 , 聂文善, 唐瑶, 雷鸣
, 聂文善, 唐瑶, 雷鸣 , 唐和清
, 唐和清中南民族大学资源与环境学院, 武汉 430074
收稿日期: 2020-04-27; 修回日期: 2020-06-13; 录用日期: 2020-06-13
基金项目: 国家自然科学基金(No.21707170);中央高校基本科研业务费专项资金项目(No.CZT20019)
作者简介: 王会敏(1994-), 女, E-mail:2802053761@qq.com
通讯作者(责任作者): 雷鸣, E-mail:leiming@mail.scuec.edu.cn
摘要:分别以Zn(CH3COO)2·2H2O、Mn(CH3COO)3·2H2O和Co(CH3COO)2·4H2O为锌源、锰源和钴源,采用溶胶-凝胶自燃烧法成功制备了ZnMnxCo2-xO4(x=0~2)复合物,并用X射线衍射和X射线光电子能谱对其进行表征.同时,还研究了Mn/Co物质的量比、催化剂用量及PMS用量对目标污染物降解的影响.结果表明,该复合物可催化活化过一硫酸钾(PMS)降解有机污染物,当催化剂中x=0.8,催化剂投加量为0.2 g·L-1,PMS用量为0.4 mmol·L-1(0.25 g·L-1)时,20 μmol·L-1(10 mg·L-1)罗丹明B(RhB)可在15 min内完全降解.ZnMn0.8Co1.2O4的高催化活性主要归功于Mn3+和Co2+的协同效应.将ZnMnxCo2-xO4-PMS体系用于亚甲基蓝、结晶紫、金橙、双酚A、4-氯酚等其他污染物的降解,也取得了较好的效果.基于电子自旋共振ESR和自由基猝灭实验的结果,可以推测该反应体系中活性物种为硫酸根自由基和羟基自由基.
关键词:ZnMnxCo2-xO4催化活化过一硫酸钾(PMS)有机污染物
Degradation of organic contaminants by peroxymonosulfate activated with ZnMnxCo2-xO4 as a heterogeneous catalyst
WANG Huimin
, NIE Wenshan, TANG Yao, LEI Ming, TANG HeqingCollege of Resources and Environmental Science, South-Central University for Nationalities, Wuhan 430074
Received 27 April 2020; received in revised from 13 June 2020; accepted 13 June 2020
Abstract: ZnMnxCo2-xO4 was successfully prepared by a sol-gel autocombustion method using Zn(CH3COO)2·2H2O,Mn(CH3COO)3·2H2O and Co(CH3COO)2·4H2O as sources of zinc,manganese and cobalt,respectively. The obtained ZnMnxCo2-xO4 was characterized by X-ray diffractometry and X-ray photoelectron spectrometry. It was found that the catalysts could activate potassium peroxymonosulfate (PMS) to degrade organic contaminants. The use of 0.2 g·L-1 ZnMn0.8Co1.2O4 and 0.4 mmol·L-1(0.25 g·L-1) PMS led to the complete degradation of 20 μmol·L-1 (10 mg·L-1) RhB within 15 min. It was also investigated the effect of Mn/Co molar ratio,catalysts load,and PMS concentration on the catalytic activities of ZnMnxCo2-xO4. The highly catalytic activity of ZnMnxCo2-xO4 was ascribed to the synergistic effect between Mn3+ and Co2+. The catalytic system also degraded efficiently other pollutants including methylene blue,crystal violet,golden orange,bisphenol A,and 4-chlorophenol. Based on electron spin-resonance (ESR) measurements and the free radical quenching studies,it was confirmed that the generated active species in the catalytic system involved both sulfate radical (SO4·-) and hydroxyl radical (·OH).
Keywords: ZnMnxCo2-xO4catalytic activationPMSorganic contaminants
1 引言(Introduction)近年来, 有机污染物在地下水和地表水中越来越频繁地被检测到(Wang et al., 2020).由于它们能够通过食物链和环境生态循环进入人体并对人类健康造成不利的影响(Oulton et al., 2015;Adoamnei et al., 2018;Mota et al., 2020), 因而引起了环保工作者的广泛关注.在有机污染物的治理中, 高级氧化技术(AOPs)由于其体系能够产生氧化能力强的羟基自由基(·OH, E=2.80 eV)并降解大多数有机污染物而受到人们的青睐(G?gol et al., 2018).与·OH相比, 硫酸根自由基(SO4·-, E=2.5~3.1 eV)具有更强的氧化能力和更长的寿命(t1/2(SO4·-)=30~40 μs, t1/2(·OH) < 1 μs)(Hu et al., 2016), 因此, 以SO4·-为主的新型高级氧化技术(SR-AOP)在有机污染物的控制和治理中具有广泛的应用前景.
