

南京农业大学 信息科学技术学院,江苏 南京 210095
收稿日期:2018-09-30;接收日期:2018-10-29;网络出版时间:2018-12-08
基金项目:国家重点研发计划(No. 2017YFD0800204),中央高校基本科研业务费专项资金(No. KYZ201600175)资助
摘要:文中提出了一种简单有效的蛋白质亚细胞区间定位预测方法,为进一步了解蛋白质的功能和性质提供理论基础。运用稀疏编码,结合氨基酸组成信息提取蛋白质序列特征,基于不同字典大小对得到的特征进行多层次池化整合,并送入支持向量机进行分类。经Jackknife检验,在数据集ZD98、CH317和Gram1253上的预测成功率分别达到95.9%、93.4%和94.7%。实验证明基于多层次稀疏编码的分类预测算法能显著提高蛋白质亚细胞区间定位的预测精度。
关键词:稀疏编码氨基酸组成多层次池化支持向量机亚细胞区间定位
Prediction of protein subcellular localization based on multilayer sparse coding
Xingjian Chen, Xuejiao Hu, Wei Xue
School of Information Science and Technology, Nanjing Agricultural University, Nanjing 210095, Jiangsu, China
Received: September 30, 2018; Accepted: October 29, 2018; Published: December 8, 2018
Supported by: National Key Technology R & D Program of China (No. 2017YFD0800204), the Fundamental Research Funds for the Central Universities (No. KYZ201600175)
Corresponding author: Wei Xue. Tel: +86-25-84396350; E-mail: xwsky@njau.edu.cn.
Abstract: In order to provide a theoretical basis for better understanding the function and properties of proteins, we proposed a simple and effective feature extraction method for protein sequences to determine the subcellular localization of proteins. First, we introduced sparse coding combined with the information of amino acid composition to extract the feature values of protein sequences. Then the multilayer pooling integration was performed according to different sizes of dictionaries. Finally, the extracted feature values were sent into the support vector machine to test the effectiveness of our model. The success rates in data set ZD98, CH317 and Gram1253 were 95.9%, 93.4% and 94.7%, respectively as verified by the Jackknife test. Experiments showed that our method based on multilayer sparse coding can remarkably improve the accuracy of the prediction of protein subcellular localization.
Keywords: sparse codingamino acid compositionmultilayer poolingsupport vector machinesubcellular localization prediction
蛋白质作为生物体的基本组成物质,在生命活动中发挥着重要作用。蛋白质的功能与其亚细胞区间密切相关,不同蛋白质只有处于特定亚细胞区间才能发挥其功能,因而通过已有方法预测确定某种蛋白质所处的亚细胞区间,对明确蛋白质的功能和性质、认识蛋白质间的相互作用具有重要意义[1]。随着蛋白序列数据的不断增加,使用传统人工实验手段获取蛋白质亚细胞区间位置已远不能满足科研需要,这促使了机器学习在蛋白质亚细胞定位预测中的发展。
