
 , 张晓1, 程婷1, 王津南2
, 张晓1, 程婷1, 王津南21. 江苏开放大学, 环境生态学院, 南京 210019;
2. 南京大学环境学院, 污染物与资源化研究国家重点实验室, 南京 210023
收稿日期: 2020-05-20; 修回日期: 2020-06-18; 录用日期: 2020-06-18
基金项目: 江苏省高校自然科学研究项目(No.17KJD610002)
作者简介: 徐苏倩(1993-), 女, E-mail:sqxu2015@163.com
通讯作者(责任作者): 徐苏倩
摘要:采用水热合成法制备Cu-Al2O3-g-C3N4类芬顿催化剂,以扫描电镜(SEM)、X射线衍射(XRD)、X射线光电子能谱(XPS)、电子自旋共振(EPR)、拉曼光谱(Raman)对所制备的催化剂及反应过程进行表征.以染料亚甲基蓝(MB)和罗丹明B(Rh-B)及小分子有机物2,4-二氯苯氧乙酸(2,4-D)、双酚A(BPA)和苯妥英(PHT)为目标污染物,研究催化剂在初始pH=7条件下的类芬顿催化活性.同时,探讨Cu掺杂量和有机物配体g-C3N4掺杂量对体系催化性能的影响,并验证晶格氧诱发与有机配体络合两种方式对催化剂活性和稳定性提高产生的影响.DMPO-EPR自由基测定实验及Raman光谱监测催化反应过程验证表明:Cu的晶格氧掺杂诱发了靠近铜晶格O2·-的富电子Cu中心,以及靠近铝晶格O2·-的缺电子Al中心;引入的g-C3N4以阳离子π作用形式通过σ-型Cu—O—C键桥将π体系上的电子转移至Cu,形成一个新的缺电子π中心.在H2O2存在的情况下,富电子Cu中心将电子传递给H2O2,使其被还原为·OH;同时,体系中H2O的电子被缺电子中心剥夺,进而氧化为·OH.羟基自由基转化率TOFs值的进一步计算结果表明,Cu-Al2O3-g-C3N4体系中TOF值为0.516 s-1,是传统均相芬顿体系TOF值(1.53×10-2 s-1)的33倍以上.
关键词:多相芬顿催化双反应活性中心Cu-Al2O3-g-C3N4双氧水利用率有机污染物降解
The synthesis of Fenton like catalyst Cu-Al2O3-g-C3N4 with double catalytic active centers and the mechanism of selective conversion of H2O2 to hydroxyl radical
XU Suqian1
, ZHANG Xiao1, CHEN Ting1, WANG Jingnan21. School of the Environment and Ecology, Jiangsu Open University, Nanjing 210019;
2. State Key Laboratory of Pollution Control and Resource Reuse, School of the Environment, Nanjing University, Nanjing 210023
Received 20 May 2020; received in revised from 18 June 2020; accepted 18 June 2020
Abstract: A Fenton-like catalyst,Cu-Al2O3-g-C3N4,was synthesized by a high pressure hydrothermal method,and the scanning electron microscope (SEM),X-ray diffraction (XRD),X-ray photoelectron (XPS),electron paramagnetic resonance (EPR) and Roman were applied to characterizes both Cu-Al2O3-g-C3N4 and its reaction process. To further analyze the catalytic capability of Cu-Al2O3-g-C3N4 for Fenton-like reation(pH=7),methylene blue (MB),Rhodamine B (RhB),Bisphenol A (BPA),Phenytoin (PHT) and 2,4-dichlorophenoxyaceticacid (2,4-D) were selected as model pollutants. Effect of the doping amount of Cu and g-C3N4 during the preparation of Cu-Al2O3-g-C3N4 on Rh B degradation was discussed. DMPO-trapped EPR signals and Raman spectroscopy reveal that lattice substitution of Cu for Al induces the formation of electron-rich Cu center and the electron-deficient Al center. Surface Cu coordinates with the hydroxyl on the tri-s-triazine ring of g-C3N4 through Cu—O—C linker so that the orbital interactions involving electron transport from π→Cu also induces the formation of another electron-rich Cu center and a new electron-deficient π-electron conjugated system. This kind of surface structure facilitates the reduction of H2O2 to ·OH at the electron-rich Cu center and the oxidation of H2O to ·OH at the electron-deficient Al center or electron-deficient π-electron conjugated system. Turnover frequency (TOF) for the Cu-Al2O3-g-C3N4 dispersions was 0.516 s-1 under neutral conditions,which was still 33 times higher than those of the class homogeneous Fenton reaction under strongly acidic conditions (1.53×10-2 s-1).
Keywords: heterogeneous Fenton-like catalysisdual-reaction centresCu-Al2O3-g-C3N4H2O2 utilizationorganic pollutants degradation
1 引言(Introudction)作为高级氧化技术之一的传统均相芬顿催化反应被广泛用于降解水体中的有机污染物, 但也存在着pH(2~3)响应范围窄及反应过程中有铁泥产生等问题.为解决这些不足, 研究人员致力于制备能够适用较宽pH响应范围的非均相芬顿催化剂.值得注意的是, 在芬顿反应过程中, H2O2不仅可以作为电子受体氧化低价态金属(Mn+), 也能充当电子供体还原高价态金属(M(n+m)+), 实现Mn+/M(n+m)+与·OH或者HO2·之间的电子循环.经典芬顿反应体系中, H2O2还原Fe3+成为Fe2+的反应速率常数(0.001~0.02 L·mol-1·s-1)远低于Fe2+被氧化成Fe3+时的速率常数(6~76 L·mol-1·s-1), 从而成为反应限速步骤(Bokare et al., 2014).为解决芬顿反应过程中的限速问题, 研究人员广泛尝试同构型过渡金属掺杂(李民等, 2019;韩金栋等, 2020)、外加光能(光芬顿)(Song et al., 2006)、电能(电芬顿)、超声(超声芬顿)(Yuan et al., 2012)等物质或能量输入方式促进活性物种之间的价态循环, 但外加能量输入会不可避免地增加废水处理成本.
Nichela等(2013)研究发现, 过渡金属Cu也可以活化H2O2产生羟基自由基, 且相较于Fe2+/Fe3+, Cu+和Cu2+更容易与H2O2反应产生活性自由基(Cu2++H2O2→Cu++HO2·+OH-(k=4.6×102 L·mol-1·s-1), Cu++H2O2 →Cu2++HO·+HO-(k=1.0×104 L·mol-1·s-1)).此外, 他们还研究发现, 铜系均相芬顿反应比铁系均相芬顿反应表现出更加宽广的pH响应范围.因此, Cu/H2O2体系引发了研究人员对其催化反应机理的探讨(Lyu et al., 2019).