SO4·-的产生主要是通过过硫酸盐(PS)或过一氧硫酸盐(PMS)的活化, 其中, 活化PMS的方法有很多种, 包括热活化、碱活化和过渡金属活化等(Xiao et al., 2018).相较之下, 过渡金属活化是较为常见的方法, 研究证明, Cu+(Nie et al., 2019)、Mn2+(Peng et al., 2018)、Fe2+(Zhang et al., 2019)、Co2+(Chen et al., 2019)等可以活化PMS, 且在相同的条件下, Co2+对PMS的活性最高(Hu et al., 2016).然而, 在均相Co2+-PMS体系中, 一方面Co2+的投加浓度往往高于我国规定的Co2+排放浓度限值, 会对环境造成二次污染;另一方面, 均相Co2+难以被回收再利用(Ling et al., 2010;Chen et al., 2019).为了解决上述问题, 非均相钴基催化剂被广泛研究.例如, Anipsitakis等(2005)首次尝试将市售的Co3O4应用于PMS活化, 发现该体系对2, 4-二氯苯酚具有良好的降解性能.一般而言, 金属氧化物作为催化剂的性能主要取决于高表面积和窄粒度分布.因此, Chen等(2008)成功合成了纳米级Co3O4, 在PMS活化方面显示出良好的性能, 尤其是在中性pH条件下.但单一的钴基氧化物活化PMS容易出现金属离子溶出的问题.有研究表明, 将过渡金属引入到Co3O4中形成双金属纳米催化剂不仅可以强烈地促进PMS活化, 而且可以极大地限制钴的溶出(Su et al., 2013;Yao et al., 2015;刘萌等, 2018).
最近有文献报道, Zn/Co双金属氧化物在活化PMS降解BPA的过程中展现出优异的活性.例如, 刘萌等(2018)采用微波法成功制备了ZnCo2O4来活化PMS, 发现当ZnCo2O4和PMS的投加量分别为0.2 g·L-1和0.44 mmol·L-1时, 20 mg·L-1 BPA在15 min内能被完全去除;实验结果还表明, 引入Zn可以减少钴的溶出, 提高催化剂的稳定性, 但反应后催化剂表面仍有Co2+转化为Co3+.此外, 锰基催化剂也被证明能够活化PMS降解污染物, 如Chen等(2019)通过水热法成功制备了新型磁性MnO2/MnFe2O4纳米材料用于活化PMS以降解罗丹明B(RhB), 结果表明, 在PMS存在下, 5 min内物质的量比为7:1的MnO2/MnFe2O4纳米材料对RhB的降解率达到90%.另有研究表明, Mn3+能还原Co3+生成Co2+, 从而促进Co3+/Co2+的循环(Li et al., 2015).因此, 若能将Mn引入ZnCo2O4催化剂, 将有望进一步提高催化剂的活性.
溶胶-凝胶自燃烧法结合了溶胶-凝胶法与自燃烧法的特点, 可实现复杂氧化物的快速原位合成, 并在一定程度上避免团聚, 因而成为合成纳米级复杂氧化物的重要方法之一, 近年来被广泛用于多金属氧化物的合成研究.例如, Wang等(2018)使用溶胶-凝胶自燃烧法合成了一系列ZnMnxCo2-xO4, 且发现不同Mn/Co物质的量比对CO具有不同的氧化活性.基于此, 本文按照其使用的溶胶-凝胶自燃烧法在ZnCo2O4中掺入Mn, 制备ZnMnxCo2-xO4作为活化PMS的催化剂, 用于有机污染物的降解.该催化剂一方面通过Mn的掺入代替Co从而减少环境毒性较大的Co用量, 另一方面通过Mn3+促进Co2+/Co3+循环, 进一步加快其对PMS的活化.
2 材料和方法(Materials and methods)2.1 试剂二水合醋酸锌(Zn(CH3COO)2·2H2O, 纯度99.999%)、二水合醋酸锰(Mn(CH3COO)3·2H2O, 纯度97%)、四水合醋酸钴(Co(CH3COO)2·4H2O, 分析纯)均购自阿拉丁试剂, 一水合柠檬酸(C6H8O7·H2O)、盐酸、罗丹明B(RhB)、结晶紫(CV)、亚甲基蓝(MB)、金橙II(OII)、双酚A(BPA)、4-氯酚(4-CP)购自国药试剂.所有实验药品使用时均无需进一步纯化.实验过程中所用的水为超纯水.
2.2 ZnMnxCo2-xO4的制备称取适量Zn(CH3COO)2·2H2O、Mn(CH3COO)3·2H2O和Co(CH3COO)2·4H2O于100 mL烧杯中(Zn:Mn:Co物质的量比为1:x:(2-x)), 加入50 mL水, 超声分散后转移至聚四氟乙烯反应釜内胆中.在30 ℃恒温搅拌下将10 mL柠檬酸水溶液滴加到含有金属离子的混合溶液中, 并控制阳离子/柠檬酸的物质的量比为1:3.将聚四氟乙烯反应釜内胆于170 ℃下反应12 h, 自然冷却后将釜中悬浮液抽滤并用蒸馏水多次洗涤, 得到的固体于60 ℃下烘干, 然后将其研磨成粉末后在马弗炉中600 ℃下煅烧6 h.最后经盐酸和水多次洗涤后得到ZnMnxCo2-xO4产物, 烘干后研磨成粉末并保存在保干器中.