通过对目前研究现状的分析,可将近年来使用机器学习方法对蛋白质亚细胞区间进行预测的研究方向大致分为两类,分别为序列特征提取和分类模型构建[2]。目前用于蛋白质序列的特征提取算法主要有氨基酸组成(Amino acid composition,AAC)、伪氨基酸组成(Pseudo amino acid composition,PseAAC)、基因本体(Gene ontology, GO)、位置特异性得分矩阵(Position specific scoring matrix,PSSM)和基于不同特征的融合等。如Zhou等基于Mahalanobis距离提取蛋白质序列的组分信息,使用斜变判别函数对蛋白质亚细胞区间进行预测,在Jackknife检验下ZD98数据集上的准确率约为72.5%[3];Wan等提出了GOASVM算法,基于GO注释信息与蛋白非相邻区域的同源性来表示蛋白质序列,取得了较好的效果[4];Chen等采用混合增量的方式对蛋白质序列的N端、C端以及疏水性3种特征进行融合,在ZD98和CH317数据集上的成功率分别为90.8%和82.7%[5];Zhao等提出蛋白质序列的词袋特征,将词袋模型与基于伪氨基酸组成的特征提取算法相结合,获得了较高的准确率[6]。同时,在分类预测模型方面,国内外研究者也开展了大量工作,如Wan等通过GO数据库的注释信息,提出自适应决策支持向量机,实现了对多功能膜蛋白序列的区间预测[7];Ali等提取蛋白质序列的伪氨基酸特征,采用区间投票、最邻近算法和概率神经网络等多种分类器进行对比预测,取得了较好的结果[8];除此之外还有基于逻辑回归、贝叶斯集成和长短期记忆网络等多种分类模型的预测方法[9-11]。
总结前人研究成果可发现,能否准确描述蛋白质序列特征直接影响了最终分类器的预测效果。由于蛋白质序列中包含的信息量较大,且分属同一亚细胞区间的序列长度不等,序列特征分布不均,导致单一使用传统蛋白质序列特征提取算法的分类结果不佳。而对于一些较为复杂的特征融合及其改进算法,虽然取得了较高的准确率,但特征提取过程复杂,且最终得到的特征向量维数较大,造成分类器的时间和空间复杂度过高。因此,本研究结合氨基酸组分信息,提出一种基于多层次稀疏编码的蛋白质序列特征提取算法,该算法能够基于简单的AAC方法对蛋白质序列进行稀疏表示,进而提取序列底层特征;根据不同字典大小对特征进行多层次池化整合,能有效增加序列特征的区分性;将得到的特征向量经主成分分析(Principal component analysis, PCA)降维,能在选取有效特征的同时降低算法的计算量。最后将得到的特征向量送入分类器进行分类。实验结果表明,本方法不仅能简化特征提取过程,降低分类器的时间及空间复杂度,也能更加全面地反映序列特征,提高分类性能。
1 材料与方法1.1 数据集为了对本文算法进行客观评价,方便与同类算法进行对比,采用近年来相关领域中使用最多且国际公认有效的ZD98和CH317作为实验基准数据集[12-19],其中ZD98由Zhou和Doctor[3]构建,共有98条蛋白质序列,分为4个亚细胞定位类别,分别是细胞质蛋白(Cytoplasmic proteins,Cy) 43条、线粒体蛋白(Mitochondrial proteins, Mi) 13条、细胞膜蛋白(Membrane proteins, Me) 30条和其他类蛋白(Other) 12条。CH317是由Chen和Li[5]构建,分为6个亚细胞定位类别,共有317条蛋白质序列,分别是分泌蛋白(Secreted proteins,Se) 17条、细胞核蛋白(Nuclear proteins,Nu) 52条、细胞质蛋白(Cytoplasmic proteins,Cy) 112条、内质网蛋白(Endoplasmic reticulum proteins,En) 47条、膜蛋白(Membrane proteins,Me) 55条和线粒体蛋白(Mitochondrial proteins,Mi) 34条。
考虑到上述数据集构建时间较长,参考Wang等的方法对ZD98和CL317数据集进行了更新[12],删除了部分重复及错误序列,其具体方法不再复述。经处理后ZD98数据集剩余96条蛋白质序列,CH317数据集剩余314条蛋白质序列。此外,为了对算法进行进一步评估,除了上述两个数据集外,本研究还采用了Xue等按照同样标准构建的Gram1253数据集进行测试[20]。Gram1253数据集共包含1 253条蛋白质序列,分为Me、Cy、Nu、Se及细胞周质(Periplasm,Pe)等5个亚细胞定位类别。以上3种数据集中的所有蛋白质序列均来自最新版本的UniProt数据库(Release 2018_08),其具体区间分布如表 1所示。
表 1 三种数据集中不同区间的蛋白质序列条数Table 1 Numbers of protein sequences in different class of 3 datasets
| Dataset | Number of sequences in each class | Total | ||||
| ZD98 | Cy | Me | Mi | Other | 96 | |
| 43 | 28 | 13 | 12 | |||
| CL317 | Cy | En | Me | Mi Nu | Se | 314 |
| 110 | 47 | 55 | 34 51 | 17 | ||
| Gram1253 | Me | Cy | Pe | Se Nu | 1 253 | |
| 166 | 443 | 423 | 199 22 | |||
表选项
1.2 序列特征编码将稀疏编码引入蛋白质亚细胞区间定位预测中,目的是在每条蛋白序列与相应的数值向量间建立一种能够更为准确表达此条蛋白序列特征的映射关系。基于多层次稀疏编码的特征提取算法主要包括局部特征提取、稀疏编码和多层次池化等3个步骤。首先对蛋白质序列进行分割处理得到若干个序列单词,使用传统蛋白质特征提取算法对序列单词进行特征编码得到特征单词,然后选取部分特征单词作为局部特征块学习字典,用字典对原始序列特征进行稀疏表示。采用平均池化的方法对稀疏矩阵降维,将基于不同字典大小得到的特征向量进行组合,即为蛋白质序列的最终特征表示。其提取流程如图 1所示。
