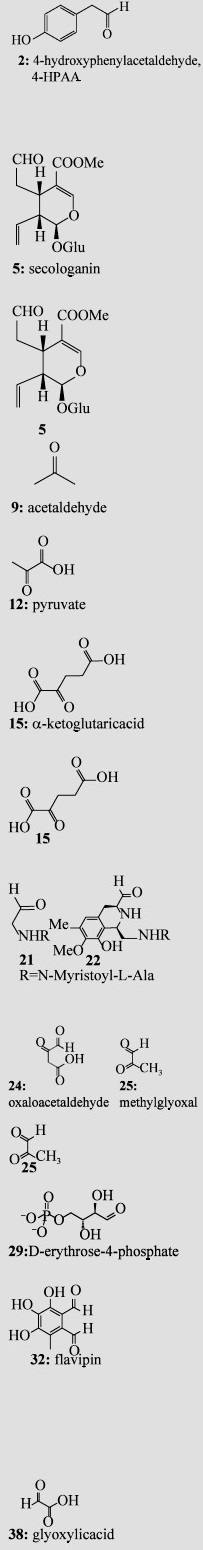
随后,Ueda等将kslB在异源宿主S. avermitilis SUKA22中进行表达,继而分离得到了一个四氢β-咔啉类化合物Kitasetalic acid (19),而在培养基中添加L-色氨酸(18)时,能够显著提高Kitasetalic acid的产量[69-70],Kitasetalic acid能够抑制多个肿瘤细胞系的葡萄糖调节蛋白78(GRP78)的表达,后续有可能作为与抗癌药物共同治疗的先导化合物进行研究[69-70]。这间接证实KslB是一个P-S酶,可能催化L-色氨酸(18)和α-酮戊二酸(15)进行缩合形成Kitasetalic acid,继而作为Kitasetaline生物合成的中间体。随后,Ueda等设计了不同基因组合的异源表达实验,证实kslA、kslB和kslC三个基因便足以生物合成Kitasetaline和JBIR-133,而仅有kslA和kslB时只能够形成JBIR-133,这说明JBIR-133可能是形成Kitasetaline的中间体,而Kitasetalic acid可能是KslA的底物,后经生物转化实验证实KslA可以将Kitasetalic acid转化成JBIR-133。Ueda等再经过巧妙的文献论证推测Kitasetaline的生物合成中可能涉及到放线硫醇介导的外源解毒体系,在Kitasetaline的野生宿主K. setae中确实存在相关的生物合成基因和放线硫醇解毒蛋白(Mycothiol- S-conjugate amidase,Mca)编码基因mca[69-70]。鉴于此,在S. avermitilis SUKA22的野生型菌株和敲除了mca的突变株中分别导入ksl相关基因,质谱检测发现,前者可以正常生产Kitasetaline,而后者则不能,但后者积累了Mycothiol-S-conjugated kitasetaline (图 2)。因此,作者推测,Mca可以水解Mycothiol-S-conjugated kitasetaline而生成Kitasetaline,mca是Kitasetaline生物合成必需的一个基因。最后通过前体介导的生物合成,喂养了5-氟-色氨酸和6-氟-色氨酸,分别得到了相应位置被氟取代的Kitasetalic acid和JBIR-133,并测定了其生物学活性,间接证实KslB酶的底物谱宽泛[69-70]。
 |
| 图 2 Kitasetaline可能的生物合成路径[69-70] Fig. 2 Proposed biosynthetic pathway of kitasetaline in K. setae NBRC 14216T[69-70]. |
| 图选项 |
3.3 能催化P-S反应的非核糖体聚肽合成酶(Nonribosomal peptide synthetase,NRPS) SfmCSfmC是一个NRPS,有4个结构域,分别为缩合(Condensation,C)结构域、腺苷酰化结构域(Adenylation,A)、肽基载体蛋白(Peptidyl carrier protein,PCP)以及还原(Reductase,RE)结构域,其中C结构域能够负责相应前体和对应醛的P-S缩合反应形成TIQ骨架结构,因此SfmC也是一个具有催化P-S反应的功能蛋白[71]。Koketsu等研究发现sfmC在E. coli中表达时以同源二聚体(Homodimer)形式存在,SfmC分子量约为163 kDa;在4个结构域同时存在的情况下、缺失了C结构域的情况下以及缺失了RE结构域的情况下,分别进行酶反应,结合相应产物的形成检测情况,证实该非核糖体肽合成酶SfmC的C结构域可催化相应底物经过两步P-S反应形成番红霉素A (Saframycin A,23)的五环刚性骨架结构[71]。番红霉素A是TIQ家族中最为典型的抗生素,具有抗肿瘤活性,其生物合成基因簇含有30个基因,其中包括sfmC。SfmC是微生物来源的非核糖体肽合成酶中的缩合结构域催化P-S反应的首例报道,而且是由同一个非核糖体肽合成酶经重复催化循环完成,在次级代谢产物的生物合成中非常罕见[71-72]。