2.3 催化剂表征方法XRD表征:将待测样品平压于载玻片上, 用D8 ADVANCE型X射线衍射仪对其进行表征, 其中, 射线源为Cu Kα靶射线, 石墨单色器滤波, 扫描范围为10°~80°.
扫描电镜(SEM)表征:采用Hitachi S-4800型扫描电镜, 将样品固定在样品台上, 观测图像.
透射电镜(TEM)表征:采用TECNAI G20型透射式电子显微镜, 工作电压为200 kV.将样品分散到无水乙醇中, 超声均匀, 用吸管吸取少量滴到铜网上, 阴干后观测形貌.
X-射线光电子能谱(XPS)表征:采用MULT1LAB2000型X-射线光电子能谱仪对催化剂的元素价态进行表征.测试条件为:真空优于1×10-8 Torr, 铝/镁靶, 高压14.0 kV, 功率300 W, 通能93.9 eV, 以C 1s=284.6 eV为基准进行结合能校正.
2.4 催化降解实验将ZnMnxCo2-xO4催化剂(0.2 g·L-1)分散在50 mL含有一定量污染物的水溶液中, 吸附30 min后取样测污染物浓度并记为c0, 再向溶液中加入一定量的PMS同时开始计时, 每隔一定时间取出1 mL溶液并加入100 μL 0.1 mol·L-1 NaN3猝灭, 离心后取上清液通过紫外可见分光光度计测定其在最大吸收波长处的吸光度值或通过高效液相色谱测定其峰面积.
2.5 污染物的定量分析2.5.1 染料定量分析UV-vis分光光度法:在预定的时间间隔内, 取出1.0 mL水样立即加入100 μL NaN3摇匀, 离心后取上清液放入石英比色皿中测其在最大吸收波长(RhB:554 nm, CV:590 nm, MB:664 nm, OII:484 nm)处的吸光度值.
2.5.2 BPA及4-CP的定量分析HPLC分析:在预定的时间间隔内, 取出1.0 mL水样立即加入100 μL NaN3摇匀, 离心后取上清液进行HPLC测试.具体测试条件如下:色谱柱为Thermo C18-P柱(5 μm, 4.6 mm×150 mm), 流动相为V(甲醇):V(水)= 60:40, 流速为1.0 mL·min-1, 进样量为20 μL;DAD检测器, 检测波长分别为220 nm(BPA)、280 nm(4-CP);柱温30 ℃.
2.6 活性物种的鉴定2.6.1 猝灭实验反应开始前分别向体系中加入0.4 mol·L-1的叔丁醇和甲醇作为捕获剂, 然后加入PMS开始反应, 定时取点分析.
2.6.2 电子自旋共振测试在浓度为0.04 g·L-1 ZnMn0.6Co1.4O4反应体系中加入0.42 mol·L-1 DMPO溶液, 加入2 mmol·L-1 PMS 5 min后用毛细管吸取少量溶液放入检测仪中检测.测试条件为:接收增益1×105, 调制幅度1 G, 微波功率10 mW, 调制频率100 kHz.
3 结果与讨论(Results and discussion)3.1 催化剂的表征图 1所示为ZnMnxCo2-xO4尖晶石氧化物的XRD图谱.由图 1可见, 当x=0时, 所得样品的XRD衍射峰与ZnCo2O4的标准图谱(JCPDS 23-1390)一致:2θ为31.2°、36.8°、38.5°、44.8°、55.6°、59.3°及65.1°处分别对应ZnCo2O4的(220)、(311)、(222)、(440)、(422)、(511)和(440)晶面, 这也与文献报道的一致(Sharma et al., 2007);除此之外, 未观察到其他杂质峰, 说明当x=0时, 产物为纯净的ZnCo2O4.当x从0增大到1时, ZnCo2O4的XRD衍射峰逐渐变宽, 说明随着Mn掺入量的增多, ZnCo2O4的晶型变差.且在谱图中未观察到锰氧化物的特征衍射峰, 说明成功制得ZnMnxCo2-xO4, Mn掺杂入到ZnCo2O4中.
图 1(Fig. 1)
 |
| 图 1 ZnMnxCo2-xO4的X射线衍射图谱 Fig. 1XRD patterns of ZnMnxCo2-xO4 |
通过SEM和TEM分析研究了ZnMn0.8Co1.2O4的形貌特征(图 2), 发现采用溶胶-凝胶自燃烧法制得的样品呈球状结构, 直径分布在7~25 nm左右.