 |
| 图 1 稀疏编码特征提取流程 Fig. 1 The feature extraction of sparse coding. The solid black line represents the progress of the training set, and the dotted black line represents the flow of the test set |
| 图选项 |
1.2.1 局部特征提取在进行稀疏编码过程之前,首先需要提取蛋白质序列的局部特征作为特征块,组成训练样本构造字典。由于每条蛋白质序列长度不等,其主要特征可能分布在序列的不同位置,因此参考Zhao等的方法[6]采用滑动窗口分割法对原始蛋白质序列进行切分得到序列单词。滑动窗口分割法即按照一定长度对每条蛋白质序列进行切片,通过设定窗口大小和滑动间距得到若干个序列单词,经特征提取后得到特征单词集合形成构建字典的基础。这种方法能完整地保留蛋白质序列的全部信息。本研究取滑动间隔为1,滑动窗口大小决定序列单词长度,需满足以下条件:
 | (1) |
切分后每条蛋白质序列被表示为若干个长度相等的序列单词,运用已有的蛋白质序列特征提取算法统计序列单词的组分信息,即可得到相应的特征单词。Nakashima和Nishikawa[21]最早将氨基酸组成和蛋白质亚细胞区间定位预测联系起来,提出AAC编码方式,统计每个氨基酸残基在蛋白质序列中出现的频率,其定义如下:
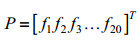 | (2) |
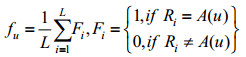 | (3) |
通过AAC算法计算蛋白质序列P的序列单词特征,将每条蛋白质序列的所有特征单词进行组合,则每条序列都被表示为一个片段特征矩阵,如公式(4)所示:
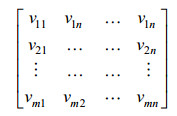 | (4) |
1.2.2 稀疏编码得到由蛋白质序列的局部特征组成的训练样本之后,下一步即是对这些训练样本进行稀疏编码。稀疏编码是一种无监督的机器学习算法,通过在高维数据中寻找一组超完备的基向量来对样本进行稀疏表示,主要分为字典学习和稀疏重构两个过程。其公式表示如下:
 | (5) |
字典学习是指通过学习局部特征,从中找到最能够为有效重构训练样本的元素组合。在字典学习过程中,为了确保能够按照约束条件自适应地训练出超完备字典,一般要求字典中的原子个数较大,即若字典D的大小为K*n,则需使K > n,这样得到的字典更容易学习到训练样本深层次的特征。同时训练样本的稀疏系数ui需满足以下条件:
 | (6) |

本研究采用K-SVD算法训练字典。K-SVD算法是由Aharon等提出的一种基于K-means算法扩展而来的字典学习算法[22],其实质是迭代交替学习字典原子并优化其相应的稀疏系数。该算法要经过K次迭代,每次迭代时都需要对误差项进行奇异值分解,采用逐列更新的方式对字典进行优化,每次只更新其中的一个原子和其对应的稀疏系数,选择使重构误差最小的分解项作为新的元素值,经过不断迭代得到最优化的解。K-SVD算法主要分为以下几个步骤:(1)随机初始化字典D,设置迭代终止条件;(2)固定字典D,求解稀疏矩阵U;(3)固定稀疏矩阵U,求解字典D;(4)交替执行步骤(2)和(3),直至迭代结束。
得到字典后,通常使用正交匹配追踪(Orthogonal matching pursuit,OMP)算法,求得原始样本的稀疏矩阵。OMP算法的核心思想是在每次迭代过程中使用最小二乘法对原始样本进行稀疏逼近,选择字典中最匹配的基元对其进行稀疏重构,求出残差并继续选择下一个最匹配的基元。这种更新方式能保证在下一次迭代过程中不会重复选择相同基元,在一定程度上加快了算法的收敛速度,克服了传统匹配追踪(Matching pursuit,MP)算法容易陷入局部最优解的问题。
使用OMP算法对原始蛋白质序列的片段特征矩阵进行稀疏表示,则每一条的蛋白质序列可被表示为一个m*K稀疏矩阵Z,m即为每一条蛋白质序列切割得到的片段条数,K为字典的原子数目。此过程即为蛋白质序列的稀疏编码。
1.2.3 多层次池化经稀疏编码后所得到的稀疏矩阵维度较高,如果直接展开进行串接表示数据量过大,训练分类器时的内存和时间消耗代价过高。所以需要对特征矩阵进行降维,通常使用池化方法。池化是指把特征向量集映射为单个向量的过程,对不同位置的特征进行聚合统计,能提取有效特征,减少计算量。常用的池化算法有最大池化(Max-pooling)[23]和平均池化(Mean-pooling)[24]。Max-pooling即对邻域内的特征点取最大值,能更多地保留矩阵的边缘信息。而Mean-pooling则是对邻域内特征点求平均值,能更多地保留矩阵的背景信息。考虑到序列数据的特殊性,本研究选择Mean-pooling作为最终的池化方法。公式表示如下:
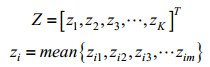 | (7) |
这里根据不同字典大小对特征矩阵进行多层次池化,可以分别帮助提取蛋白质序列的整体和局部特征。将几种池化的结果融合在一起作为蛋白质序列的最终特征,可使其表述更具有代表性。具体方法如下:在稀疏编码的字典学习过程中,将字典原子数目分别设置为K1、K2和K3,经K-SVD算法后得到3种不同层次大小的字典,然后针对不同字典使用OMP算法对原始蛋白质序列的片段特征矩阵进行稀疏表示,将得到的稀疏矩阵分别进行平均池化得到不同层次的特征向量。将每个池化块内的特征向量进行串接融合在一起,得到一个K1+K2+K3维的向量作为最终特征向量。K值分别设置为30、50和70,最后生成一个150维的向量,经PCA进行特征选择后送入分类器完成分类。