3.4 McbB和NscbBMcbB是Marinacarbolines生物合成路径中的一个负责β-咔啉骨架合成的关键酶,已经进行了系统的酶学及酶结构功能相关性研究[73-77]。Marinacarbolines是Huang等从南海深海泥沉积物来源的Marinactinospora thermotolerans SCSIO 00652中分离得到的β-咔啉生物碱类化合物,该类化合物对疟原虫多重耐药株Dd2及敏感株3D7具有显著的抑制作用但不显示细胞毒活性,是优良的抗疟药物先导化合物[73-74]。鉴于此,研究团队在分析其化学结构特点的基础上筛选并鉴定了其生物合成基因簇,大小约为5.6 kb,包含4个基因,预测功能分别为编码酰胺键合成酶的mcbA,编码未知功能蛋白的mcbB,编码脱羧酶的mcbC和编码O-甲基转移酶的mcbD;其中mcbABC连续同向排布,为主基因簇,mcbD独立存在于主基因簇之外[75]。随后,发现mcbB在E. coli BL21中表达时,产生一个含β-咔啉生物碱骨架的主产物1-酰基-3-羧基-β-咔啉(26)和两个中间体结构衍生物1-酰基-β-咔啉(27)和1-酰基-3-羟基-β-咔啉,从而间接证实McbB是一个负责β-咔啉骨架形成的P-S酶。接着通过喂养氟代前体化合物和同位素标记化合物的实验,证实这两个β-咔啉分子骨架(26和27)的形成以L-色氨酸(18)和三酸酸循环途径中的草酰乙醛(24)作为生物合成的前体。进一步对McbB及其同源序列进行保守位点分析,证实Glu97与其活性密切相关,可能是McbB的催化活性中心[75]。
随后Wang和Mori等几乎同时开展了McbB的结构与功能研究,前者仅表征了该酶晶体学特征[76-77],后者更深入系统进行了酶学及酶结构功能相关性研究,通过体外实验证实草酰乙醛(24)可作为醛基供体,与L-色氨酸(18)在McbB的催化下形成1-酰基-3-羧基-β-咔啉(26)和1-酰基-β-咔啉(27);McbB具有完全不同于其他P-S酶的三维折叠特征(PDB ID 3X27),与底物L-色氨酸的共结晶结构结合关键残基定点突变分析证实,催化活性中心Glu97残基是McbB酶反应的“酸碱”催化剂[76]。底物宽泛性研究表明,McbB能够识别5-甲基-DL-色氨酸、7-甲基-DL-色氨酸、丙酮醛(25)、甲醛、乙醛、丙醛和异丁醛;McbB的双点突变株(His87Ala/Arg72Ala)能够识别非天然的醛基供体苯乙二醛,并与L-色氨酸发生P-S反应[76]。
笔者所在研究团队通过基因组挖掘技术,以McbB为探针,从一株分离自肾脏移植病人的线团拟诺卡氏菌Nocardiopsis synnemataformans DSM 44143中挖掘并鉴定了一个新的P-S酶NscbB,它能够催化L-色氨酸(18)和丙酮醛(25)产生1-酰基-3-羧基-β-咔啉(26)和1-酰基-β-咔啉(27)[78-79]。NscbB与McbB具有66%的一致性和80%的同源性,同样有着对应的Glu97,因此,推测它们可能有着类似的催化机制,动力学常数表征表明NscbB的kcat/Km值比McbB约高30倍,而Km值则相差不太明显,但热稳定性相对较差[78]。
综上,推测McbB和NscbB的催化机制为(图 3):在酶的作用下,Glu97作为活性中心催化草酰乙醛或丙酮醛与L-色氨酸经过P-S缩合形成席夫碱中间体,继而形成关键中间体26a;26a中的六元氮杂环经两重氧化反应芳构化形成26,或是六元氮杂环在脱羧和芳构化后氧化形成化合物27[75, 78]。
 |
| 图 3 NscbB和McbB催化P-S反应的机制推导图[75, 78] Fig. 3 Proposed mechanism of the P-S reaction applied to the NscbB and McbB enzymatic reaction[75, 78]. |
| 图选项 |
3.5 StnK2StnK2是链黑菌素生物合成中的一个P-S酶,能够催化(2S, 3S)-β-甲基色氨酸(28)和D-赤藓糖-4-磷酸(29)形成特殊的四氢-β-咔啉骨架(30),而不是彻底芳香化的β-咔啉[80-81]。与McbB底物谱宽泛不同的是StnK2对醛基要求较为严格,仅识别D-赤藓糖-4-磷酸(29),且具有严格的立体选择性,仅能识别L-色氨酸中的(S)-3-甲基和(S)-2-氨基丙酸结构单元,特异性产生四氢- β-咔啉骨架中的(R)-C-1。与McbB类似的是,二者都只能识别L-色氨酸,不能识别D-色氨酸,StnK2对L-色氨酸的结构类似物具有宽泛性,可以识别4-氟-L-色氨酸、5-氟-L-色氨酸、6-氟-L-色氨酸以及对应的β-甲基化产物,如4-氟-(2S, 3S)- β-甲基-色氨酸、5-氟-(2S, 3S)-β-甲基-色氨酸、6-氟-(2S, 3S)-β-甲基-色氨酸。酶促反应动力学实验表明,5-氟-(2S, 3S)-β-甲基-色氨酸和6-氟-(2S, 3S)-β-甲基-色氨酸对StnK2的亲和力比(2S, 3S)-β-甲基色氨酸要高一些,这提示后续可以利用前体介导的生物合成法得到氟代的链黑菌素,为后活性筛选提供更多的化合物实体分子[81]。此外,StnK2还能够识别5-羟基-L-色氨酸,转化率较低;StnK2不能识别5-羟基-色胺[81]。