上述传统芬顿系统体系中, 大部分H2O2充当电子供体并最终被分解为O2和O2·-, 导致H2O2利用率很低.为提高反应过程中H2O2选择性转化为HO·, 研究人员创造性地借鉴原电池效应来实现H2O2还原为HO·.Lyu等(2016)制备出晶格掺杂二氧化硅纳米球复合材料并在非均相芬顿反应中应用, 通过研究发现, 具有不同电负性(Ti、Cu、Al)的金属掺杂后可诱发体系电子极化分布, 使得H2O2大部分被利用在降解有机污染物上, 从而提高了H2O2的利用率.
阳离子-π作用是一种存在于阳离子和芳香性体系之间的相互作用, 普遍存在于生物体系中, 对分子识别、蛋白质和核酸的结构与功能起着十分重要的作用.在这种体系下, 金属与芳香环体系之间的电荷转移中, 阳离子均处于π体系的上方, 也能够成为一种诱发电子极化分布的手段.石墨相氮化碳(g-C3N4)是常温常压状态下氮碳化合物最稳定的同素异形体, 是一种以三嗪(C3N3)环为基本单元结构和以3-s-三嗪(C6N7)环为基本单元结构, 其C和N原子均发生sp2杂化, 通过pz轨道上的孤对电子形成类似于苯环结构的π键共轭结构;由于此共轭结构的优良导电性, 能够提高电子转移效率, 因而被广泛应用在光催化领域(Wang et al., 2017).Ma等(2017)合成了Fe-g-C3N4/GMC类芬顿催化剂, 考查其在pH=4~10范围内降解染料AR 73的催化活性时发现, 高度分散在g-C3N4网格上并且与N络合的Fe为主要活性位点(Fe-N), 伏安循环结果表明石墨化多孔碳化物能够加速Fe3+与Fe2+之间的电子循环. Lai等(2017)研究了OH-CCN/CuCo-Al2O3芬顿体系下, Cu与g-C3N4表面羟基络合后的H2O2电子转移路径, 证明了阳离子-π作用下诱发电子极化分布进而提高H2O2利用率的可行性.但目前仍然缺乏将晶格氧诱发和表面有机配体络合两种方式结合在同一体系中用于芬顿体系的报道.
本研究以构建双反应活性中心为出发点, 通过水热法与煅烧法合成制备Cu-Al2O3-g-C3N4类芬顿催化剂.采用SEM、TEM、XRD、XPS、EPR、Raman光谱对Cu-Al2O3-g-C3N4的结构组分及反应过程进行表征;以CuO、Al2O3、Fe-Al2O3为对照组探究Cu-Al2O3-g-C3N4类芬顿催化剂的催化效果及优越性;通过固体EPR表征活性物种周围单电子情况, 结合液体EPR揭示反应过程中的活性物种、电子转移路径及作用机理;通过计算·OH转化率TOFs值和H2O2利用率衡量催化剂对体系中氧化剂的选择性转化和氧化剂的利用率.以期为非均相芬顿体系相关理论研究及在实际废水处理中的应用提供理论支撑.
2 材料和方法(Materials and methods)2.1 试验材料与试剂葡萄糖(C6H12O6·H2O, 99%)、九水合硝酸铝(Al(NO3)3·9H2O, 99%)、硝酸铜(Cu(NO3)2·3H2O, 99%)、过氧化氢(H2O2, 30%, 质量分数)购买于国药集团化学试剂北京有限公司.辣根过氧化氢酶(POD, 99%)、N, N-二乙基对苯二胺(DPD, 98%)、5, 5-二甲基1-吡咯啉N-氧化物(DMPO)、对苯二甲酸(TPA, 99%)、罗丹明B(RhB)、亚甲基蓝(MB)、2, 4-二氯苯氧乙酸(2, 4-D, ≥99%)、苯妥英(PHT, 99%)和双酚A(BPA, ≥99%)购买于Sigma-Aldrich(St. Louis, USA)公司.氢氧化钠(NaOH)、盐酸(HCl)、硝酸(HNO3)及其他所有化学试剂均购于南京化学试剂股份有限公司.反应溶液体系pH用1、0.1 mol·L-1盐酸和氢氧化钠溶液调节.超纯水(电阻率为18.2 M·cm)由Milli-Q超纯水仪制得.
2.2 表征方法2.2.1 场发射扫描电镜(FESEM)扫描电镜用来表征和展示催化剂表面的微观结构特征.本研究中的测试仪器是FESEM(QUANTA FEG 250), 放大倍数为5000~50000倍.
2.2.2 比表面积(BET)氮气吸附/脱附等温线可以用来测定催化剂的孔容、比表面积、孔径分布等参数.测定仪器为NOVA2000e比表面积及孔径测试分析仪.比表面积用Brunauer-Emmett-Teller(BET)方法计算, 孔径由Barret-Joyner-Halenda(BJH)方法计算获得.
2.2.3 傅里叶红外光谱(FT-IR)傅里叶红外光谱可以表征催化剂表面的官能团, 测定仪器为NEXUS870 FT-IR检测器, 样品采用溴化钾压片制得, 扫描范围为400~4000 cm-1.
2.2.4 电子能谱仪(XPS)电子能谱仪用来表征催化剂中金属元素的价态和表面元素组成, 测定仪器型号为PHI 5000 VersaProbe, Al Kα radiation (225 W, 15 mA, 15 kV), 用C1s=284.60 eV作为修正参数.
2.2.5 拉曼光谱(Raman)本文中固体拉曼采用LabRAM Aramis仪器测定, 激发波长为310 nm, 扫描范围为280~550 nm, 持续时间为10 s.
2.2.6 电子顺磁共振(EPR)EPR研究的对象是在分子轨道中出现未配对电子(或称单电子)的物质, 通过共振谱线的研究, 可以获测定分子、原子及离子中未偶电子的状态及其周围环境方面的信息, 进一步得到相关物质结构和化学键的信息.本研究通过比较g||和g⊥的变化来判断相应物种的电子周边环境, 进而揭示该物种的配位情况等其他信息.EPR的测试仪器为EMX-10/12电子顺磁共振检测仪.
