 , 王子琨1, 王雪青1, 苏春景1, 黎欢毅1, 石雪风1, 黄皓旻1,2,3, 付名利1,2,3, 胡芸1,2,3, 叶代启1,2,3, 吴军良1,2,3
, 王子琨1, 王雪青1, 苏春景1, 黎欢毅1, 石雪风1, 黄皓旻1,2,3, 付名利1,2,3, 胡芸1,2,3, 叶代启1,2,3, 吴军良1,2,3
1. 华南理工大学环境与能源学院, 广州 510006;
2. 挥发性有机物污染治理技术与装备国家工程实验室, 广州 510006;
3. 广东省大气环境与污染控制重点实验室, 广州 510006
收稿日期: 2021-03-30; 修回日期: 2021-05-06; 录用日期: 2021-05-06
基金项目: 国家重点研发计划(No.2016YFC0204200);国家自然科学基金(No.51678245,51578245);广东省自然科学基金(No.2020A1515010929);广州市科技计划项目(No.201804020026)
作者简介: 毛梦绮(1995-), 女, E-mail: 904271063@qq.com
通讯作者(责任作者): 吴军良, 男, 博士, 教授, 硕士生导师, 主要研究方向为挥发性有机物污染控制材料、技术与装备, 包括环境吸附与催化领域. E-mail: ppjl@scut.edu.cn
摘要:在等离子体催化系统中,不同催化剂常表现出显著的性能差异.本文通过表面氧物种表征及臭氧氧化原位反应,探究了α-MnO2与CeO2两种常用催化剂在等离子体催化系统中催化氧化甲醇性能差异的本质原因.等离子体催化氧化甲醇的性能评价结果显示,在相同输入功率下CeO2对甲醇的转化率和CO2选择性均高于α-MnO2.氧气程序升温脱附(O2-TPD)和X射线光电子能谱(XPS)等表征手段及甲醇常温催化、O3分解和O3催化氧化实验结果表明,CeO2比α-MnO2拥有更多的表面活性氧,能吸附活化更多的甲醇分子;同时,CeO2能更有效地利用臭氧分解产生的活性氧物种对甲醇进行深度氧化.进一步通过原位DRIFTS实验研究两种催化剂协同臭氧催化氧化甲醇反应中间产物的变化,结果表明,CeO2催化剂氧化甲醇的表面副产物较少,而α-MnO2表面则会累积大量的副产物碳酸盐,从而影响其催化性能.
关键词:等离子体催化α-MnO2CeO2挥发性有机物表面氧物种
Exploration on the difference in performance of manganese oxide and cerium oxide in synergistic plasma-catalytic oxidation of methanol
MAO Mengqi1
, WANG Zikun1, WANG Xueqing1, SU Chunjing1, LI Huanyi1, SHI Xuefeng1, HUANG Haomin1,2,3, FU Mingli1,2,3, HU Yun1,2,3, YE Daiqi1,2,3, WU Junliang1,2,31. School of Environment and Energy, South China University of Technology, Guangzhou 510006;
2. National Engineering Laboratory for VOCs Pollution Control Technology and Equipment, Guangzhou 510006;
3. Guangdong Provincial Key Laboratory of Atmospheric Environment and Pollution Control(SCUT), Guangzhou 510006
Received 30 March 2021; received in revised from 6 May 2021; accepted 6 May 2021
Abstract: Different types of catalysts usually exhibit significant activity differences in plasma-catalytic systems. In this paper, the performance of α-MnO2 and CeO2 in the plasma-catalytic oxidation of methanol was explored by characterizing the surface oxygen species and investigating the in-situ ozone oxidation. At the same input power, CeO2 demonstrated higher methanol conversion and CO2 selectivity than α-MnO2. By characterizing the catalysts with temperature-programmed desorption of oxygen (O2-TPD) and X-ray photoelectron spectroscopy (XPS), and performing catalytic methanol oxidation, O3 decomposition and methanol ozonation experiments at ambient temperature, we revealed that CeO2, as compared to α-MnO2, possessed more surface oxygen species that are highly beneficial for activating methanol molecules. Besides, CeO2 might utilize more effectively the active oxygen species generated by ozone decomposition for complete methanol degradation. In-situ DRIFTS was performed to track the formation of intermediates in catalytic methanol oxidation by ozone over the two catalysts, and suggests that, while a large amount of carbonate by-products accumulate on the surface of α-MnO2, CeO2 generated less surface by-products.
Keywords: plasma catalysisα-MnO2CeO2volatile organic compoundssurface oxygen species
1 引言(Introduction)挥发性有机物(VOCs)是近地面臭氧(O3)、细颗粒物(PM2.5)和光化学烟雾形成的关键前体物(Shao et al., 2016), 对区域大气环境质量具有严重影响.低温等离子体技术可在常温常压下去除VOCs, 具有广阔的应用前景, 但该技术存在能耗高、反应选择性差及有副产物O3产生等缺点.近年来, 等离子体耦合多相催化(简称等离子体催化)技术应运而生, 该技术结合了等离子体反应启动快速、条件温和与催化反应选择性高、反应彻底的优点, 有效地提高了污染物转化率和CO2选择性, 降低了能耗并减少副产物的生成(Wang et al., 2018).