1.3 支持向量机为了方便与其他算法进行对比,选择支持向量机(Support vector machine, SVM)建立分类模型。SVM是Vapnik领导的AT & T Bell实验室在1995年提出的一种基于统计学习理论的分类方法,通过核函数将输入样本从原空间非线性映射到高维特征空间,利用线性方法解决非线性问题,在高维特征空间中构造最优分类超平面,在解决小样本、非线性及高维模式识别问题中表现出强大的泛化能力[25]。蛋白质序列经特征编码后,使用LIBSVM通用软件包,基于一对一算法(One-versus-one)构造SVM多类分类器,在训练阶段为任意两类样本设计一个SVM,则k个类别的数据集就需要设计k(k–1)/2个SVM。当对一个未知样本进行分类时,最后取得票最多的类别为该未知样本的类别。运用SVM进行蛋白质亚细胞区间定位预测的流程图如图 2所示。
 |
| 图 2 基于SVM的亚细胞定位预测流程 Fig. 2 Subcellular localization prediction process based on SVM. In the training process of SVM, it is necessary to manually set parameters according to different kinds of kernel functions, that is the parameter selection |
| 图选项 |
将数据集中的样本分为训练样本和预测样本,先送入训练样本的特征向量,设定输出为相应的亚细胞定位yi,训练SVM,确定模型参数,再送入预测样本的特征向量,SVM分类器会给出一个预测结果,用xi表示,若xi = yi,则预测正确,若xi≠yi,则预测错误,最后统计整个数据集的预测准确率作为蛋白质亚细胞区间定位的评价指标。
2 结果与分析2.1 测试方法为了验证方法的有效性,采用Jackknife进行假设检验。Jackknife是蛋白质亚细胞定位预测研究中公认且使用最多的一种测试方法[12-20],每次仅用一条序列作为测试集进行验证,其余全部序列作为训练集送入分类器进行训练,以此类推直至所有序列均预测完毕,是一种客观有效的假设检验方法[26]。为了便于比较实验结果,同时对预测方法进行有效评估,引入敏感性(Sensitivity, Se)、特异性(Specificity, Sp)和相关系数(Matthews correlation coefficient, MCC)等3个评价指标,并统计总的预测准确率(Overall accuracy, OA),定义如下[27]:
 | (8) |
 | (9) |
 | (10) |
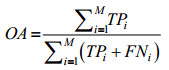 | (11) |
2.2 参数选择在使用PCA对最终的特征向量进行选择时,维数D的设置对于整个算法的准确度存在一定影响。选取的维数越多,包含的特征就越多,但可能造成分类器的训练时间过长;维数越小,则越有可能丢失一些真正有意义的特征,影响分类效果。因此需要通过实验寻求一个最优的D值。图 3显示了数据集ZD98、CH317和Gram1253在PCA进行特征选择过程中分别取不同的D值所对应的预测准确率。在特征向量的维数较低时,3个数据集的预测准确率也相对较低,在维数高于某一确定值时,预测准确率也随之降低。在维数取60到70之间时,在ZD98、CH317和Gram1253数据集上的预测准确率均达到最大且趋于稳定,当前的D值即为最优值。本研究使用的3种数据集的最优D值分别为60、65和65。
 |
| 图 3 基于不同维度的预测准确率 Fig. 3 The prediction accuracies based on different dimensions. The abscissa represents the dimensions of feature vectors and the ordinate represents the total accuracy |
| 图选项 |
2.3 结果分析将本方法在ZD98、CH317和Gram1253数据集上采用Jackknife进行实验的预测结果列于表 1中,为了进一步说明本文方法的有效性,表中分别列出了3个数据集在各个亚细胞区间进行预测得到的不同实验结果。
表 1 数据集实验结果Table 1 The experimental results of data sets
| Dataset | Jackknife test (%) | ||||
| Location | Se (%) | Sp (%) | MCC (%) | OA (%) | |
| ZD98 | Cy | 100 | 95.6 | 95.9 | 95.9 |
| Me | 96.7 | 96.7 | 95.2 | ||
| Mi | 84.6 | 91.7 | 86.4 | ||
| Other | 91.7 | 91.7 | 90.5 | ||
| CL317 | Cy | 94.6 | 93.8 | 90.9 | 93.4 |
| Me | 92.7 | 92.7 | 91.1 | ||
| Mi | 97.1 | 97.1 | 96.7 | ||
| Se | 76.5 | 92.9 | 83.4 | ||