2.3 Cu-Al2O3-g-C3N4的制备g-C3N4的合成:将一定量的尿素放入带有盖子的刚玉坩埚中, 然后在马弗炉中以5 ℃·min-1的升温速率加热至550 ℃并保温3 h后, 取出自然冷却到室温, 得到的淡黄色产物即为g-C3N4粉末(Zhang et al., 2013).
Cu-Al2O3-g-C3N4的合成:将7.5 g Al(NO3)3·9H2O、5.0 g C6H12O6、0.604 g Cu(NO3)2·3H2O和0.619 g已经烧制而成的g-C3N4溶解到60 mL去离子水中, 30 ℃下搅拌0.5 h后转移到100 mL聚四氟乙烯反应釜中, 180 ℃下密闭高温水热反应20 h, 待自然冷却到室温后, 将所得的固体产物离心过滤, 并用去离子水和乙醇洗涤数次, 在70 ℃下高温烘干.最后在马弗炉中焙烧, 550 ℃下保持5 h, 获得Cu-Al2O3-g-C3N4.基于上述合成方法, 制备出不同g-C3N4含量(1.5%、3.0%、4.5%、6.0%和9.0%)的样品(后面的催化降解罗丹明B实验结果表明, g-C3N4含量为4.5%的样品表现出最好的催化活性和稳定性).
2.4 实验方法2.4.1 催化剂活性及稳定性评价方法催化剂的活性评价实验方法如下:将定量的催化剂粉末和一定浓度的有机污染物溶液(200 mL)混合后置于250 mL锥形瓶中, 然后将锥形瓶放入恒温水浴磁力搅拌器中, 保持温度30 ℃, 搅拌30 min使催化剂和污染物之间达到吸附-脱附平衡, 取样后加入一定量的H2O2, 开始类芬顿催化反应.反应过程中每隔一定时间取样, 并立即过孔径为0.45 μm的可拆式滤膜, 同时为保证检测数据的准确性, 待分析的样品用亚硫酸钠等物质猝灭H2O2.滤液进行有机物浓度检测、金属离子析出量检测、总有机碳(TOC)分析、H2O2剩余浓度分析等.
反应过程中通过金属离子的释放情况和催化剂的重复活性实验来确定催化剂稳定性.溶液中金属离子通过原子吸收光谱仪(ICE)测定.催化剂的重复活性实验方法如下:在类芬顿反应结束后, 用可回收性滤头将催化剂粉末滤出, 去离子水洗涤数次后放入80 ℃烘箱中干燥, 然后将收集的所有催化剂用于下一次类芬顿反应, 如此反复循环, 实现对催化剂的稳定性评价实验.
2.4.2 过氧化氢分解实验方法反应过程中H2O2分解实验测定方法如下:将200 mL超纯水加入到250 mL锥形瓶中, 加入定量的H2O2然后将其置于30 ℃恒温水浴磁力搅拌器中充分搅拌.取原始样(初始浓度C0)后加入定量的催化剂连续搅拌, 启动芬顿反应的催化分解H2O2过程.反应过程中每隔一定时间取样后立刻过孔径为0.45 μm的滤膜, 并立即测定其滤液中残余的H2O2浓度, 测定方法为文献报道(Schick et al., 1997)的DPD-POD法.将测定出的不同反应时间下的H2O2浓度绘制成线即可得到H2O2随时间的分解曲线.
2.4.3 分析及计算方法① 紫外-可见光分光光度法(UV-Vis)分析:染料类污染物(亚甲基蓝、罗丹明B等)可以通过紫外可见分光光度计(UV-vis 1800 spectrometer)在各种染料的最大吸收波长处测定其吸光度, 比照标准曲线即可得出不同反应时间段下溶液中染料的浓度.亚甲基蓝的最大吸收波长为664 nm, 罗丹明B的最大吸收波长为554 nm.
② 高效液相色谱(HPLC)分析:在芬顿反应过程中, 对于大多数有机物, 如内分泌干扰素、医药类等有机物都可以用液相色谱(HPLC, 1200 series, Agilent)进行分析测定.色谱柱为C18column(5 μm, 4.6 mm×150 mm;Agilent), 测定不同有机物的流动相也有所不同(有机相为甲醇或乙腈, 水相为超纯水或缓冲盐体系), 柱温为30 ℃, 进样量为10 μL, 流量为1 mL·min-1, 紫外检测波长依据具体污染物设定.
③ 原子吸收光谱ICE分析:催化过程中从固体催化剂析出到溶液中的金属离子通过ICP-OES(Thermo ICE3500)测定.待测水样经0.45 μm滤膜过滤后再进行测定;为防止沉淀, pH较高的样品需预先酸化处理.
④ H2O2浓度分析:反应过程中每隔一定时间进行取样, 样品经0.45 μm滤膜过滤后, 立即取其滤液进行H2O2浓度测定, 方法采用前人文献报道的DPD/POD法(Schick et al., 1997).测定过程中保持每个样品与DPD和POD的作用时间在45 s左右.另外, 用超纯水和H2O(30%, 质量分数)配制不同浓度梯度的H2O2标样, 按照上述相同方法进行测定并绘制标准H2O2曲线.
⑤ 总有机碳(TOC)分析:总有机碳是指水体中有机物所含碳的总量, 本研究采用OI-1030D型总有机碳分析仪通过测定总碳(TC)和无机碳(IC), 然后获得TOC值(TOC=TC-IC).样品测定前需过0.45 μm滤膜.
⑥ 电子顺磁共振(EPR)分析:采用EMX-10/12型电子顺磁共振波谱仪进行EPR分析, 具体操作步骤如下:将1 g固体DMPO溶于8.8 mL超纯水中, 配制成待用的DMPO溶液, 并于4 ℃下保存, 防止失效;将0.05 g固体催化剂加入到1 mL水或甲醇中, 混匀后加入到100 μL H2O2和10 μL DMPO溶液中;然后将混合液吸入毛细管内, 放进EPR测量管中进行测试.值得注意的是, 本文中EPR分析不仅可以用来证明·OH和O2·-自由基的产生和存在, 而且还可以用于判断催化剂表面电子的存在状态.
⑦ 原位拉曼(in situ Raman)光谱分析:原位拉曼光谱在一定反应条件下可检测到附着于催化剂表面的过氧离子的存在, 有机物也会表现出明显的特征峰, 因此, 将拉曼分析引入到同时含有催化剂、H2O2及有机污染物的多相芬顿体系可以有效地研究其界面相互作用.