目前等离子体催化体系中常用的催化剂有贵金属和金属氧化物两类.以氧化锰和氧化铈为代表的金属氧化物因价格相对低廉, 且协同等离子体反应时表现出良好的催化性能而受到广泛关注.已报道的研究表明, 氧化锰催化剂通常表现出较高的VOCs转化率, 而氧化铈催化剂则表现出较高的CO2选择性.例如, Norsic等(2016)将MnO2和CeO2负载在γ-Al2O3上对比其协同等离子体氧化甲醇的性能, 结果表明, MnO2/Al2O3和CeO2/Al2O3分别表现出更高的甲醇转化率和CO2选择性, 且同时负载Mn和Ce的MnO2/CeO2/Al2O3催化剂具有最高的甲醇转化率(100%)和CO2选择性(52%).Zhu等(2017)将La0.8M0.2MnO3(M=Ba、Ca、Ce、Mg和Sr)催化剂用于等离子体降解乙酸乙酯反应, 发现LaMnO3的乙酸乙酯转化率高于掺Ba、Ca和Mg的La0.8M0.2MnO3催化剂, 掺杂Ce的La0.8Ce0.2MnO3催化剂显示出最高的CO2选择性.虽然氧化锰和氧化铈协同等离子体催化氧化VOCs的性能存在明显差异, 但两种催化剂在等离子体场内降解VOCs性能差别的内在原因尚不清楚.因此, 有必要通过实验研究两种催化剂在等离子体场内作用的本质原因, 以期为高效催化剂的选择与设计合成提供理论指导.
热催化研究表明, 在锰基和铈基催化剂中α-MnO2和CeO2纳米棒催化氧化VOCs活性较高(Liang et al., 2008; 廖银念, 2013; Peng et al., 2016).在等离子体催化的研究中也有类似的结论, 例如, Wang等(2017)和Li等(2014)分别考察了不同晶型氧化锰协同等离子体对甲苯和乙醛的催化性能, 发现α-MnO2比β-MnO2和γ-MnO2拥有更好的甲苯和乙醛氧化性能.Wang等(2019)通过水热法合成不同形貌的CeO2催化剂用于等离子体降解甲醇, 结果表明, CeO2纳米棒的甲醇转化率和CO2选择性最高.因此, 本文选择α-MnO2纳米棒和CeO2纳米棒两种催化剂探究其在等离子体系统中降解VOCs的表面催化作用.
目前, 等离子体催化降解VOCs的相关研究多以工业上广泛使用的溶剂物质(如甲苯)作为目标污染物, 但甲苯分子结构较为复杂, 在等离子体催化反应中产生的副产物种类和数量众多(Chang et al., 2018), 不利于了解反应过程中催化剂表面的物质变化, 难以准确阐明催化剂在反应中的具体作用.针对这一问题, 本研究选择小分子VOCs—甲醇作为目标污染物, 其衍生的中间产物和副产物相对较少, 有助于清晰观测反应过程中催化剂表面物质变化, 了解催化剂作用本质.
本研究采用水热法合成α-MnO2和CeO2纳米棒催化剂, 分别考察两种催化剂在等离子体系统中催化氧化甲醇的性能, 并通过XPS、O2-TPD等表征手段及甲醇常温催化、O3分解和O3催化氧化实验, 探究α-MnO2和CeO2表面活性氧物种含量和臭氧分解利用能力对甲醇催化氧化性能的影响;同时, 采用原位DRIFTS进一步跟踪甲醇降解过程中催化剂表面产物的变化, 探究两种催化剂催化氧化甲醇性能差异的原因.
2 实验部分(Experimental)2.1 材料合成α-MnO2纳米棒的制备:采用水热法(Cheng et al., 2010), 取0.4 g MnSO4·H2O和1 g KMnO4溶解于纯水中, 以450 r·min-1的搅拌速度进行30 min磁力搅拌, 待完全溶解后转移到反应釜内, 在140 ℃下进行12 h的水热合成.
CeO2纳米棒的制备:采用水热法(廖银念, 2013), 将5 mmol醋酸铈分散于20 mL去离子水中, 在室温下以450 r·min-1的搅拌速度进行磁力搅拌, 待完全溶解后加入55 mL NaOH溶液, 继续搅拌30 min.然后将得到的紫色混合液转移到反应釜中, 在130 ℃下进行5 h的水热合成.
2.2 材料表征SEM通过MERLIN扫描电子显微镜(ZEISS, 德国)进行观测;XRD测试所用仪器为D8 Advance X衍射仪(Bruker-AXS, 德国), 测试条件:2θ=10°~90°;BET采用ASAP 2020 M全自动表面分析仪(Micromeritics, 美国)测定, 测试前在150 ℃下对样品进行6 h脱气处理;XPS测试所用仪器为EscaLab 250Xi型X射线光电子能谱仪(Thermo Scientific, 美国), 以284.8 eV为C 1s校准结合能;O2-TPD使用AutoChem 2920全自动程序升温化学吸附仪(Micromeritics, 美国)进行测定, 测试前样品在He气氛, 300 ℃下进行1 h吹扫, 待温度降至60 ℃时, 通入20% O2/He进行1 h吸附, 最后在He气流吹扫条件下记录升温脱附的O2信号;红外光谱分析采用美国Thermo Fisher公司Nicolet iS50R型原位漫反射红外光谱仪(In-situ DRIFTS), 所用检测器为MCT, 分辨率为4 cm-1, 扫描范围为850~4000 cm-1, 扫描次数为32.
2.3 等离子体催化活性评价等离子体催化反应系统由配气装置、等离子体反应器、电源和检测装置组成, 活性评价装置如图 1所示.等离子体反应器采用的是介质阻挡(DBD)反应器, 催化剂装填在放电区末端, 以玻璃石英管为反应管, 不锈钢金属棒作内电极连接高压交流电源(频率1.968 kHz, 南京苏曼), 不锈钢金属网缠绕在石英管外壁作接地极.使用示波器(DS-1052E, 普源)测试等离子体放电特性, 反应器的输入功率采用李萨如方法计算.