图 2(Fig. 2)
 |
| 图 2 催化剂的形貌(a.SEM, b.TEM) Fig. 2Morphology of catalyst(a.SEM, b. TEM) |
为了进一步证明Mn被成功掺入到ZnCo2O4, 本研究对ZnMn0.8Co1.2O4进行了XPS表征.如图 3a所示, 结合能在290、535、658、811及1053 eV处分别对应C 1s、O 1s、Mn 2p、Co 2p及Zn 2p(Wang et al., 2018).该结果进一步说明Mn成功掺入到ZnCo2O4中.另外, ZnMn0.8Co1.2O4表面的Mn、Co物质的量比测试值为0.28, 小于理论值(n(Mn):n(Co)=0.67), 说明该催化剂表面暴露了大量的Co活性位点.
图 3(Fig. 3)
 |
| 图 3 ZnMn0.8Co1.2O4的X-射线光电子能谱(a.全谱, b.Mn 2p, c.Co 2p, d.Zn 2p) Fig. 3XPS spectra of ZnMnxCo2-xO4(a.wide survey, b.Mn 2p, c.Co 2p, d.Zn 2p) |
图 3b给出了催化剂表面的Mn 2p图谱, 如图 3b所示, 结合能为653.9 eV和642.3 eV处对应于Mn 2p1/2和Mn 2p3/2(Wang et al., 2018).对Mn 2p3/2进行分峰拟合发现, 结合能641.5 eV和643.3 eV处分别归属于Mn3+和Mn4+(Wang et al., 2018), 且Mn4+与Mn3+的峰面积之比为51.3:48.7.说明在催化剂的形成过程中, Mn3+部分氧化形成了Mn4+.
图 3c给出了Co 2p图谱, 在结合能为799.4 eV和779.5 eV处分别归属于Co 2p1/2和Co 2p3/2(Joshi et al., 2018;Wang et al., 2018).以780.2 eV和795.3 eV为中心的峰归属于Co2+, 并伴有结合能为790.0 eV的卫星峰出现.以781.1 eV和796.4 eV为中心的峰归属于Co3+, 并伴有以805.5 eV为中心的卫星峰.对Co 2p3/2分峰拟合发现, Co2+和Co3+的峰面积之比为50.9:49.1.图 3d给出的是Zn 2p图谱, 结合能为1043.8 eV和1020.8 eV处是典型的+2价Zn的位置(Joshi et al., 2018), 说明催化剂表面的Zn的价态全部为+2价.
综合催化剂的XRD和XPS实验结果, 说明Mn成功掺入到ZnCo2O4, 并且在催化剂形成过程中发生了Mn、Co的部分氧化, 导致其表面Mn和Co的价态分别为+3、+4和+2、+3.
3.2 不同体系中RhB的降解效果图 4为在不同反应体系下RhB的降解效果对比.从图 4中可以发现, 单独使用PMS时, RhB浓度未发生明显变化, 说明PMS难以直接氧化RhB.另外, 在单独的ZnCo2O4、ZnMn0.8Co1.2O4或ZnMn2O4体系中, RhB在这些催化剂表面的吸附去除率均小于3%, 同时也观察不到RhB降解, 说明催化剂对RhB的吸附作用很弱, 也不能够直接降解RhB.ZnCo2O4-PMS、ZnMn0.8Co1.2O4-PMS、ZnMn2O4-PMS对RhB的降解均表现出一定的催化活性, 其中, ZnCo2O4-PMS和ZnMn2O4-PMS在15 min内对RhB的降解率分别为91%和19%.与之相比, ZnMn0.8Co1.2O4-PMS表现出更高的催化降解RhB的能力, 其降解率达到98%.这说明ZnMnxCo2-xO4能够活化PMS产生活性物种快速降解RhB.
图 4(Fig. 4)
 |
| 图 4 不同体系对RhB降解效果的对比(RhB 20 μmol·L-1, PMS 0.4 mmol·L-1, ZnMnxCo2-xO4 0.2 g·L-1) Fig. 4Comparison of RhB degradation over different systems |
通过拟一级动力学模型ln (ct/c0)=-kt拟合RhB的降解速率, 其中, k为表观速率常数(min-1), t为反应时间(min), c0和ct分别代表初始和t时刻污染物的浓度.根据拟合结果可知, ZnMn0.8Co1.2O4-PMS体系的降解速率常数为0.25 min-1, 分别是ZnCo2O4-PMS和ZnMn2O4-PMS体系的1.7和16.7倍.