原位拉曼的样品制备方法为:将清洁的固体样品加入到去离子水或有机污染物溶液中, 再加入一定量的H2O2, 混合均匀后用胶头滴管取出滴在载玻片上, 然后迅速放置于载物台上, 结合具体实验要求测定其拉曼信号.本文中原位拉曼光谱分析采用共聚焦激光显微拉曼光谱仪(Reinishaw, UK), 激光激发波长为532 nm, 40 mW, 扫描范围为400~2000 nm, 分辨率为1 cm-1, 持续时间为120 s.
⑧ 过氧化氢(H2O2)利用率计算:过氧化氢利用率(η)是指矿化有机污染物所需的的理论H2O2消耗量与反应过程中实际测得的H2O2消耗量之间的比值([ΔH2O2]S/[ΔH2O2]A).其中, [ΔH2O2]S通过测定污染物在特定时间下TOC的降解率结合理论计算得到, 而[ΔH2O2]A则通过DPD/POD法实际测量得出反应体系中剩余H2O2量进而转换得到实际消耗H2O2量而获得.
3 结果与讨论(Results and discussion)3.1 Cu-Al2O3-g-C3N4的表征采用XRD、FTIR、EPR、ICE、SEM、in situ Raman表征分析Cu-Al2O3-g-C3N4、Cu-Al2O3、Al2O3等材料的物理化学结构及其催化机理.图 1A所示为球状介孔Cu-Al2O3的扫描电镜图片, 可见煅烧后的催化剂表面凹凸不平, 由大量互相交错组合的片状单体物组成, 整体呈现结构独立完整且颗粒大小较为均一的花球状.值得注意的是, 原位掺杂氮化碳之后, Cu-Al2O3-g-C3N4表面(图 1B)相较于Cu-Al2O3表面呈现出部分蓬松的区域, 并且Cu-Al2O3-g-C3N4的EDS元素分布中出现了新增元素N, 表明原位掺杂的g-C3N4在催化剂自组装过程中均匀地分布在催化剂结构中.
图 1(Fig. 1)
 |
| 图 1 Cu-Al2O3(A)和Cu-Al2O3-g-C3N4(B)的扫描电镜和EDS元素分布图(C, O, N, Al和Cu) Fig. 1Micrographs images of Cu-Al2O3 and element distribution of C, O, Al and Cu(A) and Cu-Al2O3-g-C3N4 and element distribution of C, O, N, Al and Cu(B) |
为进一步了解催化剂表面的官能团及催化剂的晶体结构, 对催化剂进行傅利叶红外(FT-IR)表征.分析图 2可知, 几种样品的FT-IR大体上呈现相似的特征峰分布, 在1710 cm-1处的峰归结为羰基CO的收缩振动峰, 在1513 cm-1附近的峰可以归结为碳碳双键(CC), 在1000~1450 cm-1处的峰是C—O弯曲振动峰(Sakaki et al., 1996; Baccile et al., 2017);同时, 位于3450 cm-1附近的峰是吸附在催化剂表面的水分子的羟基的伸缩振动峰.比较不同样品的峰值发现, Cu-Al2O3的OH[ν(OH)]峰位于3460 cm-1处, 而此峰在Cu-Al2O3-g-C3N4图谱上向低波数处偏移至3455 cm-1, 根据前人的报道(Miti et al., 2009), 这样的偏移证明Cu与g-C3N4表面C连接的羟基通过σ键合的形式进行成键, 形成Cu—O—C键桥.
图 2(Fig. 2)
 |
| 图 2 Al2O3、Cu-Al2O3和Cu-Al2O3-g-C3N4的傅里叶红外光谱 Fig. 2FTIR spectra of Al2O3, Cu-Al2O3 and Cu-Al2O3-g-C3N4 |
表 1(Table 1)
| 表 1 各样品的比表面积、孔容和孔径分布 Table 1 BET, pore volume and pore diameter for different samples | ||||||||||||
表 1 各样品的比表面积、孔容和孔径分布 Table 1 BET, pore volume and pore diameter for different samples
| ||||||||||||
球花型介孔Cu-Al2O3-g-C3N4的氮气吸脱附曲线和孔径分布如图 3所示.由图 3a可知, 样品的N2吸脱附等温线有两个明显的滞后环, 第一个滞后环出现在相对压力P/P0=0.4~0.8范围内, 表明合成的样品中主要存在的是介孔;第二个较小的滞后环出现在相对压力P/P0=0.8~1.0之间, 表明催化剂中存在一小部分较大的介孔.由Cu-Al2O3-g-C3N4的孔径分布图(图 3b)可知, 催化剂中的介孔孔径主要分布在3.8 nm左右, 且由氮气吸脱附等温线计算得到的Cu-Al2O3-g-C3N4比表面积为284.07 m2·g-1, 孔径为0.254 cm3·g-1.比较Cu-Al2O3的相关参数可知, 掺杂的g-C3N4同时存在于催化剂的内部表面和外部表面, 部分覆盖在催化剂原本的孔道中, 使得两者的孔容变小, 孔径分布集中数值范围偏低.
图 3(Fig. 3)
 |
| 图 3 Cu-Al2O3(a)和Cu-Al2O3-g-C3N4(c)的N2吸附-脱附等温线及Cu-Al2O3(b)和Cu-Al2O3-g-C3N4(d)的BJH孔径分布曲线 Fig. 3N2 adsorption/desorption isotherms for Cu-Al2O3(a) and Cu-Al2O3-g-C3N4(c) and pore-size distribution curves of Cu-Al2O3(b) and Cu-Al2O3-g-C3N4(d) |
为进一步探究催化剂表面的化学物质价态和元素键合情况, 对各元素进行XPS分析, 结果见图 4.由图 4a和4b可知, C 1s中位于284.6 eV和288.2 eV附近的峰分别为C—C和>C=O, 值得注意的是, Cu-Al2O3-g-C3N4中位于286.1 eV附近的峰为苯环结构中的C原子与羟基键合的特征峰(C—O—H/C—O—金属).由此得出, Cu-Al2O3-g-C3N4中在Cu与g-C3N4之间存在着C—O—Cu键桥形式.这与图 2中Cu-Al2O3-g-C3N4的FT-IR中[ν(OH)]峰的偏移情况相符合.Cu-Al2O3-g-C3N4在Cu 2p轨道上的XPS图谱均呈现3个特征峰(图 4 c), 其中, 位于932.1 eV附近的峰归结为Cu+的特征峰(Piyong et al., 2017), 而位于943.1~934.9 eV处的峰归为Cu2+, 位于941.2 eV附近的峰为Cu2+的卫星峰.俄歇动能谱可更进一步确认催化材料中Cu的物种价态(因为在结合能谱中, Cu+和Cu0的结合能峰差别很小).图 4 d为Cu-Al2O3-g-C3N4中Cu的俄歇峰谱图, 570.2~573.7 eV处的峰证明体系中确实存在Cu+而非零价铜.Al 2p轨道中75.3~75.4 eV处的结合能峰为Al—O—Cu键的特征峰, 表明Cu掺杂到了Al2O3骨架中, 此结论与Al2O3和Cu-Al2O3的分峰结果相对应.根据各元素的XPS分峰图谱分析可得, 在Cu-Al2O3-g-C3N4中, 活性物种Cu主要以二价和一价的形式存在于体系中, 并且掺杂的Cu与Al2O3之间形成了Cu—O—Al键, 而Cu又分别与g-C3N4通过Cu—O—C键桥的形式连接在一起, 为催化过程中电子的转移提供了理论支撑.