图 1(Fig. 1)
 |
| 图 1 实验装置示意图 Fig. 1Schematic illustration of the experimental setup |
反应进气中的甲醇浓度为571 mg·m-3, 气体流量为100 mL·min-1, 催化剂用量为0.1 g.反应器进出口气体中的有机组分、CO和CO2采用气相色谱仪(上海凡伟)进行检测分析, 采用基于紫外吸收原理的臭氧分析仪(106 M, 美国2B)检测O3浓度.甲醇转化率(Xmethanol)和反应产物选择性(SP)计算公式如下:
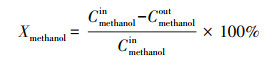 | (1) |
 | (2) |
2.4 α-MnO2和CeO2的催化作用差异研究为了探究α-MnO2和CeO2催化剂的表面活性氧物种差异对甲醇催化性能的影响, 进行了甲醇常温催化实验, 将两种催化剂装填在反应管中, 用N2吹扫30 min后, 在常温下通入571 mg·m-3的甲醇/N2混合气进行反应.为了研究等离子体放电产生的臭氧对于两种催化剂氧化甲醇的贡献, 设计了O3分解和O3催化氧化实验.O3分解实验即分别向装填有两种催化剂的反应管中通入1221 mg·m-3的O3/(O2+N2)混合气, 该混合气由单独等离子体在0.5 W的输入功率和干空背景气氛下放电产生(这一放电条件与等离子体催化氧化甲醇反应一致), 实际测量所得浓度即1221 mg·m-3, 检测反应器出口浓度, 按式(3)计算催化剂的O3分解性能(XO3).O3催化氧化实验则是在常温下将928 mg·m-3 O3/(O2+N2)和414 mg·m-3甲醇/N2的混合气通入装填有两种催化剂的反应管中进行反应, 其中, 反应混合气(928 mg·m-3 O3/(O2+N2)和414 mg·m-3甲醇/N2)由干空放电产生的1221 mg·m-3 O3/(O2+N2)与571 mg·m-3甲醇/N2混合所得.为了探讨α-MnO2和CeO2催化氧化甲醇反应路径的差异, 通过原位DRIFTS实验来跟踪两种催化剂与臭氧协同氧化甲醇反应过程中表面有机物质的变化.实验时先将样品在100 ℃、N2条件下预处理60 min去除表面的吸附水, 待温度降到30 ℃时采集背景信号, 随后通入571 mg·m-3甲醇/N2的混合气, 吸附一定时间后通入1221 mg·m-3 O3/(O2+N2)的混合气进行反应并采集红外信号.
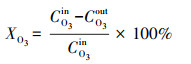 | (3) |
图 2(Fig. 2)
 |
| 图 2 α-MnO2和CeO2催化剂的XRD谱图 Fig. 2XRD patterns of α-MnO2 and CeO2 catalysts |
两种催化剂的N2吸附结果如表 1所示, 可知两种催化剂均为介孔材料, 且相比于α-MnO2, CeO2的比表面积和孔容更大.CeO2和α-MnO2都是等离子体场内性能优良的催化剂活性相, CeO2更大的比表面积和孔容, 将有利于VOCs的吸附及等离子体产生的活性物种与催化剂表面的接触和反应.
表 1(Table 1)
| 表 1 α-MnO2和CeO2催化剂的N2吸附结果 Table 1 The results of N2 adsorption of α-MnO2 and CeO2 catalysts | ||||||||||||
表 1 α-MnO2和CeO2催化剂的N2吸附结果 Table 1 The results of N2 adsorption of α-MnO2 and CeO2 catalysts
| ||||||||||||
图 3为α-MnO2和CeO2催化剂的SEM图, 可见所合成的两种催化剂均为纳米棒状结构, α-MnO2纳米棒和CeO2纳米棒的尺寸分别为1.2 μm×100 nm和600 nm×(30±5) nm.
图 3(Fig. 3)
 |
| 图 3 α-MnO2(a)和CeO2(b)催化剂的SEM图 Fig. 3SEM images of α-MnO2(a) and CeO2(b) catalysts |
通过XPS分析可以了解α-MnO2和CeO2催化剂表面O元素的化学状态.图 4是α-MnO2和CeO2的O 1s谱图, 采用Avantage软件对两种样品谱图进行分峰拟合, 得到了两个拟合峰, 分别对应于两类氧物种, 低结合能528.6 eV处的特征峰归属为晶格氧OLat, 530.3~530.7 eV处的特征峰归属于表面氧OSur(Hu et al., 2016; Yu et al., 2018).其中, 表面氧OSur中包含的活性氧物种(如O22-、O2-)对VOCs具有较强的氧化能力(Hu et al., 2016; Yao et al., 2018), 这可能有利于甲醇的氧化.对两个样品的峰面积进行积分, 并将计算得到的反映活性氧物种比例的OSur/(OSur+OLat)比值在图 4中标出, 可知CeO2的OSur/(OSur+OLat)比值为69.5%, 远大于α-MnO2的14.9%, 说明CeO2样品比α-MnO2含有更多的表面活性氧物种.
图 4(Fig. 4)
 |
| 图 4 α-MnO2和CeO2催化剂O 1s的XPS谱图 Fig. 4XPS spectra of O 1s for α-MnO2 and CeO2 samples |
通过O2-TPD测试可以了解两种催化剂在升温过程中脱附出的氧物种类型和数量差异, 其结果如图 5所示.α-MnO2样品在60~700 ℃之间出现了3个脱附峰, 其中, 103 ℃处的峰归属于物理吸附氧或表面活性氧的脱附(Zheng et al., 2016), 451 ℃处的肩峰和532 ℃处的高峰则归属于次表面晶格氧的脱附(Durán et al., 2009).CeO2样品在同样温度区间出现了两个脱附峰, 112 ℃处的峰归属于物理吸附氧的脱附, 309 ℃附近平而宽的峰可归为表面氧(如O22-、O2-)脱附(Huang et al., 2014; Chen et al., 2018).表面氧脱附量越高说明催化剂的表面活性氧物种越多, 对比两个样品的脱附峰面积, 可以发现CeO2在180~500 ℃之间表面活性氧脱附量比α-MnO2的表面活性氧(<300 ℃)更多, 这与XPS的结果一致, 进一步证明CeO2催化剂拥有更多的表面活性氧物种.