| Nu | 92.3 | 90.1 | 89.6 | ||
| En | 95.7 | 90.0 | 91.5 | ||
| Gram1253 | Cy | 99.3 | 97.1 | 96.7 | 94.7 |
| Me | 94.0 | 90.7 | 90.2 | ||
| Pe | 98.9 | 93.2 | 93.4 | ||
| Se | 85.6 | 98.6 | 80.5 | ||
| Nu | 86.4 | 83.2 | 85.7 | ||
表选项
由表 1可知,本方法在3个数据集上均获得了较好的实验结果,总的准确率分别达到了95.9%、93.4%和94.7%,实验证明本方法能有效增加蛋白质亚细胞区间定位预测的准确率。同时为了方便进行对比,将部分同领域内基于蛋白质序列特征提取的改进算法得到的实验结果也一并列出。
从表 2可以看出,在ZD98数据集上本文算法相比DCC、OF和DE等复杂的特征融合算法在总体预测精度上最大提升了约7个百分点,在Cyto这一亚细胞类上的预测准确率达到了100%,预测全部正确,且整体预测准确率方面也均优于其他方法。将本方法与BOW、GA和OA等改进算法的实验结果进行对比,在相同数据集上的准确率也都提高了约2到5个百分点,实验表明本文算法较基于传统蛋白质序列特征提取的改进算法仍具有显著优势。通过表 3的比较可以看出,在CH317数据集上,本文算法在Mito这一亚细胞类上的预测准确率最高达到了97.1%,相比其他算法最大提升了约14.7个百分点,在Nucl这一亚细胞类上的准确率最高也提升了12.3个百分点,这一实验结果也充分说明了本文算法对少数类别序列进行特征提取的有效性,使得序列底层特征更加具有区分性。对比BOW、IAC和CF等改进算法,在总预测准确率上均提升了2–4个百分点,进一步表明通过多层次池化分别提取序列的整体和局部信息,能有效提高蛋白质亚细胞定位预测精度。对于较大数据集Gram1453而言,本文引用了文献[20]中基于不同蛋白质序列特征提取算法的实验结果进行对比,如AAC、Dipe和PseAAC等,同时也基于PSSM特征进行了相关的对比实验,如PSSM_SVM等,表 4结果表明,本方法在各个区间类别的预测率上均有一定提高,且相较于传统算法,如PSSM_SVM和BLAST_KNN等,本文方法不需要依靠复杂工具实现,在算法的可移植性上也具有明显优势。
表 2 数据集预测结果比较Table 2 Comparison of the accuracy of ZD98 data set
| Methods | Jackknife test (%) | References | ||||
| Cyto | Memb | Mito | Other | OA (%) | ||
| DCC_SVM | 93.0 | 86.7 | 92.3 | 75.0 | 88.9 | [28] |
| OF_SVM | 97.7 | 86.3 | 92.3 | 66.7 | 90.8 | [16] |
| DE_SVM | 95.4 | 93.3 | 76.9 | 83.3 | 90.8 | [17] |
| BOW_SVM | 97.7 | 92.9 | 76.9 | 83.3 | 91.7 | [6] |
| GA_SVM | 95.4 | 90.0 | 92.3 | 83.3 | 91.8 | [19] |
| OA_SVM | 95.3 | 88.9 | 97.4 | 91.7 | 93.2 | [13] |
| This study | 100 | 96.7 | 84.6 | 91.7 | 95.9 | – |
表选项
表 3 CH317数据集预测结果比较Table 3 Comparison of the accuracy of CH317 data set
| Methods | Jackknife test (%) | References | ||||||
| Cyto | Memb | Mito | Secr | Nucl | Endo | OA (%) | ||
| DCC_SVM | 91.1 | 92.7 | 82.4 | 76.5 | 80.0 | 93.6 | 88.3 | [28] |
| GA_SVM | 92.9 | 89.1 | 82.4 | 76.5 | 84.6 | 93.6 | 89.0 | [19] |
| BOW_SVM | 94.6 | 87.3 | 82.4 | 82.4 | 84.3 | 91.5 | 89.2 | [6] |
| IAC_SVM | 96.4 | 94.5 | 82.4 | 76.5 | 80.8 | 93.6 | 90.5 | [18] |
| EI_SVM | 94.6 | 95.7 | 92.7 | 82.4 | 90.4 | 70.6 | 91.1 | [14] |
| CF_SVM | 96.4 | 90.9 | 92.3 | 95.7 | 82.4 | 64.7 | 91.5 | [29] |
| This study | 94.6 | 92.7 | 97.1 | 76.5 | 92.3 | 95.7 | 93.4 | – |
表选项
表 4 Gram1253数据集预测结果比较Table 4 Comparison of the accuracy of Gram1253 data set
| Methods | Jackknife test (%) | |||||
| Cyto | Memb | Peri | Secr | Nucl | OA (%) | |