图 4(Fig. 4)
 |
| 图 4 Cu-Al2O3-g-C3N4 (a.C 1s, c.Cu 2p, e.Al 2p)和Cu-Al2O3(b.C1 s, f.Al 2p)在不同轨道上的XPS图及Cu-Al2O3-g-C3N4中Cu的俄歇能谱(d) Fig. 4XPS spectra in C 1s(a), Cu 2p (c) and Al 2p (e) for Cu-Al2O3-g-C3N4 and XPS spectra in C 1s (b) and Al 2p(f) for Cu-Al2O3 and Cu-Al2O3-g-C3N4, LMM X-ray induced Auger kinetic energy for Cu (d) |
如图 5a和5b所示, Cu-Al2O3-g-C3N4和Cu-Al2O3显示出超精细耦合结构的EPR信号, 这是自旋为I=3/2的Cu(Ⅱ)的典型特征.Cu-Al2O3-g-C3N4样品的g因子和A值如表 2所示, 其中, g|| > g⊥> 2.0023 (ge), 表明催化剂表面存在的未成对电子位于Cu(Ⅱ)的dx2-y2轨道上, 并且g因子所在数值范围及Cu-Al2O3-g-C3N4和Cu-Al2O3的EPR信号形状符合处于六配位的八面体几何结构中的Cu(Ⅱ)存在形式.通过对两种催化剂的固体EPR信号比较可知(图 5c), Cu-Al2O3-g-C3N4的信号强度低于Cu-Al2O3.这是因为在Cu-Al2O3-g-C3N4中, g-C3N4的引入使得催化剂表面的电子在阳离子π作用(电子转移路径:π→Cu)的诱发影响下进一步呈现极化分布, 促使富电子Cu周围的自由电子数量增加;由于EPR信号强度反映的是Cu周围带自由电子数量的情况, 这样, 基于Cu-Al2O3-g-C3N4的EPR信号强度均小于未掺杂的Cu-Al2O3.由此可以得出结论, Cu-Al2O3-g-C3N4中Cu周围的自由电子数量更多, 表现出更加明显的还原态.
图 5(Fig. 5)
 |
| 图 5 Cu-Al2O3-g-C3N4 (a)和Cu-Al2O3(b)的固体EPR图谱及EPR放大图谱(c) Fig. 5Solid EPR spectra of Cu-Al2O3-g-C3N4(a) and Cu-Al2O3(b) and enlargement of solid EPR spectra(c) |
表 2(Table 2)
| 表 2 EPR测定的各样品的g和A值 Table 2 EPR-measured g and A values for Cu(Ⅱ) species in various samples | ||||||||||||
表 2 EPR测定的各样品的g和A值 Table 2 EPR-measured g and A values for Cu(Ⅱ) species in various samples
| ||||||||||||
3.2 Cu-Al2O3-g-C3N4的活性和稳定性评价通过在中性pH条件下降解罗丹明B(Rh-B), 评价所合成催化剂的芬顿催化活性.在未加入H2O2的条件下, 催化反应经历30 min的吸附平衡, 分别有24.2%、24.7%和25.5%的罗丹明B(10 mg·L-1)被吸附在Al2O3、Fe-Al2O3和Cu-Al2O3表面.当加入氧化剂H2O2之后, 如图 6a所示, 在2 h内Al2O3类芬顿体系中并没有明显的罗丹明B降解, 在Fe-Al2O3体系中取得40%的降解效果, 而在Cu-Al2O3体系中, 经过90 min的降解, 罗丹明B的去除率达到90%以上, 在120 min后10 mg·L-1的罗丹明B被完全降解.同时, 掺杂不同数量Cu物种后Cu-Al2O3降解罗丹明B的曲线如图 6b所示.由图可知, 随着Cu含量由3.0%增加到7.8%, Cu-Al2O3对罗丹明B的降解效率逐渐增大, 当Cu含量增加到9.0%时, 其类芬顿降解效率反而下降.这也证明存在最佳的Cu掺杂量范围, 并且掺杂进入骨架内的Cu物种的催化效率要比骨架外的Cu物种催化效率高.综上, 本研究最终选择Cu含量为7.8%为最优掺杂比例, 并且以下实验中皆以此掺杂比例形成的花球状Cu-Al2O3为类芬顿催化剂.