图 5(Fig. 5)
 |
| 图 5 α-MnO2和CeO2催化剂的O2-TPD谱图 Fig. 5O2-TPD profiles of α-MnO2 and CeO2 catalysts |
3.2 等离子体催化活性评价装填α-MnO2和CeO2催化剂的等离子体催化系统降解甲醇的活性评价结果如图 6所示.从图中可以看出, 在单独等离子体系统中甲醇转化率、CO2选择性和COx选择性都较低, 且有较多的甲酸甲酯和高浓度副产物O3生成.在单独等离子体系统中, 主要是通过甲醇分子与等离子体放电产生的活性物种在等离子体场内碰撞实现甲醇氧化, 反应选择性差, 导致副产物多且能量效率低(J?gi et al., 2014).在引入两种催化剂之后, 甲醇的氧化性能显著提高, 同时副产物O3的排放浓度明显降低, 说明等离子体引发的催化剂表面催化反应对甲醇氧化起到了积极贡献.此外, CeO2催化剂协同等离子体降解甲醇的性能优于α-MnO2, 在相同的输入功率下CeO2的甲醇转化率和CO2选择性分别达到84%和85%, 而装填α-MnO2的等离子体催化系统的甲醇转化率和CO2选择性分别只有58%和17%, 并且产生了很多甲酸甲酯, 表明在等离子体催化系统中CeO2更能促进甲醇完全氧化.文献结果显示氧化锰催化剂往往具有更高的VOCs转化率, 但在本研究中, CeO2催化剂的转化率、选择性都比α-MnO2高, 这可能是由于本研究制备的两种催化剂具有特定晶型与微观形貌, 而文献普遍用浸渍或焙烧等方法制备的催化剂无特定微观形貌.
图 6(Fig. 6)
 |
| 图 6 单独等离子体和等离子体催化系统的甲醇降解性能 (a.甲醇转化率、CO2、COx和CH3COOH选择性;b.出口O3浓度)(温度30 ℃, 催化剂用量0.1 g, 气体流量100 mL·min-1, 甲醇浓度571 mg·m-3, 输入功率0.5 W) Fig. 6Methanol oxidation in plasma catalytic systems and plasma alone (a.methanol conversion, CO2, COx and CH3COOH selectivity; b.outlet O3 concentration) |
在等离子体催化反应体系中, 催化剂与等离子体之间存在相互作用, 使得影响甲醇降解性能的因素较多.α-MnO2和CeO2等离子体催化氧化甲醇性能的提高可能是催化剂改变了等离子体的放电特性, 增强了等离子体气相碰撞反应, 也可能是催化剂引发的表面催化作用的贡献.
为此, 研究了α-MnO2和CeO2催化剂对等离子体放电特性的影响, 图 7显示了单独等离子体及装填α-MnO2和CeO2的等离子体催化系统的电压电流特性曲线.图 7a的电流尖峰表明单独等离子体系统为典型的丝状放电, 电流尖峰长短不一, 说明是不稳定的等离子体微放电状态.从图 7b可以看出, 装填α-MnO2之后电流尖峰放电丝的长度变短, 同时放电细丝的分布更加密集, 说明装填α-MnO2的等离子体催化系统中同时存在丝状放电和表面放电.当介电常数较高的催化剂填充在DBD反应器中时, 催化剂颗粒之间的接触点附近会形成更强的局部非均匀电场, 使得等离子体的部分空间间隙放电转变为局部放电(Koen et al., 2017; Song et al., 2018; Wang et al., 2019; Wu et al., 2021), 从而改变等离子体的放电状态.MnO2具有较高的介电常数(>80)(Wang et al., 2019), 由此可以推测α-MnO2对等离子体放电特性的改变是由于其介电常数较高, 使得α-MnO2纳米棒之间的接触点周围产生了较强的局部非均匀电场, 等离子体部分空间间隙放电转变为局部放电所致.表面放电提高了电离和分子之间碰撞解离的速率, 从而产生更多的反应活性物种来提高VOCs的降解率(杨仕玲, 2020).因此, α-MnO2协同等离子体氧化甲醇性能的提高有一部分原因可能是由等离子体气相碰撞反应增强所引起的.这种增强作用增加了放电空间内反应活性物种的数量, 使等离子体区域中有更多的甲醇转化为甲醛和甲酸, 接着生成了较多的甲酸甲酯.图 7c是装填CeO2的等离子体催化系统的电压电流特性曲线, 可以看出装填CeO2之后电流尖峰放电丝的分布及长度几乎不变, 其图形与单独等离子体的相似, 说明CeO2催化剂的引入对等离子体的放电特性基本没有影响, CeO2协同等离子体氧化甲醇性能的提高则主要是表面催化反应的作用.文献报道结果显示, CeO2的介电常数相对较小约为24(Kim et al., 2015; Kumar et al., 2016), 因此, 其对等离子体放电特性的影响不明显.