3.3 影响RhB降解的因素3.3.1 不同Mn/Co比对RhB降解效果的影响图 5给出了不同Mn/Co比的ZnMnxCo2-xO4活化PMS降解RhB的速率.如图所示, 当x=0即ZnCo2O4时, 反应体系的速率常数为0.15 min-1;当x=0.2即催化剂为ZnMn0.2Co1.8O4时, 反应速率常数为0.09 min-1, 说明在ZnCo2O4掺入少量Mn时, 体系对PMS的催化活性降低, 这是因为在ZnCo2O4掺入Mn后, 其表面的Co活性位点减少, 导致其活性有所降低.进一步增加Mn的掺入量(x=0.2~0.8), 该反应体系的速率常数由0.09 min-1增大到0.25 min-1, 这是因为随着Mn的掺入量增大, 催化剂表面Mn/Co之间的相互作用增强, 促进Co3+/Co2+的循环, 加快对PMS的活化, 说明Mn/Co在活化PMS时有协同作用.继续增加Mn的掺入量(x=1.0~2.0), 该反应体系的速率常数开始减小(0.23~0.01 min-1), 这是因为Mn的掺入量过大, 导致催化剂表面的Co活性位点过少, 从而导致其对PMS的活性减弱;当x=2.0时(即催化剂为ZnMn2O4), 该反应体系的速率常数只有0.01 min-1, 说明该催化剂中起主要催化作用的为Co2+.综上可知, 在ZnMnxCo2-xO4-PMS反应体系中存在Mn/Co协同作用, 其中, 催化剂表面的Co为主要活性中心, 且当Mn与Co物质的量比为2:3即x=0.8时, 对PMS的催化活性达到最高.
图 5(Fig. 5)
 |
| 图 5 Mn/Co比对ZnMnxCo2-xO4-PMS降解RhB速率的影响(RhB 20 μmol·L-1, PMS 0.4 mmol·L-1, ZnMnxCo2-xO4 0.2 g·L-1) Fig. 5Effect of Mn/Co ratios on degradation of RhB rate by ZnMnxCo2-xO4-PMS |
3.3.2 PMS浓度对RhB降解效果的影响在该体系中, PMS浓度是影响RhB在ZnMn0.8Co1.2O4-PMS体系中降解的一个重要因素.图 6所示为不同PMS浓度对RhB在ZnMnxCo2-xO4-PMS体系中降解速率的影响.如图所示, 当体系中只存在0.2 g·L-1 ZnMn0.8Co1.2O4时, RhB的降解速率几乎为0(0.001 min-1), 这表明ZnMn0.8Co1.2O4依靠自身的氧化性或吸附性能引起的RhB降解非常微弱;当PMS浓度增大时, RhB的降解速率也逐渐增大, 当PMS浓度达到0.4 mmol·L-1时, 该反应体系的速率常数达到0.25 min-1, 这是因为当体系中的PMS浓度增大, 其与催化剂表面的活性位点接触发生反应的几率增大, 从而增加了反应体系中活性物种的浓度, 进而提高了RhB的降解速率.当PMS的浓度继续增加至0.6 mmol·L-1时, RhB的降解速率增大并不明显(0.26 min-1), 这可能是因为当PMS的浓度增加至0.6 mmol·L-1时, 0.2 g·L-1 ZnMn0.8Co1.2O4催化剂表面提供的活性位点不够, 所以其与0.6 mmol·L-1 PMS催化反应的速率与0.4 mmol·L-1 PMS差别不大.因此, 在催化剂浓度为0.2 g·L-1时, PMS的最佳浓度确定为0.4 mmol·L-1.
图 6(Fig. 6)
 |
| 图 6 PMS浓度对RhB在ZnMn0.8Co1.2O4-PMS体系中降解的影响(RhB 20 μmol·L-1, ZnMn0.8Co1.2O4 0.2 g·L-1) Fig. 6Effect of PMS concentrations on the degradation of RhB in ZnMn0.8Co1.2O4-PMS system |
3.3.3 催化剂浓度对RhB降解效果的影响与PMS相同, ZnMn0.8Co1.2O4投加量也是影响RhB在ZnMn0.8Co1.2O4-PMS体系中降解的一个重要参数.图 7所示为不同ZnMn0.8Co1.2O4投加量对RhB降解速率的影响.当反应体系中只有0.4 mmol·L-1 PMS而没有投加ZnMn0.8Co1.2O4时, RhB的降解速率为0.007 min-1, 几乎不降解, 这表明单独的PMS对RhB的氧化降解较弱.逐渐增大ZnMn0.8Co1.2O4的投加量(0~0.3 g·L-1), RhB的降解速率逐渐增加(0.007~0.39 min-1).对此解释为:ZnMn0.8Co1.2O4的投加量增大实际上是增加了反应体系中的活性位点(Mn、Co), 从而产生了更多的活性物种, 最终导致RhB降解速率增大.
图 7(Fig. 7)
 |
| 图 7 ZnMn0.8Co1.2O4浓度对RhB在ZnMn0.8Co1.2O4-PMS体系中降解的影响(RhB 20 μmol·L-1, PMS 0.4 mmol·L-1) Fig. 7Effect of ZnMn0.8Co1.2O4 load on the degradation of RhB in ZnMn0.8Co1.2O4-PMS system |
3.4 催化剂的重复利用性催化剂的重复利用性是评价催化剂性能的重要指标之一.图 8显示了ZnMn0.8Co1.2O4在循环使用时的活性变化.每一次ZnMn0.8Co1.2O4-PMS体系降解RhB 15 min之后将反应液过滤洗涤, 再将催化剂分散到新的反应体系中继续反应.由图 8可知, 在第1次使用时, 15 min后RhB的去除率达到了98%, 在重复使用该催化剂活化PMS降解RhB 8次时, 仍可使RhB的去除率达到90%, 说明ZnMn0.8Co1.2O4具有优良的可循环性.