图 6(Fig. 6)
 |
| 图 6 Al2O3、Fe-Al2O3和Cu-Al2O3类芬顿降解罗丹明B曲线(a)及Cu-Al2O3中不同Cu掺杂量(b)、Cu-Al2O3-g-C3N4(Cu-7.8%)中不同g-C3N4掺杂量(c)对罗丹明B降解效果的影响 (反应初始pH=7, 罗丹明B初始浓度10 mg·L-1, H2O2投加量12.5 mmol·L-1, 催化剂投加量0.5 g·L-1) Fig. 6Fenton degradation of Rh B in Al2O3, Fe-Al2O3, Cu-Al2O3 suspensions(a) and effect of the amount of the introduced Cu during the preparation of Cu-Al2O3(b) and the amount of the g-C3N4 during the preparation of Cu-Al2O3-g-C3N4(Cu-7.8%)(c) on RhB degradation (Initial pH 7, initial RhB concentration 10 mg·L-1, initial H2O2 concentration 12.5 mmol·L-1, catalyst 0.5 g·L-1) |
图 6c为不同g-C3N4掺杂量下催化剂对初始浓度为10 mg·L-1罗丹明B的降解效果.由图可知, 随着g-C3N4掺杂量由1.5%提高到6.0%, Cu-Al2O3-g-C3N4对罗丹明B的降解效果呈上升趋势, 但当掺杂量提高到9.0%时, 降解效果反而降低.这是因为过量的g-C3N4会包裹覆盖在活性物质Cu上, 从而阻碍H2O2与之接触产生活性自由基.根据降解效果最终选择含量为4.5%的g-C3N4作为最优筛选催化剂, 并且以下实验若无特殊说明, 均以此含量的Cu-Al2O3-g-C3N4(4.5%)作为反应催化剂.图 6d所示为合成的Cu-Al2O3-g-C3N4对亚甲基蓝(MB)、2, 4-二氯苯氧乙酸(2, 4-D)、双酚A(BPA)、罗丹明B(RhB)和苯妥英(PHT)这5种污染物在初始pH=7、初始污染物浓度10 mg·L-1、催化剂投加量0.5 g·L-1和H2O2浓度12.5 mmol·L-1反应条件下的降解效果曲线.由图可知, 在90 min的类芬顿降解过程, 5种污染物都表现出较好的去除率, 表明合成的催化剂能够有效地降解不同类型的难降解有机污染物.
为检验合成的催化剂Cu-Al2O3-g-C3N4的稳定性, 本研究检测了在反应pH=7条件下, 两种催化剂在不同时间段内降解BPA时的离子析出量.图 7a结果表明, Cu-Al2O3-g-C3N4在整个反应过程中Cu离子和Al离子的析出量均低于0.2 mg·L-1, 两者数据值均低于美国和欧盟在此方面的限值(2 mg·L-1).本研究同时考察了Cu2+浓度为0.35 mg·L-1时对10 mg·L-1 BPA的均相芬顿降解速率(图 7b), 经过120 min的降解, 只有不到20%的BPA被去除.这也证明在整个类芬顿反应过程中, 起主要降解作用的是固态化催化剂与双氧水发生的界面化学反应, 而均相芬顿在体系中只发挥了很小一部分作用.
图 7(Fig. 7)
 |
| 图 7 Cu-Al2O3和Cu-Al2O3-g-C3N4在降解BPA过程中Cu离子和Al离子随时间变化的析出量(a)及Cu2+浓度为0.35 mg·L-1时的均相芬顿降解10 mg·L-1 BPA的速率曲线(b) Fig. 7Metal leaching during the degradation of BPA in the Cu-Al2O3 and Cu-Al2O3-g-C3N4 suspension(a) and Fenton degradation of BPA in Cu2+(0.35 mg·L-1) (b) |
3.3 Cu-Al2O3-g-C3N4的界面电子转移机制图 8a和8b显示了在不加H2O2和污染物的情况下, DMPO捕获的各样品悬浮液中HO2·/O2·-和·OH的EPR信号.如图 8b所示, 空白和Al2O3、CuO(商用)悬浮液体系(甲醇/水体积比为9:1)没有出现任何EPR信号, 而在Cu-Al2O3、Cu-Al2O3-g-C3N4溶液体系中观测到了DMPO-HO2·/O2·-信号明显的6个特征峰(强度比1:1:1:1:1:1), 这说明Cu-Al2O3中具有大量单电子的富电子Cu中心可将溶液中的溶解氧(O2)还原为O2·-.同时, 图谱中也出现了与超氧自由基重叠在一起的峰, 这是捕获剂DMPO与O2·-反应产生的碳中心自由基特征峰.
图 8(Fig. 8)
 |
| 图 8 DMPO自由基捕获实验(a.在甲醇体系中捕获HO2·/O2·-(没有H2O2), b.在水溶液中捕获·OH(没有H2O2), c.在H2O2存在的条件下在水溶液中捕获·OH, d.在H2O2存在的条件下在甲醇体系中捕获HO2·/O2·-) Fig. 8DMPO spin-trapping EPR spectra for HO2·/O2·- in various methanol dispersions without H2O2(a), ·OH in various methanol dispersions(b), ·OH in various aqueous suspensions(c) and HO2·/O2·- in various methanol dispersions in the presence of H2O2(d) |
值得注意的是, 如图 8b所示, 采用EPR检测催化剂在水溶液中的DMPO-·OH信号(不含H2O2和污染物), Cu-Al2O3、Cu-Al2O3-g-C3N4均显示出明显的·OH信号(强度比为1:2:2:1), 而此特征峰没有在其他体系中出现.可以推测在没有加入H2O2和污染物情况下的Cu-Al2O3、Cu-Al2O3-g-C3N4水溶液体系中, 溶液的O2从富电子Cu中心获取电子的同时, 就已经存在物质可以充当电子供体来维持催化剂中电子的得失平衡.由于体系中只存在催化剂、H2O和水中溶解氧, 催化剂充当了传递电子的载体, 水中溶解氧充当着电子的受体, 那么电子的供体源只能是H2O, 这也就说明Cu-Al2O3、Cu-Al2O3-g-C3N4的缺电子中心可以将水氧化为·OH.
图 8c和8d显示了加入H2O2后, DMPO捕获的各样品悬浮液中·OH和HO2·/O2·-的EPR信号.在水体系中, 羟基自由基信号大小呈现Cu-Al2O3-g-C3N4>Cu-Al2O3>CuO的顺序, 而在甲醇和水体系中, ·OH和HO2·/O2·-的信号呈现相反的顺序, 为CuO>Cu-Al2O3>Cu-Al2O3-g-C3N4.另外, 除·OH之外的6条等高线根据出峰的相对位置确认为·OH进攻含碳化合物(DMPO)时产生的碳中心自由基.这种现象表明, 在存在双反应催化活性中心的3个催化反应体系中, H2O2取代了水中溶解的O2而主要被富电子Cu中心的未成对电子选择性还原为·OH, 仍然有小部分的HO2·/O2·-被检测到可能是部分O2在铜中心还原或者少量H2O2在缺电子中心被氧化所致.而在CuO体系中, H2O2主要遵从于经典芬顿反应机制进行分解, 即H2O2一方面被低价态金属物种还原产生·OH, 还有部分H2O2被高价态金属氧化为O2·-和O2.值得注意的是, 在Cu-Al2O3-g-C3N4中, 由于存在着Cu与g-C3N4之间的阳离子-π作用, 使得电子从π体系转移至Cu附近(Lagutschenkov et al., 2010), 这样在Cu-Al2O3晶格氧诱发产生电子极化双反应活性中心分布的基础上, 实现了进一步诱发形成缺电子π体系和富电子Cu中心, 使得上述的H2O2选择性转化反应更加明显.因此, Cu-Al2O3-g-C3N4的信号强度相较于Cu-Al2O3更加明显.