图 7(Fig. 7)
 |
| 图 7 DBD反应器在不同体系下的放电特性 (a.单独等离子体, b.α-MnO2, c.CeO2) Fig. 7Typical voltage, current waveforms for DBD reactors (a.plasma only, b.plasma with α-MnO2, c.plasma with CeO2) |
3.3 α-MnO2和CeO2的表面催化作用在等离子体催化体系中, 催化剂自身的表面活性氧物种和臭氧分解生成的活性氧物种都可能参与了VOCs的表面催化氧化(Jia et al., 2018).为了探究α-MnO2和CeO2自身活性氧物种含量和分解利用臭氧的能力差异对甲醇催化氧化性能的影响, 分别进行了甲醇常温催化、臭氧分解和臭氧催化氧化实验.
3.3.1 甲醇常温催化实验甲醇常温催化实验可考察α-MnO2和CeO2催化剂表面活性氧的作用.图 8是反应过程中产生的CO2量随时间变化情况, 可以发现两种样品的反应过程中均有CO2生成, 说明α-MnO2和CeO2自身的表面活性氧物种在常温条件下均能氧化甲醇.许多研究表明, 甲醇在氧化锰和氧化铈催化剂表面会先吸附形成甲氧基(Finocchio et al., 2001; Wu et al., 2012), 接着转化成甲酸盐, 然后生成CO2(Zhu et al., 2016; Wang et al., 2019).从图中还可以看出, CeO2样品生成的CO2量大于α-MnO2样品, 从XPS和O2-TPD结果可推测, 这是因为CeO2具有更多的表面活性氧物种, 可以活化和氧化更多的甲醇.
图 8(Fig. 8)
 |
| 图 8 α-MnO2和CeO2常温催化甲醇反应的CO2生成量随时间变化曲线 Fig. 8Evolution of CO2 production in methanol oxidation over α-MnO2 and CeO2 at room temperature |
3.3.2 O3分解实验和O3催化氧化实验在等离子体催化系统中, O3在催化剂表面分解形成的活性氧物种可能是氧化VOCs的氧物种主要来源(王雪青等, 2019).催化剂对O3的分解效果越好, 产生的活性氧物种越多, 越有利于VOCs的氧化(Yang et al., 2018; Zhang et al., 2019).为了对比两种催化剂臭氧分解性能的差别, 进行了O3分解实验, 结果如图 9所示.α-MnO2和CeO2对臭氧的分解都可以达到100%, 说明两种催化剂都具有良好的臭氧分解能力, 但鉴于二者在等离子体场内对甲醇氧化性能的差异, 有可能α-MnO2和CeO2催化剂表面分解臭氧产生的活性氧物种对甲醇氧化的贡献是不同的, 因此, 进一步进行了O3催化氧化实验, 结果如图 10所示.α-MnO2和CeO2的甲醇转化率都为100%, 结合O3分解实验结果, 可以说明两种催化剂分解臭氧所生成的活性氧物种都能有效地实现甲醇转化.但对比二者的碳平衡数据可以发现, CeO2的CO2选择性(97%)明显高于α-MnO2(78%), 而α-MnO2的CO产量比CeO2多约15%, 可以推测甲醇的完全氧化程度可能与两种催化剂分解O3形成的活性氧物种的种类或数量有关.Oyama和Hu认为O3在MnOx表面分解形成的原子氧物种对VOCs氧化起重要作用(Reed et al., 2005; Hu et al., 2017).而臭氧在CeO2表面分解产生的活性氧物种(如O*、O22-、O2-)被认为是促进VOCs及其中间产物转化和完全氧化的关键活性物种(Egawa et al., 2018; Mao et al., 2018; Jia et al., 2018; Wu et al., 2021).Zhu等(2016)认为O3在氧化锰和氧化铈表面分解形成的活性氧的传递和利用影响了其在等离子体中的催化性能.CeO2协同臭氧催化氧化甲醇的产物CO2选择性比α-MnO2更高, 可能是因为其能更有效地利用O3分解形成的活性氧物种来促进甲醇深度氧化.
图 9(Fig. 9)
 |
| 图 9 α-MnO2和CeO2催化剂对O3的分解性能 Fig. 9Ozone decomposition on α-MnO2 and CeO2 catalysts |
图 10(Fig. 10)
 |
| 图 10 α-MnO2和CeO2催化剂协同O3催化氧化甲醇的性能 Fig. 10The performance of α-MnO2 and CeO2 catalysts for methanol oxidation with ozone |
3.4 原位DRIFTS实验采用原位DRIFTS跟踪甲醇吸附与臭氧氧化反应状态下α-MnO2和CeO2表面物质的变化, 可以提供反应路径和催化剂作用的关键信息.图 11a为甲醇在α-MnO2表面吸附过程的原位红外谱图, 可以看到在2360 cm-1和1037 cm-1处出现了两个红外峰.其中, 2360 cm-1处归属于CO2碳氧键的伸缩振动峰(Wu et al., 2012), 1037 cm-1处归属于与Mn3+桥连的甲氧基特征峰(Finocchio et al., 2001).α-MnO2上没有观察到甲基的特征峰, 说明甲醇在α-MnO2上的吸附较少.图 11b为CeO2表面的甲醇吸附过程, 可以观察到在2925、2801、1101和1032 cm-1等处出现了红外峰.其中, 2925 cm-1和2801 cm-1处归属于甲基的特征峰, 1101 cm-1和1032 cm-1处分别属于Ce4+上单齿甲氧基与双齿甲氧基的特征峰(Wu et al., 2012).此外, 1595 cm-1和1640 cm-1处的峰分别是Ce3+上的双齿甲酸盐特征峰与吸附水的伸缩和弯曲振动峰(Rousseau et al., 2010).图 11a中2360 cm-1处的CO2特征峰和图 11b中1595 cm-1和1640 cm-1处的甲酸盐与吸附水特征峰, 进一步证实了α-MnO2和CeO2自身存在的表面活性氧物种会在常温下对甲醇进行氧化.由于两种催化剂表面活性氧含量和氧物种类型存在差异, 导致甲醇在两种催化剂上吸附活化形成的表面产物也不同(Durán et al., 2009; Li et al., 2014; Wang et al., 2019).