3.6 真菌来源的P-S酶植物来源STR序列的测定和功能的鉴定为其他真核生物来源结构相似的生物碱的发现提供了可供参考的标记和标准。以STR的基因序列作为标记,Yan等从一株球毛壳菌Chaetomium globosum 1C51中激活了一个高度同源性基因fps,从而得到了一系列骨架全新的吲哚类生物碱Chaetoglines[82]。研究发现1-甲基-L-色氨酸(1-MT,31)能够特异性地激活C. globosum 1C51中的STR编码基因,使其上调5倍以上。1-MT在诱导表达出的FPS作用下,与该真菌合成的芳香醛(Flavipin,32)发生P-S反应,随后经由多种氧化还原酶的作用下形成一系列骨架全新的生物碱Chaetogline (33–36)[82]。其中Chaetoglines B(34)和F(36)的抗菌活性超过临床用药(替硝唑),可为新型抗临床(厌氧)菌的药物研发提供化合物实体;Chaetogline F(36)还具有较好的乙酰胆碱酯酶(Acetylcholinesterase,AChE)抑制活性,而AChE是老年痴呆症疾病密切相关的药物作用靶标[82]。因此,Chaetogline F(36)可以作为开发抗老年痴呆药物的起始分子[82-83]。此外,该FPS的能够识别色氨酸、5-甲基-色氨酸、5-羟基-L-色氨酸和5-氯-色氨酸,底物识别具有一定的宽泛性[82]。总体来说,通过1-MT (31)的添加,成功激活了菌株C. globosum 1C51中FPS编码基因的表达,改变了其次生代谢路径,从而得到了新的吲哚生物碱类化合物,这是真菌蕴含P-S酶的首次报道。
3.7 其他P-S酶在蓝细菌Nostoc 78-12A中蕴藏着P-S酶(对应的底物可能是37和38),但目前并未获得其氨基酸序列和相应编码基因。前期工作中,Becher等从蓝细菌Nostoc 78-12A中分离得到了一个氯取代和N-甲基取代的咔啉阳离子生物碱Nostocarboline (39),它能够显著抑制丁酰胆碱酯酶(Butyrylcholinesterase,BChE)的活性,而BChE可能是阿尔茨海默病相关治疗药物的靶标[84]。因此,Nostocarboline可能成为相应的神经药物研发的先导化合物[84]。此外,Nostocarboline还能显著地选择性抑制光能自养生物,具有抑制疟原虫Plasmodium falciparum的活性[85]。随后Portmann等采用前体介导的生物合成的方法,在蓝细菌Nostoc 78-12A中喂养氟、溴或是甲基取代的色氨酸衍生物前体,得到了相应位置被卤代或者甲基化的Nostocarboline衍生物[85]。证实了前体介导的生物合成方法的普遍适用性,同时也说明Nostoc78-12A中相应负责该P-S环化的酶对底物识别具有宽泛性。
4 总结与展望综上所述,一系列P-S酶的功能鉴定和相关生物合成、化学结构多样性等研究,不但解决了MIA、BIA以及β-咔啉生物碱核心骨架的生物合成及前体物质来源等问题,同时也对生物碱资源的发掘乃至大规模生产、应用具有实际指导意义。动物来源的1MeTIQ合酶和SAL合酶则与帕金森氏病致病机理密切相关。关键酶和效应分子的发现,使得P-S反应在帕金森氏病致病机理分析和疾病预防及治疗等方面具有重要意义。然而,在已鉴定的P-S酶中,只有NCS和STR被广泛鉴定并应用于化学-酶法合成结构新颖的生物碱。同时,由于底物的识别范围、分离方法以及酶的稳定性等方面的局限,迫切需要发掘优化更多高效P-S酶作为潜在生物催化剂进行优化开发。相关研究工作前景广阔。
因此,基于已有P-S酶的研究进展,相关研究工作未来可在以下几个方面进行拓展:(1)利用已有P-S酶的序列作为分子标记,从GenBank数据库中定向挖掘潜在的P-S酶新资源;(2)采用不同方法得到动植物来源P-S酶的序列信息,对其进行系统比较,通过序列分析,找出其同源性及功能差异性序列,为酶蛋白定向进化研究提供可靠参考;(3)借助计算机动态模拟和共结晶等手段,解析P-S酶的三维结构与催化机制的关系,为蛋白质改造奠定理论基础;(4)加强对P-S酶的蛋白质体外定向进化研究,提高P-S酶的活性、热稳定性、底物识别宽泛性等,构建新型P-S酶,使其发挥更大作用,完成更多特异性反应,最终实现工业级催化改造;(5)加强P-S酶在化学-酶法合成新颖结构生物碱及其衍生物方面的研究,从而增加生物碱家族成员的结构多样性,为活性筛选提供更多化合物实体。目前,笔者所在研究团队围绕微生物特别是放线菌开展新颖P-S酶资源的发掘,并开展相关酶学、结构生物学和天然产物化学方面的研究,已取得了阶段性进展,相关工作会在后续报道。综上所述,随着基因组挖掘、生物信息以及基因编辑等新技术的发展和广泛应用,P-S酶的发掘和鉴定工作必将进入一个崭新的局面,相关研究工作方兴未艾!