图 9展示了Cu-Al2O3-g-C3N4在不同悬浮液体系下的原位拉曼光谱图.在前人的报道中(Lyu et al., 2017), Al2O3悬浮液中无论是否加入H2O2, 都不能观测到位于875 cm-1处的H2O2的特征吸附峰.在图 9中, 只有催化剂和水或者催化剂和污染物BPA存在的悬浮液中只能检测到仪器本身的信号.但当加入H2O2之后, 在875~878 cm-1范围内都出现一个特征吸附峰, 这是由于H2O2与材料表面金属物种络合后O—O伸缩振动所导致的(Silvia et al., 2002).值得注意的是, 当悬浮液体系中加入BPA之后, 这个特征峰的强度有所降低.根据前人报道(Lyu et al., 2015), 催化剂表面的Cu(Ⅱ)能够与BPA中的酚羟基脱质子后的氧原子形成络合中间体σ-Cu2+-ligand, 电子在这中间以π→Cu(Ⅱ)(σ供体)的转移路径来实现提高芳环的电子极化率.形成的σ-Cu2+-ligand络合体会诱发H2O2与有机配位体发生反应.有机配体的π电子传递给螯合的Cu2+, 使之被还原为Cu+, 自身发生羟基化反应产生羟基自由基和羟基化产物(Lagutschenkov et al., 2010), 从而提高了体系中·OH的产生量和H2O2的利用率.因此, 加入污染物质BPA后的特征峰峰强有所降低.
图 9(Fig. 9)
 |
| 图 9 样品在Cu-Al2O3-g-C3N4不同体系中的原位拉曼图谱 Fig. 9In situ Raman spectra for various aqueous dispersions of Cu-Al2O3-g-C3N4 |
3.4 羟基自由基的高效产生图 10为Cu-Al2O3-g-C3N4的芬顿催化降解机理图.对于Cu-Al2O3-g-C3N4, 由于Cu的电负性大于Al的电负性, Cu的掺杂导致晶格O2-的电子云呈现非均匀分布, 进而导致催化剂表面电势差的产生, 形成富电子Cu中心和缺电子Al中心.同时, 由于引入的g-C3N4具有类苯环结构的C和N以sp2杂化形式相间排列, 具有π共轭体系, 能够与金属氧化物中的Cu通过阳离子-π键(C—O—Cu键桥)形成络合, 并且以“π→Cu”的电子迁移路径进一步形成电子的不均匀分布, 强化富电子Cu中心周围的电子, 并产生一个新的缺电子π体系.也就是说, 在Cu-Al2O3-g-C3N4中, 存在一个富电子中心(Cu)和两个缺电子中心(Al和π共轭体系).由于H2O2分子的特殊结构, 既可作为亲电试剂也可作为亲核试剂, 这样较大部分的H2O2选择性地吸附在这两个中心位点.在体系中存在H2O2时, 溶液中的H2O会占据催化剂缺电子中心, 失去电子形成·OH, 而在催化剂表面的富电子中心处, 更多的是发生H2O2得电子还原作用, 产生·OH.这样就实现了H2O2的选择性还原高效率产生活性物种·OH.
图 10(Fig. 10)
 |
| 图 10 Cu-Al2O3-g-C3N4的芬顿降解机理图 Fig. 10The Fenton-like reaction mechanism on the surface of Cu-Al2O3-g-C3N4 |
为了更好地评价Cu-Al2O3-g-C3N4在芬顿反应过程中H2O2的利用率和转化率, 本研究通过POD/DPD法检测反应过程中H2O2的消耗量, 通过对苯二甲酸(TPA)探针法定量测定Cu-Al2O3-g-C3N4与H2O2反应时·OH的产量(Bordiga et al., 2003).如图 11a所示, 在反应的初始120 s内, 体系产生的·OH浓度大约是H2O2消耗量的1.5倍左右, 随着时间的延长, ·OH浓度与H2O2消耗量之间的比值逐渐下降, 但在前600 s内, 该比值仍然大于1.而在600 s以后, ·OH产量呈现出急剧下降的趋势.这是因为在反应的初始阶段TPA的浓度远大于·OH的浓度, ·OH能够被高浓度的TPA大量捕获.但随着反应的进行, 产生的·OH会与体系中的其他物质(TPA被降解的中间产物、·OH本身及H2O2)发生反应, 导致测量值低于实际值.本文之前的实验已经证实了Cu-Al2O3-g-C3N4催化还原H2O2的活性位点是富电子Cu中心, 根据前人报道可知(Lyu et al., 2017), 其数量可以依据表面铜物种的浓度而确定.通过苯酚(phenol)在Cu-Al2O3-g-C3N4的基底Cu-Al2O3的单层吸附以获得表面铜物种与苯酚的络合量, 进而可以等效转化估算表面Cu中心的量.图 11b是Cu-Al2O3对于苯酚的Langmuir吸附等温线, 计算公式见式(2), 其平衡吸附量为qe=0.61×10-5 mol·g-1, 等效为Cu-Al2O3-g-C3N4和表面的Cu中心的数量N(surf.).而·OH的转化率(TOF)可以定义为催化剂表面每个活性中心上每秒内·OH的转化常数, 计算公式详见式(3), 其中, NA为阿伏伽德罗常数, C(catal.)为催化剂的浓度(mg·L-1), V(·OH)为反应前20 s的·OH浓度变化(mol·L-1·s-1).经过计算得出在Cu-Al2O3-g-C3N4和Cu-Al2O3体系中, ·OH产生的TOF分别为0.516 s-1和0.380 s-1.由于实际反应体系中存在着自由基链式反应, 根据实验检测和一系列计算得出的数值必然低于实际值, 但此数值仍然是传统均相芬顿体系TOF(1.53×10-2 s-1)的30倍以上(Huang et al., 2015).