图 11(Fig. 11)
 |
| 图 11 甲醇在α-MnO2(a)和CeO2(b)表面吸附分解产物的原位红外谱图 Fig. 11In-situ DRIFTS spectrum of methanol adsorption process over α-MnO2(a) and CeO2(b) catalysts surface |
图 12a展示了通入O3后α-MnO2表面吸附物种变化的红外谱图, 可看到在持续通入臭氧的30 min内, 1037 cm-1处甲氧基的特征峰逐渐消失, 同时2360 cm-1处CO2特征峰的峰强度随O3通入时间的增加逐渐增强;此外, 1553 cm-1和1401 cm-1处出现了两个新的特征峰, 分别归属于甲酸盐物种和碳酸氢盐物种(Vayssilov et al., 2011).说明O3在α-MnO2表面分解产生的活性氧物种将甲氧基氧化成了中间物种甲酸盐、碳酸氢盐和CO2.再继续通入臭氧, 可以发现1800~1200 cm-1范围内又出现了6个新的红外峰.其中, 1642 cm-1处归属于表面吸附水的特征峰, 1370 cm-1处是甲酸盐的碳氢键振动模式(Rousseau et al., 2010), 剩下1720、1498、1300和1202 cm-1处的特征峰均属于碳酸盐物种, 1720 cm-1和1202 cm-1处为通过两个氧中心结合的碳酸盐, 而1498 cm-1和1300 cm-1处则是通过3个氧中心结合的碳酸盐(Vayssilov et al., 2011).这表明α-MnO2表面的甲酸盐等中间产物在臭氧的作用下进一步被氧化成了碳酸盐副产物和水.然而这些碳酸盐主要通过两个或三个氧中心结合, 通常需要较高的温度才能从催化剂表面解吸脱附(Vayssilov et al., 2011), 所以逐渐积累在催化剂表面.由于这些碳酸盐副产物在常温下能稳定地存在于α-MnO2表面, 抑制了α-MnO2表面臭氧分解生成的表面活性氧物种与甲醇及其反应中间产物发生氧化反应, 从而影响了甲醇在α-MnO2表面的深度氧化.
图 12(Fig. 12)
 |
| 图 12 O3催化氧化过程中甲醇在α-MnO2(a)和CeO2(b)表面产物的原位红外谱图 Fig. 12In-situ DRIFTS spectrum of ozone catalytic oxidation over α-MnO2(a) and CeO2(b) catalysts surface |
图 12b所示为CeO2表面吸附物种的变化过程, 可以观察到通入O3后, 2925 cm-1和2801 cm-1处甲基和1101 cm-1和1032 cm-1处甲氧基的特征峰都逐渐消失, 而1595 cm-1和1640 cm-1处甲酸盐与吸附水特征峰的峰强度逐渐增强, 同时1273 cm-1处还出现了新的特征峰, 归属于单齿甲酸盐物种(Popova et al., 2012).说明CeO2表面的甲基和甲氧基都被O3分解所产生的活性氧物种氧化成了甲酸盐和水.甲酸盐在O3的作用下会继续氧化为CO2和水, 但这一过程相对缓慢, 所以有少量甲酸盐累积在催化剂表面(王雪青, 2019).值得注意的是, 与α-MnO2相比, CeO2表面没有观察到碳酸盐物种的产生, 推测可能是由于臭氧在两种催化剂表面分解形成的活性氧物种差异所致(Reed et al., 2005; Hu et al., 2017; 王佳伶等, 2020), 臭氧在CeO2表面分解形成的活性氧物种能有效促进甲醇及其反应中间产物转化为CO2(Zhu et al., 2016; Norsic et al., 2016; Norsic et al., 2018).
4 结论(Conclusions)1) 在等离子体催化氧化甲醇体系中, CeO2的甲醇降解率(84%)和CO2选择性(85%)比α-MnO2更高.CeO2基本没有改变等离子体区域的放电特征, 甲醇氧化性能的提高主要是表面催化反应的作用, 与α-MnO2相比, CeO2的表面活性氧更多, 更有利于甲醇的活化和转化.此外, CeO2能更有效地利用O3分解产生的活性氧物种促进甲醇的完全氧化.
2) 原位红外进一步证实了CeO2表面氧化甲醇的副产物少.而α-MnO2表面会生成大量的副产物碳酸盐累积, 从而限制了α-MnO2利用臭氧所分解的表面活性氧对甲醇进行深度氧化, 最终影响了催化性能的提高.