参考文献
| [1] | Amirkia V, Heinrich M. Alkaloids as drug leads-a predictive structural and biodiversity-based analysis. Phytochem Lett, 2014, 10: 98-103. |
| [2] | Roddan R, Ward JM, Keep NH, et al. Pictet-Spenglerases in alkaloid biosynthesis: future applications in biocatalysis. Curr Opin Chem Biol, 2020, 55: 69-76. DOI:10.1016/j.cbpa.2019.12.003 |
| [3] | Wang FP. Modern chemistry of natural products. Beijing: Science Press, 2009: 733-995 (in Chinese). 王锋鹏. 现代天然产物化学. 北京: 科学出版社, 2009: 733-995. |
| [4] | O'Connor SE, Maresh JJ. Chemistry and biology of monoterpene indole alkaloid biosynthesis. Nat Prod Rep, 2006, 23(4): 532-547. DOI:10.1039/b512615k |
| [5] | Hagel JM, Facchini PJ. Benzylisoquinoline alkaloid metabolism: a century of discovery and a brave new world. Plant Cell Physiol, 2013, 54(5): 647-672. DOI:10.1093/pcp/pct020 |
| [6] | Cao RH, Peng WL, Wang ZH, et al. β-Carboline alkaloids: biochemical and pharmacological functions. Curr Med Chem, 2007, 14(4): 479-500. |
| [7] | Pictet A, Spengler T. über die bildung von isochinolin-derivaten durch einwirkung von methylal auf phenyl-?thylamin, phenyl-alanin und tyrosin. Ber Dtsch Chem Ges, 1911, 44(3): 2030-2036. DOI:10.1002/cber.19110440309 |
| [8] | Cox ED, Cook JM. The pictet-spengler condensation: a new direction for an old reaction. Chem Rev, 1995, 95(6): 1797-1842. DOI:10.1021/cr00038a004 |
| [9] | St?ckigt J, Antonchick AP, Wu FR, et al. The Pictet-Spengler reaction in nature and in organic chemistry. Angew Chem Int Ed Engl, 2011, 50(37): 8538-8564. DOI:10.1002/anie.201008071 |
| [10] | Chen XC, Wang R, Arshad A, et al. Pictet-Spenglerases and their related biological characteristics. Chemistry, 2011, 74(3): 218-224 (in Chinese). 陈薛钗, 王睿, Arshad A, 等. Pictet-Spengler酶及其相关生物学特性. 化学通报, 2011, 74(3): 218-224. |
| [11] | Lin CI, McCarty RM, Liu HW. The enzymology of organic transformations: a survey of name reactions in biological systems. Angew Chem Int Ed Engl, 2017, 56(13): 3446-3489. DOI:10.1002/anie.201603291 |
| [12] | Calcaterra A, Mangiardi L, Monache GD, et al. The Pictet-Spengler reaction updates its habits. Molecules, 2020, 25(2): 414. |
| [13] | Luk LYP, Bunn S, Liscombe DK, et al. Mechanistic studies on norcoclaurine synthase of benzylisoquinoline alkaloid biosynthesis: an enzymatic Pictet-Spengler reaction. Biochemistry, 2007, 46(35): 10153-10161. DOI:10.1021/bi700752n |
| [14] | Samanani N, Facchini PJ. Purification and characterization of norcoclaurine synthase. The first committed enzyme in benzylisoquinoline alkaloid biosynthesis in plants. J Biol Chem, 2002, 277(37): 33878-33883. DOI:10.1074/jbc.M203051200 |
| [15] | Samanani N, Liscombe DK, Facchini PJ. Molecular cloning and characterization of norcoclaurine synthase, an enzyme catalyzing the first committed step in benzylisoquinoline alkaloid biosynthesis. Plant J, 2004, 40(2): 302-313. DOI:10.1111/j.1365-313X.2004.02210.x |
| [16] | Berkner H, Schweimer K, Matecko I, et al. Conformation, catalytic site, and enzymatic mechanism of the PR10 allergen-related enzyme norcoclaurine synthase. Biochem J, 2008, 413(2): 281-290. DOI:10.1042/BJ20080306 |
| [17] | Ilari A, Franceschini S, Bonamore A, et al. Structural basis of enzymatic (S)-norcoclaurine biosynthesis. J Biol Chem, 2009, 284(2): 897-904. DOI:10.1074/jbc.M803738200 |
| [18] | Lichman BR, Gershater MC, Lamming ED, et al. 'Dopamine-first' mechanism enables the rational engineering of the norcoclaurine synthase aldehyde activity profile. FEBS J, 2015, 282(6): 1137-1151. DOI:10.1111/febs.13208 |