图 8(Fig. 8)
 |
| 图 8 RhB在ZnMn0.8Co1.2O4-PMS体系中循环8次的去除率(ZnMn0.8Co1.2O4 0.2 g·L-1, PMS 0.4 mmol·L-1, RhB 10 μmol·L-1) Fig. 8Removal of RhB in the ZnMn0.8Co1.2O4-PMS system in successive eight cycles |
3.5 ZnMn0.8Co1.2O4-PMS反应体系对不同污染物的降解进一步选择3种常见染料(20 μmol·L-1 CV、20 μmol·L-1 MB和50 μmol·L-1 OII)及两种常见的酚类污染物(0.08 mmol·L-1 BPA和0.1 mmol·L-1 4-CP)作为目标污染物, 考察ZnMn0.8Co1.2O4-PMS体系对它们的去除效果.如图 9所示, ZnMn0.8Co1.2O4在30 min内达到吸附平衡时对各污染物的吸附去除率均在5%以下.达到吸附平衡后向体系加入PMS, 15 min内体系对20 μmol·L-1 RhB、CV和MB及50 μmol·L-1 OII的去除效率均能达到100%, 说明ZnMn0.8Co1.2O4-PMS对染料有很好的去除效果.另外, 该体系在15 min内对0.08 mmol·L-1 BPA和0.1 mmol·L-1 4-CP也有很好的去除效果(BPA去除率达到100%, 4-CP去除率达到93%).这些结果表明, ZnMn0.8Co1.2O4-PMS体系对多种有机污染物均能达到较好的降解效果, ZnMn0.8Co1.2O4可作为优良的活化PMS的催化剂用于多种有机污染物的处理.
图 9(Fig. 9)
 |
| 图 9 ZnMn0.8Co1.2O4-PMS体系对不同污染物的降解(ZnMn0.8Co1.2O4 0.3 g·L-1, PMS 0.4 mmol·L-1) Fig. 9Degradation of different contaminants in the ZnMn0.8Co1.2O4-PMS system |
基于上述实验结果, 本课题组构筑了一种利用高级氧化技术处理有机污染物的体系, 该体系条件如下:催化剂为ZnMn0.8Co1.2O4, 催化剂投加量为0.2 g·L-1, PMS浓度为0.4 mmol·L-1(0.25 g·L-1).表 1比较了有机污染物(RhB和MB)在不同催化剂体系中的去除率.由表 1可知, 本研究建立的氧化体系对有机污染物的氧化效率更高, 且消耗的氧化剂PMS量更少.
表 1(Table 1)
| 表 1 有机污染物在不同体系中的去除率 Table 1 Removal efficiency of organic pollutans in several reported systems | ||||||||||||||||||||||||||||||||||||||||||
表 1 有机污染物在不同体系中的去除率 Table 1 Removal efficiency of organic pollutans in several reported systems
| ||||||||||||||||||||||||||||||||||||||||||
3.6 ZnMn0.8Co1.2O4-PMS机理探究本研究通过猝灭实验和电子自旋共振(ESR)鉴定了ZnMn0.8Co1.2O4-PMS体系中的活性物种.在猝灭实验中, 将甲醇和叔丁醇作为猝灭剂加入到ZnMn0.8Co1.2O4-PMS反应体系中.甲醇通常被用来捕获SO4·-和·OH, 这是因为甲醇与这两种自由基均具有较高的反应速率, 其反应速率常数分别为3.2×106 L·mol-1·s-1和9.7×108 L·mol-1·s-1.而叔丁醇(TBA)与·OH具有高反应活性, 其反应速率常数是与SO4·-反应速率常数的418~1900倍, 因此使用TBA来捕获·OH.如图 10a所示, 在ZnMn0.8Co1.2O4-PMS反应体系中未加捕获剂时RhB的降解速率常数为0.25 min-1, 而加入0.4 mol·L-1的TBA或甲醇的体系中k值分别降为0.11、0.05 min-1, 当TBA或甲醇浓度进一步增加至2 mol·L-1时, RhB的降解速率常数进一步降低至0.09、0.01 min-1.通过添加2 mol·L-1的甲醇可以完全抑制RhB的降解, 这表明在ZnMn0.8Co1.2O4-PMS反应体系中主要活性物种为SO4·-和·OH.