参考文献
| Chang T, Shen Z, Huang Y, et al. 2018. Post-plasma-catalytic removal of toluene using MnO2-Co3O4 catalysts and their synergistic mechanism[J]. Chemical Engineering Journal, 348: 15-25. DOI:10.1016/j.cej.2018.04.186 |
| Chen X, Chen X, Yu E, et al. 2018. In situ pyrolysis of Ce-MOF to prepare CeO2 catalyst with obviously improved catalytic performance for toluene combustion[J]. Chemical Engineering Journal, 344: 469-479. DOI:10.1016/j.cej.2018.03.091 |
| Cheng F, Su Y, Liang J, et al. 2010. MnO2-based nanostructures as catalysts for electrochemical oxygen reduction in alkaline media[J]. Chemistry of Materials, 22: 898-905. DOI:10.1021/cm901698s |
| Durán F G, Barbero B P, Cadús L E, et al. 2009. Manganese and iron oxides as combustion catalysts of volatile organic compounds[J]. Applied Catalysis B: Environmental, 92: 194-201. DOI:10.1016/j.apcatb.2009.07.010 |
| Egawa Y, Hawada S, Einaga H. 2018. Contribution of catalytic performance of CeO2 in nonthermal plasma chemical reaction[J]. Joint Journal of Novel Carbon Resource Sciences & Green Asia Strategy, 5: 34-37. |
| Finocchio E, Busca G. 2001. Characterization and hydrocarbon oxidation activity of coprecipitated mixed oxides Mn3O4/Al2O3[J]. Catalysis Today, 70: 213-225. DOI:10.1016/S0920-5861(01)00419-9 |
| Hu M, Yao Z, Hui K N, et al. 2017. Novel mechanistic view of catalytic ozonation of gaseous toluene by dual-site kinetic modelling[J]. Chemical Engineering Journal, 308: 710-718. DOI:10.1016/j.cej.2016.09.086 |
| Hu Z, Liu X, Meng D, et al. 2016. Effect of ceria crystal plane on the physicochemical and catalytic properties of Pd/Ceria for CO and propane oxidation[J]. Acs Catalysis, 6: 2265-2279. DOI:10.1021/acscatal.5b02617 |
| Huang H, Dai Q, Wang X. 2014. Morphology effect of Ru/CeO2 catalysts for the catalytic combustion of chlorobenzene[J]. Applied Catalysis B: Environmental, 158-159: 96-105. DOI:10.1016/j.apcatb.2014.01.062 |
| Jia Z, Ben A, Yang D, et al. 2018. Plasma catalysis application of gold nanoparticles for acetaldehyde decomposition[J]. Chemical Engineering Journal, 347: 913-922. DOI:10.1016/j.cej.2018.04.106 |
| J?gi I, Haljaste A, Laan M. 2014. Hybrid TiO2 based plasma-catalytic reactors for the removal of hazardous gasses[J]. Surface and Coatings Technology, 242: 195-199. DOI:10.1016/j.surfcoat.2013.10.016 |
| Kim H H, Teramoto Y, Negishi N, et al. 2015. A multidisciplinary approach to understand the interactions of nonthermal plasma and catalyst: A review[J]. ChemInform, 46: 13-22. |
| Koen V L, Annemie B. 2017. How bead size and dielectric constant affect the plasma behaviour in a packed bed plasma reactor: a modelling study[J]. Plasma Sources Science and Technology, 26. DOI:10.1088/1361-6595/aa7c59 |
| Kumar P, Kumar P, Kumar A, et al. 2016. Structural, morphological, electrical and dielectric properties of Mn doped CeO2[J]. Journal of Alloys and Compounds, 672: 543-548. DOI:10.1016/j.jallcom.2016.02.153 |
| Li Y, Fan Z, Shi J, et al. 2014. Post plasma-catalysis for VOCs degradation over different phase structure MnO2 catalysts[J]. Chemical Engineering Journal, 241: 251-258. DOI:10.1016/j.cej.2013.12.036 |
| Liang S, Teng F, Bulgan G, et al. 2008. Effect of Phase Structure of MnO2 Nanorod Catalyst on the Activity for CO Oxidation[J]. The Journal of Physical Chemistry C, 112: 5307-5315. DOI:10.1021/jp0774995 |
| 廖银念. 2013. 铈基金属氧化物催化氧化甲苯的形貌及尺寸效应[D]. 广州: 华南理工大学 |
| Mao L, Chen Z, Wu X, et al. 2018. Plasma-catalyst hybrid reactor with CeO2/γ-Al2O3 for benzene decomposition with synergetic effect and nano particle by-product reduction[J]. Journal of Hazardous Materials, 347: 150-159. DOI:10.1016/j.jhazmat.2017.12.064 |
| Norsic C, Tatibou?t J-M, Batiot C, et al. 2016. Non thermal plasma assisted catalysis of methanol oxidation on Mn, Ce and Cu oxides supported on γ-Al2O3[J]. Chemical Engineering Journal, 304: 563-572. DOI:10.1016/j.cej.2016.06.091 |
| Norsic C, Tatibou?t J-M, Batiot C, et al. 2018. Methanol oxidation in dry and humid air by dielectric barrier discharge plasma combined with MnO2-CuO based catalysts[J]. Chemical Engineering Journal, 347: 944-952. DOI:10.1016/j.cej.2018.04.065 |
| Peng R, Sun X, Li S, et al. 2016. Shape effect of Pt/CeO2 catalysts on the catalytic oxidation of toluene[J]. Chemical Engineering Journal, 306: 1234-1246. DOI:10.1016/j.cej.2016.08.056 |
| Popova G Y, Chesalov Y A, Sadovskaya E M, et al. 2012. Effect of water on decomposition of formic acid over V-Ti oxide catalyst: Kinetic and in situ FTIR study[J]. Journal of Molecular Catalysis A: Chemical, 357: 148-153. DOI:10.1016/j.molcata.2012.02.005 |