| [19] | Lichman BR, Sula A, Pesnot T, et al. Structural evidence for the dopamine-first mechanism of norcoclaurine synthase. Biochemistry, 2017, 56(40): 5274-5277. DOI:10.1021/acs.biochem.7b00769 |
| [20] | Sheng X, Himo F. Enzymatic Pictet-Spengler reaction: computational study of the mechanism and enantioselectivity of norcoclaurine synthase. J Am Chem Soc, 2019, 141(28): 11230-11238. DOI:10.1021/jacs.9b04591 |
| [21] | Minami H, Dubouzet E, Iwasa K, et al. Functional analysis of norcoclaurine synthase in Coptis japonica. J Biol Chem, 2007, 282(9): 6274-6282. |
| [22] | Ruff BM, Br?se S, O'Connor SE. Biocatalytic production of tetrahydroisoquinolines. Tetrahedron Lett, 2012, 53(9): 1071-1074. DOI:10.1016/j.tetlet.2011.12.089 |
| [23] | Nishihachijo M, Hirai Y, Kawano S, et al. Asymmetric synthesis of tetrahydroisoquinolines by enzymatic Pictet-Spengler reaction. Biosci Biotechnol Biochem, 2014, 78(4): 701-707. |
| [24] | Roddan R, Gygli G, Sula A, et al. Acceptance and kinetic resolution of α-methyl-substituted aldehydes by norcoclaurine synthases. ACS Catal, 2019, 9(10): 9640-9649. |
| [25] | Lichman BR, Zhao JX, Hailes HC, et al. Enzyme catalysed Pictet-Spengler formation of chiral 1, 1'-disubstituted- and spiro-tetrahydroisoquinolines. Nat Commun, 2017, 8: 14883. DOI:10.1038/ncomms14883 |
| [26] | Zhao JX, Lichman BR, Ward JM, et al. One-pot chemoenzymatic synthesis of trolline and tetrahydroisoquinoline analogues. Chem Commun, 2018, 54(11): 1323-1326. |
| [27] | Lichman BR, Lamming ED, Pesnot T, et al. One-pot triangular chemoenzymatic cascades for the syntheses of chiral alkaloids from dopamine. Green Chem, 2015, 17(2): 852-855. |
| [28] | Wang Y, Tappertzhofen N, Méndez-Sánchez D, et al. Design and use of de novo cascades for the biosynthesis of new benzylisoquinoline alkaloids. Angew Chem Int Ed, 2019, 58(30): 10120-10125. |
| [29] | Ma YT, Liu ZN, Liu X, et al. Advances in production of plant isoquinoline alkaloids in heterologous microbes. China Biotechnol, 2019, 39(11): 123-131 (in Chinese). 马雅婷, 刘珍宁, 刘雪, 等. 微生物异源合成植物异喹啉生物碱的新进展. 中国生物工程杂志, 2019, 39(11): 123-131. |
| [30] | Cravens A, Payne J, Smolke CD. Synthetic biology strategies for microbial biosynthesis of plant natural products. Nat Commun, 2019, 10: 2142. DOI:10.1038/s41467-019-09848-w |
| [31] | Galanie S, Thodey K, Trenchard IJ, et al. Complete biosynthesis of opioids in yeast. Science, 2015, 349(6252): 1095-1100. |
| [32] | Li YR, Smolke CD. Engineering biosynthesis of the anticancer alkaloid noscapine in yeast. Nat Commun, 2016, 7: 12137. DOI:10.1038/ncomms12137 |
| [33] | Li YR, Li SJ, Thodey K, et al. Complete biosynthesis of noscapine and halogenated alkaloids in yeast. Proc Natl Acad Sci USA, 2018, 115(17): E3922-E3931. |
| [34] | DeLoache WC, Russ ZN, Narcross L, et al. An enzyme-coupled biosensor enables (S)-reticuline production in yeast from glucose. Nat Chem Biol, 2015, 11(7): 465-471. |
| [35] | Trenchard IJ, Siddiqui MS, Thodey K, et al. De novo production of the key branch point benzylisoquinoline alkaloid reticuline in yeast. Metab Eng, 2015, 31: 74-83. |
| [36] | Matsumura E, Nakagawa A, Tomabechi Y, et al. Microbial production of novel sulphated alkaloids for drug discovery. Sci Rep, 2018, 8: 7980. |
| [37] | Lechner H, Soriano P, Poschner R, et al. Library of norcoclaurine synthases and their immobilization for biocatalytic transformations. Biotechnol J, 2018, 13(3): 1700542. DOI:10.1002/biot.201700542 |