| AAC_SVM | 98.4 | 94.6 | 97.6 | 70.0 | 40.9 | 92.1 |
| Dipe_SVM | 97.5 | 93.4 | 96.7 | 70.0 | 40.9 | 91.3 |
| PseAAC_SVM | 98.4 | 94.6 | 97.6 | 70.9 | 59.1 | 92.6 |
| PSSM_SVM | 98.4 | 93.4 | 95.6 | 77.2 | 65.3 | 92.8 |
| BLAST_KNN | 98.6 | 94.6 | 98.3 | 68.8 | 86.4 | 93.1 |
| Our | 99.3 | 94.0 | 98.9 | 85.6 | 86.4 | 94.7 |
表选项
与传统蛋白质序列特征提取及其改进方法相比,本文算法时间及空间复杂度低,在较简单的AAC特征下也能取得较好的效果,且通过平均池化提取特征序列特征矩阵的背景信息,将不同层次特征进行整合后经PCA降维,得到一种低维向量的形式反映序列特征的分布规律,能显著提高大数据处理的效率。
3 讨论蛋白质亚细胞定位预测一直是国内外生物信息学专家研究的热点方向。本研究在传统蛋白质序列特征提取算法AAC的基础上,提出了一种基于多层次稀疏编码的蛋白质序列特征提取算法对序列特征进行优化整合。相比其他算法,本方法提取过程简单,不需要经过复杂的特征融合步骤也能得到较高的预测准确率,且最后使用PCA对特征向量进行降维,在提高准确率的同时也降低了分类器的时间及空间复杂度。算法的主要流程包括:首先使用滑动窗口分割法对蛋白质序列进行切分提取序列单词,结合传统蛋白质特征提取算法对序列单词进行特征编码;采用K-SVD算法对序列单词特征进行字典学习,再通过OMP算法对序列特征矩阵进行稀疏表示;基于不同字典大小对特征矩阵进行多层次平均池化,分别帮助提取稀疏矩阵的整体信息和局部信息;使用SVM多类分类器对蛋白的亚细胞区间位置进行分类预测。实验表明,本文算法能在绝大部分亚细胞区间的预测成功率上获得较好的效果,对提升传统蛋白质序列特征提取算法的特征表达能力方面具有重要指导意义,是一种较为有效的蛋白质亚细胞区间预测方法。算法的源代码和所用数据集均可从https://github.com/Multisc/Multi_sc_subloc/tree/master获取。
参考文献
| [1] | Xu YY, Yang F, Shen HB. Incorporating organelle correlations into semi-supervised learning for protein subcellular localization prediction.Bioinformatics, 2016, 32(14): 2184–2192.DOI: 10.1093/bioinformatics/btw219 |
| [2] | Wei L, Ding Y, Su R, et al. Prediction of human protein subcellular localization using deep learning.Journal of Parallel & Distributed Computing, 2018, 117: 212–217. |
| [3] | Zhou GP, Doctor K. Subcellular location prediction of apoptosis proteins.Proteins, 2003, 50(1): 44–48. |
| [4] | Wan SB, Mak MW, Kung SY. GOASVM: a subcellular location predictor by incorporating term-frequency gene ontology into the general form of Chou's pseudo-amino acid composition.J Theor Biol, 2013, 323: 40–48.DOI: 10.1016/j.jtbi.2013.01.012 |
| [5] | Chen YL, Li QZ. Prediction of the subcellular location of apoptosis proteins.J Theor Biol, 2007, 245(4): 775–783.DOI: 10.1016/j.jtbi.2006.11.010 |
| [6] | Zhao N, Zhang L, Xue W, et al. Application of bag of words model in the prediction of protein subcellular location.J Food Sci Biotechnol, 2017, 36(3): 296–301.(in Chinese). 赵南, 张梁, 薛卫, 等. 词袋模型在蛋白质亚细胞定位预测中的应用.食品与生物技术学报, 2017, 36(3): 296-301.DOI:10.3969/j.issn.1673-1689.2017.03.011 |
| [7] | Wan SB, Mak MW, Kung SY. Mem-ADSVM: a two-layer multi-label predictor for identifying multi-functional types of membrane proteins.J Theor Biol, 2016, 398: 32–42.DOI: 10.1016/j.jtbi.2016.03.013 |