图 10(Fig. 10)
 |
| 图 10 甲醇或叔丁醇对ZnMn0.8Co1.2O4-PMS反应体系中RhB降解的影响(a)及ESR波谱(b)(RhB 20 μmol·L-1, PMS 0.4 mmol·L-1, ZnMn0.8Co1.2O4 0.2 g·L-1) Fig. 10Effect of methanol or tert-butyl alcohol on the degradation of RhB in ZnMn0.8Co1.2O4-PMS reaction system(a) and ESR spectrum(b) |
此外, 利用ESR进一步证实上述自由基的存在及其在RhB降解中的作用.如图 10b所示, 在ZnMn0.8Co1.2O4或PMS单一存在下没有检测到任何自由基的信号, 但在ZnMn0.8Co1.2O4-PMS体系中存在显著的ESR信号, 对应于DMPO-·OH产物, 证明在ZnMn0.8Co1.2O4-PMS体系中生成了·OH.图 10b没有观察到SO4·-的ESR信号, 这归因于反应条件下SO4·-向·OH的快速转化(反应(1)和(2)).一些关于活化PMS的文献也报道了相似的结果(Zou et al., 2013;Qi et al., 2017;Chen et al., 2019).SO4·-和·OH对RhB降解的贡献率通过TBA和Methanol存在下RhB的反应速率常数计算(Pan et al., 2020).由于体系中只存在两种自由基即SO4·-和·OH, 当添加2 mol·L-1 TBA时·OH被完全猝灭, 此时只有SO4·-对RhB的降解起作用.因此, RhB的降解速率常数(0.09 min-1)与不加猝灭剂时的降解速率常数(0.25 min-1)之比即为SO4·-的贡献率, 其值为36%, 进而得出·OH的贡献率为64%.
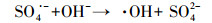 | (1) |
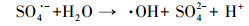 | (2) |
 | (3) |
 | (4) |
 | (5) |
 | (6) |
参考文献
| Adoamnei E, Mendiola J, Vela-Soria, et al. 2018. Urinary bisphenol A concentrations are associated with reproductive parameters in young men[J]. Environmental Research, 161: 122-128. DOI:10.1016/j.envres.2017.11.002 |
| Anipsitakis G P, Stathatos E, Dionysiou D D. 2005. Heterogeneous activation of oxone using Co3O4[J]. The Journal of Physical Chemistry B, 109(27): 13052-13055. DOI:10.1021/jp052166y |
| Chen G, Zhang X, Gao Y, et al. 2019. Novel magnetic MnO2/MnFe2O4 nanocomposite as a heterogeneous catalyst for activation of peroxymonosulfate (PMS) toward oxidation of organic pollutants[J]. Separation and Purification Technology, 213: 456-464. DOI:10.1016/j.seppur.2018.12.049 |
| Chen M T, Zhu L H, Tang H Q, et al. 2019. Efficient degradation of organic pollutants by low-level Co2+catalyzed homogeneous activation of peroxymonosulfate[J]. Journal of Hazardous Materials, 371: 456-462. DOI:10.1016/j.jhazmat.2019.03.002 |
| Chen X Y, Chen J W, Qiao X L, et al. 2008. Performance of nano-Co3O4/peroxymonosulfate system:Kinetics and mechanism study using Acid Orange 7 as a model compound[J]. Applied Catalysis B, 80(1/2): 116-121. |
| Du Y, Ma W, Liu P, Zou B, et al. 2016. Magnetic CoFe2O4 nanoparticles supported on titanate nanotube (CoFe2O4/TNTs) as a novel heterogeous catalyst for peroxymonosulfate activation an degradation of organic pollutants[J]. Journal of Hazardous Materials, 308: 58-66. DOI:10.1016/j.jhazmat.2016.01.035 |
| Fan Y, Ma W, He J, et al. 2017. CoMoO4 as a novel heterogeneous catalysts of peroxymonosulfate activation for the degradation of organic dyes[J]. RSC Advances, 7: 36193-36200. DOI:10.1039/C7RA04761D |
| G?gol M, Przyjazny A, Boczkaj G. 2018. Wastewater treatment by means of advanced oxidation processes based on cavitation-A review[J]. Chemical Engineering Journal, 338: 599-627. DOI:10.1016/j.cej.2018.01.049 |
| Hu P D, Long M C. 2016. Cobalt-catalyzed sulfate radical-based advanced oxidation:A review on heterogeneous catalysts and applications[J]. Applied Catalysis B:Environmental, 181: 103-117. DOI:10.1016/j.apcatb.2015.07.024 |
| Joshi N, da Silva L F, Jadhav H S, et al. 2018. Yolk-shelled ZnCo2O4 microspheres:Surface properties and gas sensing application[J]. Sensors and Actuators B-Chemical, 257: 906-915. DOI:10.1016/j.snb.2017.11.041 |
| Ling S K, Wang S, Peng Y. 2010. Oxidative degradation of dyes in water using Co2+/H2O2 and Co2+/peroxymonosulfate[J]. Journal of Hazardous Materials, 178: 385-389. DOI:10.1016/j.jhazmat.2010.01.091 |
| 刘萌, 胡莉敏, 张广山, 等. 2018. Co/Zn双金属氧化物活化过一硫酸盐降解双酚A的性能研究[J]. 环境化学, 37(4): 753-760. |
| Li W, Wu P X, Zhu Y J, et al. 2015. Catalytic degradation of bisphenol A by CoMnAl mixed metal oxides catalyzed peroxymonosulfate:Performance and mechanism[J]. Chemical Engineering Journal, 279: 93-102. DOI:10.1016/j.cej.2015.05.001 |