| [38] | Nishibe S, Tsukamoto H, Kinoshita H, et al. Alkaloids from embryo of the seed of Nelumbo nucifera. J Nat Prod, 1986, 49(3): 547-548. |
| [39] | Brown S, Clastre M, Courdavault V, et al. De novo production of the plant-derived alkaloid strictosidine in yeast. Proc Natl Acad Sci USA, 2015, 112(11): 3205-3210. |
| [40] | Ma XY, Panjikar S, Koepke J, et al. The structure of Rauvolfia serpentina strictosidine synthase is a novel six-bladed β-propeller fold in plant proteins. Plant Cell, 2006, 18(4): 907-920. |
| [41] | Eger E, Simon A, Sharma M, et al. Inverted binding of non-natural substrates in strictosidine synthase leads to a switch of stereochemical outcome in enzyme-catalyzed Pictet-Spengler reactions. J Am Chem Soc, 2020, 142(2): 792-800. |
| [42] | Treimer JF, Zenk MH. Purification and properties of strictosidine synthase, the key enzyme in indole alkaloid formation. Eur J Biochem, 1979, 101(1): 225-233. DOI:10.1111/j.1432-1033.1979.tb04235.x |
| [43] | McKnight TD, Roessner CA, Devagupta R, et al. Nucleotide sequence of a cDNA encoding the vacuolar protein strictosidine synthase from Catharanthus roseus. Nucleic Acids Res, 1990, 18(16): 4939. |
| [44] | de Waal A, Meijer AH, Verpoorte R. Strictosidine synthase from Catharanthus roseus: purification and characterization of multiple forms. Biochem J, 1995, 306(2): 571-580. |
| [45] | McCoy E, Galan MC, O'Connor SE. Substrate specificity of strictosidine synthase. Bioorg Med Chem Lett, 2006, 16(9): 2475-2478. DOI:10.1016/j.bmcl.2006.01.098 |
| [46] | Maresh JJ, Giddings LA, Friedrich A, et al. Strictosidine synthase: mechanism of a Pictet-Spengler catalyzing enzyme. J Am Chem Soc, 2008, 130(2): 710-723. DOI:10.1021/ja077190z |
| [47] | Pressnitz D, Fischereder EM, Pletz J, et al. Asymmetric synthesis of (R)-1-alkyl-substituted tetrahydro-β-carbolines catalyzed by strictosidine synthases. Angew Chem Int Ed, 2018, 57(33): 10683-10687. DOI:10.1002/anie.201803372 |
| [48] | Wu FR, Zhu HJ, Sun LL, et al. Scaffold tailoring by a newly detected Pictet-Spenglerase activity of strictosidine synthase: from the common tryptoline skeleton to the rare piperazino-indole framework. J Am Chem Soc, 2011, 134(3): 1498-1500. |
| [49] | Cai YR, Zhu HJ, Alperstein Z, et al. Strictosidine synthase triggered enantioselective synthesis of N-substituted (S)-3, 14, 18, 19-tetrahydroangustines as novel topoisomerase Ⅰ inhibitors. ACS Chem Biol, 2017, 12(12): 3086-3092. DOI:10.1021/acschembio.7b00740 |
| [50] | De-Eknamkul W, Ounaroon A, Tanahashi T, et al. Enzymatic condensation of dopamine and secologanin by cell-free extracts of Alangium lamarckii. Phytochemistry, 1997, 45(3): 477-484. |
| [51] | De-Eknamkul W, Suttipanta N, Kutchan TM. Purification and characterization of deacetylipecoside synthase from Alangium lamarckii Thw. Phytochemistry, 2000, 55(2): 177-181. DOI:10.1016/S0031-9422(00)00260-0 |
| [52] | Chen XC, Wang R, Mao J, et al. Activity assay of salsolinol synthase using high performance liquid chromatography-electrochemical detection. Chemistry, 2010, 73(10): 938-942 (in Chinese). 陈薛钗, 王睿, 毛健, 等. Salsolinol合成酶的高效液相色谱-电化学活性检测方法研究. 化学通报, 2010, 73(10): 938-942. |
| [53] | Deng YL, Maruyama W, Dostert P, et al. Determination of the (R)- and (S)-enantiomers of salsolinol and N-methylsalsolinol by use of a chiral high-performance liquid chromatographic column. J Chromatogr B Biomed Sci Appl, 1995, 670(1): 47-54. DOI:10.1016/0378-4347(95)00136-7 |
| [54] | Origitano T, Hannigan J, Collins MA. Rat brain salsolinol and blood-brain barrier. Brain Res, 1981, 224(2): 446-451. DOI:10.1016/0006-8993(81)90876-3 |