| [8] | Ali F, Hayat M. Classification of membrane protein types using Voting feature interval in combination with Chou's pseudo amino acid composition.J Theor Biol, 2015, 384: 78–83.DOI: 10.1016/j.jtbi.2015.07.034 |
| [9] | Wan SB, Mak MW, Kung SY. mPLR-Loc: an adaptive decision multi-label classifier based on penalized logistic regression for protein subcellular localization prediction.Anal Biochem, 2015, 473: 14–27.DOI: 10.1016/j.ab.2014.10.014 |
| [10] | Sáez-Atienzar S, Martínez-Gómez J, Alonso-Barba JI, et al. Automatic quantification of the subcellular localization of chimeric GFP protein supported by a two-level Naive Bayes classifier.Expert Syst Appl, 2015, 42(3): 1531–1537.DOI: 10.1016/j.eswa.2014.09.052 |
| [11] | S?nderby SK, S?nderby CK, Nielsen H, et al. Convolutional LSTM networks for subcellular localization of proteins//2nd International Conference on Algorithms for Computational Biology.Mexico City, Mexico: Springer, 2015: 68–80. |
| [12] | Wang X, Li H, Zhang QW, et al. Predicting subcellular localization of apoptosis proteins combining go features of homologous proteins and distance weighted KNN classifier.BioMed Res Int, 2016, 2016: 1793272. |
| [13] | Zhang SL, Duan X. Prediction of protein subcellular localization with oversampling approach and Chou's general PseAAC.J Theor Biol, 2018, 437: 239–250.DOI: 10.1016/j.jtbi.2017.10.030 |
| [14] | Xiang QL, Bo L, Li XH, et al. Subcellular localization prediction of apoptosis proteins based on evolutionary information and support vector machine.Artif Intell Med, 2017, 78: 41–46.DOI: 10.1016/j.artmed.2017.05.007 |
| [15] | Dai Q, Ma S, Hai YB, et al. A segmentation based model for subcellular location prediction of apoptosis protein.Chemom Intell Lab Syst, 2016, 158: 146–154.DOI: 10.1016/j.chemolab.2016.09.005 |
| [16] | Zhang SL, Jin J. Prediction of protein subcellular localization by using λ-order factor and principal component analysis.Lett Org Chem, 2017, 14(9): 717–724. |
| [17] | Liang YY, Zhang SL. Prediction of apoptosis protein's subcellular localization by fusing two different descriptors based on evolutionary information.Acta Biotheor, 2018, 66(1): 61–78.DOI: 10.1007/s10441-018-9319-x |
| [18] | Zhang SL, Liang YY. Predicting apoptosis protein subcellular localization by integrating auto-cross correlation and PSSM into Chou's PseAAC.J Theor Biol, 2018, 457: 163–169.DOI: 10.1016/j.jtbi.2018.08.042 |