| Mota H P, Quadrado R F N, Iglesias B A, et al. 2020. Enhanced photocatalytic degradation of organic pollutants mediated by Zn(Ⅱ)-porphyrin/poly(acrylic acid) hybrid microparticles[J]. Applied Catalysis B:Environmental, 277: 119208. DOI:10.1016/j.apcatb.2020.119208 |
| Nie W S, Mao Q H, Tang H Q, et al. 2019. Highly efficient catalysis of chalcopyrite with surface bonded ferrous species for activation of peroxymonosulfate toward degradation of bisphenol A:A mechanism study[J]. Journal of Hazardous Materials, 364: 59-68. DOI:10.1016/j.jhazmat.2018.09.078 |
| Oulton R, Haase J P, Kaalberg S, et al. 2015. Hydroxyl radical formation during ozonation of multiwalled carbon nanotubes:performance optimization and demonstration of a reactive CNT filter[J]. Environmental Science & Technology, 49(6): 3687-3697. |
| Pan C, Fu L, Ding Y, et al. 2020. Homogeneous catalytic activation of peroxymonosulfate and heterogenous reductive regeneration of Co2+ by MoS2:The pivotal role of pH[J]. Science of the Total Environment, 712: 136447. DOI:10.1016/j.scitotenv.2019.136447 |
| Peng Q, Ding Y B, Tang H Q, et al. 2018. Fast and complete degradation of norfloxacin by using Fe/Fe3C@NG as a bifunctional catalyst for activating peroxymonosulfate[J]. Separation and Purification Technology, 202: 307-317. DOI:10.1016/j.seppur.2018.03.049 |
| Qi C D, Liu X T, Li Y, et al. 2017. Enhanced degradation of organic contaminants in water byperoxydisulfate coupled with bisulfite[J]. Journal of Hazardous Materials, 328: 98-107. DOI:10.1016/j.jhazmat.2017.01.010 |
| Sharma Y, Sharma N, Subba Rao G V, et al. 2007. Nanophase ZnCo2O4 as a high performance anode material for Li-Ion batteries[J]. Advanced Functional Materials, 17: 2855-2861. DOI:10.1002/adfm.200600997 |
| Su S N, Guo W L, Leng Y Q, et al. 2013. Heterogeneous activation of Oxone by CoxFe3-xO4 nanocatalysts for degradation of rhodamine B[J]. Journal of Hazardous Materials, 244-245: 736-742. DOI:10.1016/j.jhazmat.2012.11.005 |
| Wang S X, Gao S S, Tian J Y, et al. 2019. A stable and easily prepared copper oxide catalyst for degradation of organic pollutants by peroxymonosulfate activation[J]. Journal of Hazardous Materials, 387: 121995. |
| Wang T, Sun Y, Zhou Y, et al. 2018. Identifying influential parameters of octahedrally coordinated cations in spinel ZnMnxCo2-xO4 oxides for the oxidation reaction[J]. ACS Catalysis, 8(9): 8568-8577. DOI:10.1021/acscatal.8b02376 |
| Xiao R, Luo Z, Wei Z, et al. 2018. Activation of peroxymonosulfate/persulfate by nanomaterials for sulfate radical-based advanced oxidation technologies[J]. Current Opinion in Chemical Engineering, 19: 51-58. DOI:10.1016/j.coche.2017.12.005 |
| Yao Y J, Cai Y M, Wu G D, et al. 2015. Sulfate radicals induced from peroxymonosulfate by cobalt manganese oxides (CoxMn3-xO4) for Fenton-like reaction in water[J]. Journal of Hazardous Materials, 296: 128-137. DOI:10.1016/j.jhazmat.2015.04.014 |
| Zhang G L, Ding Y B, Tang H Q, et al. 2019. Efficient degradation of drug ibuprofen through catalytic activation of peroxymonosulfate by Fe3C embedded on carbon[J]. Journal Environmental Science, 78: 1-12. DOI:10.1016/j.jes.2018.10.002 |
| Zhang W, Su Y, Zhang X, et al. 2016. Facile synthesis of porous NiCo2O4 nanoflakes as magnetic recoverable catalysts towards the efficient degradation of RhB[J]. RSC Advances, 6: 64626-64633. DOI:10.1039/C6RA12706A |
| Zou J, Ma J, Chen L W, et al. 2013. Rapid acceleration of ferrous iron/peroxymonosulfate oxidation of organic pollutants by promoting Fe(Ⅲ)/Fe(Ⅱ) cycle with hydroxylamine[J]. Environmental Science & Technology, 47: 11685-11691. |