| [55] | Naoi M, Maruyama W, Dostert P, et al. A novel enzyme enantio-selectively synthesizes (R)salsolinol, a precursor of a dopaminergic neurotoxin, N-methyl(R)salsolinol. Neurosci Lett, 1996, 212(3): 183-186. DOI:10.1016/0304-3940(96)12807-X |
| [56] | Chen XC, Arshad A, Qing H, et al. Enzymatic condensation of dopamine and acetaldehyde: a salsolinol synthase from rat brain. Biologia, 2011, 66(6): 1183-1188. |
| [57] | Chen XC, Chen Y, Wu GS, et al. Existence and characterization of salsolinol synthase in neuronal cells and rat brain. Neurochem J, 2013, 7(3): 192-197. DOI:10.1134/S1819712413030045 |
| [58] | Chen XC. Purification and characterization of salsolinol synthetase, the Parkinson's disease related protein[D]. Beijing: Beijing Institute of Technology, 2011 (in Chinese). 陈薛钗.帕金森病相关蛋白Salsolinol合成酶的分离纯化及其酶学性质研究[D].北京: 北京理工大学, 2011. |
| [59] | Feng CC, Chen XC, Liu KF, et al. Cloning and expression of Salsolinol synthase. Sci Technol Rev, 2015, 33(8): 73-76 (in Chinese). 冯程程, 陈薛钗, 刘可夫, 等. Salsolinol合成酶的克隆和表达. 科技导报, 2015, 33(8): 73-76. |
| [60] | Yamakawa T, Kotake Y, Fujitani M, et al. Regional distribution of parkinsonism-preventing endogenous tetrahydroisoquinoline derivatives and an endogenous parkinsonism-preventing substance-synthesizing enzyme in monkey brain. Neurosci Lett, 1999, 276(1): 68-70. |
| [61] | Yamakawa T, Ohta S. Biosynthesis of a parkinsonism-preventing substance, 1-methyl-1, 2, 3, 4-tetrahydroisoquinoline, is inhibited by parkinsonism-inducing compounds in rat brain mitochondrial fraction. Neurosci Lett, 1999, 259(3): 157-160. DOI:10.1016/S0304-3940(98)00913-6 |
| [62] | Absi E, Parrado J, Ayala A, et al. Decrease of 1-methyl-1, 2, 3, 4-tetrahydroisoquinoline synthesizing enzyme activity in the brain areas of aged rat. Brain Res, 2002, 955(1/2): 161-163. |
| [63] | Brevet J, Borowski D, Tempé J. Identification of the region encoding opine synthesis and of a region involved in hairy root induction on the T-DNA of cucumber-type Ri plasmid. Mol Plant Microbe Interact, 1988, 1(2): 75-70. DOI:10.1094/MPMI-1-075 |
| [64] | Suzuki K, Tanaka N, Kamada H, et al. Mikimopine synthase (mis) gene on pRi1724. Gene, 2001, 263(1/2): 49-58. |
| [65] | Kovacova V, Zluvova J, Janousek B, et al. The evolutionary fate of the horizontally transferred agrobacterial mikimopine synthase gene in the genera Nicotiana and Linaria. PLoS One, 2014, 9(11): e113872. DOI:10.1371/journal.pone.0113872 |
| [66] | Xu Y, Jia JF, Zheng GC. A simple and efficient method for the detection of opine synthase activities in plant tissues. Hereditas (Beijing), 1987, 9(5): 41-43 (in Chinese). 许耀, 贾敬芬, 郑国錩. 植物组织中冠瘿碱合成酶活性检测的一种简便有效方法. 遗传, 1987, 9(5): 41-43. |
| [67] | Aroonsri A, Kitani S, Ikeda H, et al. Kitasetaline, a novel β-carboline alkaloid from Kitasatospora setae NBRC 14216T. J Biosci Bioeng, 2012, 114(1): 56-58. DOI:10.1016/j.jbiosc.2012.02.027 |
| [68] | Aroonsri A, Kitani S, Hashimoto J, et al. Pleiotropic control of secondary metabolism and morphological development by KsbC, a butyrolactone autoregulator receptor homologue in Kitasatospora setae. Appl Environ Microbiol, 2012, 78(22): 8015-8024. DOI:10.1128/AEM.02355-12 |
| [69] | Ueda S, Kitani S, Namba T, et al. Engineered production of kitasetalic acid, a new tetrahydro-β-carboline with the ability to suppress glucose-regulated protein synthesis. J Antibiot, 2018, 71(10): 854-861. |