| Reed C, Xi Y, Oyama S T. 2005. Distinguishing between reaction intermediates and spectators: A kinetic study of acetone oxidation using ozone on a silica-supported manganese oxide catalyst[J]. Journal of Catalysis, 235: 378-392. DOI:10.1016/j.jcat.2005.08.014 |
| Rousseau S, Marie O, Bazin P, et al. 2010. Investigation of methanol oxidation over Au/Catalysts using operando ir spectroscopy: determination of the active sites, intermediate/spectator species, and reaction mechanism[J]. Journal of the American Chemical Society, 132: 10832-10841. DOI:10.1021/ja1028809 |
| Shao P, An J, Xin J, et al. 2016. Source apportionment of VOCs and the contribution to photochemical ozone formation during summer in the typical industrial area in the Yangtze River Delta, China[J]. Atmospheric Research, 176-177: 64-74. DOI:10.1016/j.atmosres.2016.02.015 |
| Song H, Hu F, Peng Y, et al. 2018. Non-thermal plasma catalysis for chlorobenzene removal over CoMn/TiO2 and CeMn/TiO2: Synergistic effect of chemical catalysis and dielectric constant[J]. Chemical Engineering Journal, 347: 447-454. DOI:10.1016/j.cej.2018.04.018 |
| Vayssilov G N, Mihaylov M, Petkov P S, et al. 2011. Reassignment of the vibrational spectra of carbonates, formates, and related surface species on ceria: a combined density functional and infrared spectroscopy investigation[J]. The Journal of Physical Chemistry C, 115: 23435-23454. DOI:10.1021/jp208050a |
| Wang B, Xu X, Xu W, et al. 2018. The mechanism of non-thermal plasma catalysis on volatile organic compounds removal[J]. Catalysis Surveys from Asia, 22: 73-94. DOI:10.1007/s10563-018-9241-x |
| Wang H, Chen S, Wang Z, et al. 2019. A novel hybrid Bi2MoO6-MnO2 catalysts with the superior plasma induced pseudo photocatalytic-catalytic performance for ethyl acetate degradation[J]. Applied Catalysis B: Environmental, 254: 339-350. DOI:10.1016/j.apcatb.2019.05.018 |
| 王佳伶, 毛梦绮, 石雪风, 等. 2020. Pt颗粒负载对CeO2臭氧催化氧化甲苯的增强作用[J]. 环境科学学报, 40(5): 1629-1639. |
| Wang T, Chen S, Wang H, et al. 2017. In-plasma catalytic degradation of toluene over different MnO2 polymorphs and study of reaction mechanism[J]. Chinese Journal of Catalysis, 38: 793-803. DOI:10.1016/S1872-2067(17)62808-0 |
| Wang X, Wu J, Wang J, et al. 2019. Methanol plasma-catalytic oxidation over CeO2 catalysts: Effect of ceria morphology and reaction mechanism[J]. Chemical Engineering Journal, 369: 233-244. DOI:10.1016/j.cej.2019.03.067 |
| 王雪青. 2019. 低温等离子体催化降解甲醇反应系统中CeO2的形貌效应[D]. 广州: 华南理工大学 |
| 王雪青, 黎欢毅, 王邦芬, 等. 2019. 等离子体场内CeO2催化降解甲醇的表面活性氧物种来源与作用研究[J]. 环境科学学报, 39(8): 2725-2734. |
| Wu K, Sun Y, Liu J, et al. 2021. Nonthermal plasma catalysis for toluene decomposition over BaTiO3-based catalysts by Ce doping at A-sites: The role of surface-reactive oxygen species[J]. Journal of Hazardous Materials, 405: 124156. DOI:10.1016/j.jhazmat.2020.124156 |
| Wu Z, Li M, Mullins D R, et al. 2012. Probing the Surface Sites of CeO2 Nanocrystals with Well-Defined Surface Planes via Methanol Adsorption and Desorption[J]. Acs Catalysis, 2: 2224-2234. DOI:10.1021/cs300467p |
| Yang Y, Jia J, Liu Y, et al. 2018. The effect of tungsten doping on the catalytic activity of α-MnO2 nanomaterial for ozone decomposition under humid condition[J]. Applied Catalysis A: General, 562: 132-141. DOI:10.1016/j.apcata.2018.06.006 |
| 杨仕玲. 2020. 低温等离子体-纳米后催化协同降解挥发性有机化合物基础研究[D]. 杭州: 浙江大学 |
| Yao X, Li Y, Fan Z, et al. 2018. Plasma catalytic removal of hexanal over Co-Mn solid solution: Effect of preparation method and synergistic reaction of ozone[J]. Industrial & Engineering Chemistry Research, 57: 4214-4224. |
| Yu L, Peng R, Chen L, et al. 2018. Ag supported on CeO2 with different morphologies for the catalytic oxidation of HCHO[J]. Chemical Engineering Journal, 334: 2480-2487. DOI:10.1016/j.cej.2017.11.121 |
| Zhang Y, Chen M, Zhang Z, et al. 2019. Simultaneously catalytic decomposition of formaldehyde and ozone over manganese cerium oxides at room temperature: Promotional effect of relative humidity on the MnCeOx solid solution[J]. Catalysis Today, 327: 323-333. DOI:10.1016/j.cattod.2018.04.027 |
| Zheng Y, Wang W, Jiang D, et al. 2016. Amorphous MnOx modified Co3O4 for formaldehyde oxidation: improved low-temperature catalytic and photothermocatalytic activity[J]. Chemical Engineering Journal, 284: 21-27. DOI:10.1016/j.cej.2015.08.137 |
| Zhu X, Liu S, Cai Y, et al. 2016. Post-plasma catalytic removal of methanol over Mn-Ce catalysts in an atmospheric dielectric barrier discharge[J]. Applied Catalysis B: Environmental, 183: 124-132. DOI:10.1016/j.apcatb.2015.10.013 |
| Zhu X, Tu X, Chen M, et al. 2017. La0.8M0.2MnO3 (M=Ba, Ca, Ce, Mg and Sr) perovskite catalysts for plasma-catalytic oxidation of ethyl acetate[J]. Catalysis Communications, 92: 35-39. DOI:10.1016/j.catcom.2016.12.013 |