| [19] | Liang YY, Liu SY, Zhang SL. Geary autocorrelation and DCCA coefficient: application to predict apoptosis protein subcellular localization via PSSM.Phys A, 2017, 467: 296–306.DOI: 10.1016/j.physa.2016.10.038 |
| [20] | Xue W, Wang XF, Zhao N, et al. Prediction of protein subcellular locations by ensemble of improved K-nearest neighbor.Chin J Biotech, 2017, 33(4): 683–691.(in Chinese). 薛卫, 王雄飞, 赵南, 等. 集成改进KNN算法预测蛋白质亚细胞定位.生物工程学报, 2017, 33(4): 683-691. |
| [21] | Nakashima H, Nishikawa K. Discrimination of intracellular and extracellular proteins using amino acid composition and residue-pair frequencies.J Mol Biol, 1994, 238(1): 54–61. |
| [22] | Aharon M, Elad M, Bruckstein A. rmK-SVD: an algorithm for designing overcomplete dictionaries for sparse representation.IEEE Trans Signal Process, 2006, 54(11): 4311–4322.DOI: 10.1109/TSP.2006.881199 |
| [23] | Liu YH, Cheng JY, Ma YM, et al. Protein secondary structure prediction based on two dimensional deep convolutional neural networks//2017 3rd IEEE International Conference on Computer and Communications.Chengdu, China: IEEE, 2017: 1995–1999. |
| [24] | Chen YH. Long sequence feature extraction based on deep learning neural network for protein secondary structure prediction//2017 IEEE 3rd Information Technology and Mechatronics Engineering Conference.Chongqing, China: IEEE, 2017: 843–847. |
| [25] | Silva MFM, Leijoto LF, Nobre CN. Algorithms analysis in adjusting the SVM parameters: an approach in the prediction of protein function.Appl Artif Intell, 2017, 31(4): 316–331.DOI: 10.1080/08839514.2017.1317207 |
| [26] | Ding H, Liang ZY, Guo FB, et al. Predicting bacteriophage proteins located in host cell with feature selection technique.Comput Biol Med, 2016, 71: 156–161.DOI: 10.1016/j.compbiomed.2016.02.012 |
| [27] | Xu YY, Yao LX, Shen HB. Bioimage-based protein subcellular location prediction: a comprehensive review.Front Comput Sci, 2018, 12(1): 26–39.DOI: 10.1007/s11704-016-6309-5 |
| [28] | Liang YY, Liu SY, Zhang SL. Detrended cross-correlation coefficient: application to predict apoptosis protein subcellular localization.Math Biosci, 2016, 282: 61–67.DOI: 10.1016/j.mbs.2016.09.019 |
| [29] | Chen HW, Chen X, Hu QM, et al. Predicting protein subcellular location based on a novel sequence numerical model.J Comput Theor Nanosci, 2015, 12(1): 82–87.DOI: 10.1166/jctn.2015.3701 |