| [70] | Ueda S, Ikeda H, Namba T, et al. Identification of biosynthetic genes for the β-carboline alkaloid kitasetaline and production of the fluorinated derivatives by heterologous expression. J Ind Microbiol Biotechnol, 2019, 46(5): 739-750. |
| [71] | Koketsu K, Watanabe K, Suda H, et al. Reconstruction of the saframycin core scaffold defines dual Pictet-Spengler mechanisms. Nat Chem Biol, 2010, 6(6): 408-410. DOI:10.1038/nchembio.365 |
| [72] | Tang MC, Tang GL. Biosynthetic progress of the tetrahydroisoquinoline antitumor antibiotics. Chin J Org Chem, 2012, 32(9): 1568-1576 (in Chinese). 唐满成, 唐功利. 抗肿瘤活性四氢异喹啉类抗生素的生物合成研究进展. 有机化学, 2012, 32(9): 1568-1576. |
| [73] | Tian XP, Tang SK, Dong JD, et al. Marinactinospora thermotolerans gen. nov., sp. nov., a marine actinomycete isolated from a sediment in the northern South China Sea. Int J Syst Evol Micr, 2009, 59(5): 948-952. |
| [74] | Huang HB, Yao YL, He ZX, et al. Antimalarial β-carboline and indolactam alkaloids from Marinactinospora thermotolerans, a deep sea isolate. J Nat Prod, 2011, 74(10): 2122-2127. DOI:10.1021/np200399t |
| [75] | Chen Q, Ji CT, Song YX, et al. Discovery of McbB, an enzyme catalyzing the β-carboline skeleton construction in the marinacarboline biosynthetic pathway. Angew Chem Int Ed Engl, 2013, 52(38): 9980-9984. |
| [76] | Mori T, Hoshino S, Sahashi S, et al. Structural basis for β-carboline alkaloid production by the microbial homodimeric enzyme McbB. Chem Biol, 2015, 22(7): 898-906. |
| [77] | Wang H, Zhang HD, Mi YL, et al. Expression, crystallization and preliminary X-ray analysis of McbB, a multifunctional enzyme involved in β-carboline skeleton biosynthesis. Acta Crystallogr F Struct Biol Commun, 2014, 70(10): 1402-1405. DOI:10.1107/S2053230X14018743 |
| [78] | Chen Q, Zhang SF, Xie YC. Characterization of a new microbial Pictet-Spenglerase NscbB affording the β-carboline skeletons from Nocardiopsis synnemataformans DSM 44143. J Biotechnol, 2018, 281: 137-143. |
| [79] | Yassin AF, Rainey FA, Burghardt J, et al. Description of Nocardiopsis synnemataformans sp. nov., elevation of Nocardiopsis alba subsp. prasina to Nocardiopsis prasina comb. nov., and designation of Nocardiopsis antarctica and Nocardiopsis alborubida as later subjective synonyms of Nocardiopsis dassonvillei. Int J Syst Bacteriol, 1997, 47(4): 983-988. DOI:10.1099/00207713-47-4-983 |
| [80] | Xu F, Kong DK, He XY, et al. Characterization of streptonigrin biosynthesis reveals a cryptic carboxyl methylation and an unusual oxidative cleavage of a N-C bond. J Am Chem Soc, 2013, 135(5): 1739-1748. DOI:10.1021/ja3069243 |
| [81] | Wang XZ, Kong DK, Huang TT, et al. StnK2 catalysing a Pictet-Spengler reaction involved in the biosynthesis of the antitumor reagent streptonigrin. Org Biomol Chem, 2018, 16(47): 9124-9128. DOI:10.1039/C8OB02710B |
| [82] | Yan W, Ge HM, Wang G, et al. Pictet-Spengler reaction-based biosynthetic machinery in fungi. Proc Natl Acad Sci USA, 2014, 111(51): 18138-18143. |
| [83] | Ge HM, Zhu CH, Shi DH, et al. Hopeahainol A: an acetylcholinesterase inhibitor from Hopea hainanensis. Chemistry, 2008, 14(1): 376-381. DOI:10.1002/chem.200700960 |
| [84] | Becher PG, Beuchat J, Gademann K, et al. Nostocarboline: isolation and synthesis of a new cholinesterase inhibitor from Nostoc 78-12A. J Nat Prod, 2005, 68(12): 1793-1795. |
| [85] | Portmann C, Prestinari C, Myers T, et al. Directed biosynthesis of phytotoxic alkaloids in the cyanobacterium Nostoc 78-12A. ChemBioChem, 2009, 10(5): 889-895. DOI:10.1002/cbic.200800837 |