图 11(Fig. 11)
 |
| 图 11 TPA法检测不同反应体系中产生的·OH浓度(a)及Cu-Al2O3的对苯酚的吸附等温线图(b) Fig. 11The measurement of the generation of ·OH using the TPA method in different catalytic system (a) and adsorption isotherms of phenol on Cu-Al2O3(b) |
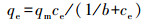 | (2) |
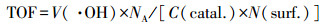 | (3) |
| 表 3 不同样品在类芬顿反应中矿化20 mg·L-1的BPA实际消耗和理论计算所需的H2O2量及H2O2利用率 Table 3 Actual H2O2 consumption and stoichiometric H2O2 consumption for mineralizing BPA (20 mg·L-1) and the utilization of H2O2 during the Fenton-like reaction | ||||||||||||||||||||||||||
表 3 不同样品在类芬顿反应中矿化20 mg·L-1的BPA实际消耗和理论计算所需的H2O2量及H2O2利用率 Table 3 Actual H2O2 consumption and stoichiometric H2O2 consumption for mineralizing BPA (20 mg·L-1) and the utilization of H2O2 during the Fenton-like reaction
| ||||||||||||||||||||||||||
H2O2利用率是指芬顿反应中矿化污染物所需要的理论H2O2消耗量值与实际测定值之间的比值.在120 min的降解时间内, Cu-Al2O3-g-C3N4、Cu-Al2O3和CuO对BPA的矿化度依次为72.3%、59.7%和23.8%.根据矿化度计算得出几种催化剂在芬顿催化过程中H2O2的利用率分别为64%、47%和28%.比较可发现, 具有双反应活性中心的Cu-Al2O3对H2O2的利用率明显高于CuO, 表明形成的电子密集区域和电子缺乏区域确实实现了对H2O2的选择性转化;而通过引入g-C3N4以π→阳离子(C—O—Cu键桥)形式强化了富电子Cu中心, 伴随新的缺电子π中心的产生.因此, 在有机配体络合的强化作用下, Cu-Al2O3-g-C3N4对H2O2选择性还原的性能和H2O2的有效利用率也进一步提升.
4 结论(Conclusions)1) 采用SEM、XRD、XPS、EPR、Raman等手段对Cu-Al2O3-g-C3N4、Cu-Al2O3等颗粒进行表征, 结果表明, Cu-Al2O3-g-C3N4具有大比表面积的球花状完整结构, 催化剂单体颗粒尺寸在10 μm左右, 活性物质铜以晶格氧掺杂形式主要存在于体相中, 与Al2O3键合形成Cu—O—Al键(此为第一个双反应活性中心).原位引入的g-C3N4以有机配体的形式与Cu以σ型Cu—O—C键合(形成第二个双反应活性中心).两种表面电子极化手段使得Cu中心周围产生单电子, 与之对应的产生缺电子Al中心和缺电子π体系.
2) 以罗丹明B为探针筛选出Cu-Al2O3-g-C3N4中最佳g-C3N4掺杂比为4.5%.以BPA为指示剂探究不同催化材料的类芬顿反应活性, 分别构建CuO/H2O2、Cu-Al2O3/H2O2和Cu-Al2O3-g-C3N4/H2O2体系, 对5种目标污染物的降解实验证明, Cu-Al2O3-g-C3N4能够有效地降解不同类型的难降解有机污染物.
3) 采用DMPO-EPR捕获自由基实验探究体系中电子转移路径, 结果表明, 在没有加入H2O2和污染物情况下, 在Cu-Al2O3、Cu-Al2O3-g-C3N4水溶液体系中, 富电子Cu中心将自由电子传递给O2(还原为HO2·/O2·-), H2O作为电子供体被催化剂缺电子中心氧化为·OH, 从而实现催化剂中电子的得失平衡;在H2O2存在情况下, H2O2取代溶解O2而主要被富电子Cu中心选择性还原为·OH, 并且比较·OH信号强度可知, 有机配体络合强化双反应活性中心可进一步促进羟基自由基的产生.
4) ·OH的转化率(TOF)实验结果发现, Cu-Al2O3-g-C3N4和Cu-Al2O3体系的TOF分别为0.516 s-1和0.380 s-1, 远高于传统均相芬顿体系;H2O2利用率计算结果表明, Cu-Al2O3-g-C3N4、Cu-Al2O3和CuO的H2O2利用率分别为72.3%、59.7%和23.8%.这表明构建双反应活性中心的确可以提升对H2O2的选择性还原性能和有效利用.
5) 对构建双反应活性中心Cu-Al2O3-g-C3N4类芬顿反应催化活性作用机制进行分析与总结:催化剂固相介孔结构发达、活性物质暴露较多且分布均匀.晶格氧掺杂和表面有机配体络合两种方式强化了催化剂表面电子极化分布, 使得H2O2主要以其亲电端吸附在富电子Cu中心上, 获得电子并被还原为·OH;并在一定程度上避免了H2O2的氧化而促进其还原反应, 体系中H2O2被较大限度地用于降解有机污染物.
参考文献
| Baccile N, Laurent G, Coelho C, et al. 2011. Structural insights on nitrogen-containing hydrothermal carbon using solid-state magic angle spinning 13C and 15N nuclear magnetic resonance[J]. Journal of Physical Chemistry C, 115(18): 8976-8982. DOI:10.1021/jp2015512 |
| Bokare A, Choi W. 2014. Review of iron-free Fenton-like systems for activating H2O2 in advanced oxidation processes[J]. Journal of Hazardous Materials, 275: 121-135. DOI:10.1016/j.jhazmat.2014.04.054 |
| Bordiga S, Damin A, Bonino F, et al. 2003. Resonance Raman effects in TS-1:the structure of Ti(IV) species and reactivity towards H2O, NH3 and H2O2:an in situ study[J]. Physical Chemistry Chemical Physics, 5(20): 4390-4393. DOI:10.1039/b306041c |
| Huang X, Hou X, Zhao J, et al. 2016. Hematite facet confined ferrous ions as high efficient fenton catalysts to degrade organic contaminants by lowering H2O2 decomposition energetic span[J]. Applied Catalysis B:Environmental, 181: 127-137. DOI:10.1016/j.apcatb.2015.06.061 |
| 韩金栋, 蒋进元, 李君超, 等. 2020. 纳米Fe/Co催化降解土霉素效果及影响因素研究[J]. 环境科学研究, 33(10): 1001-6929. |
| 李民, 王颖, 冉刚, 等. 2019. 微波强化铁碳-双氧水体系处理填埋场渗滤液膜滤浓缩液研究[J]. 环境科学学报, 39(7): 2143-2151. |
| Lagutschenkov A, Sinha R K, Maitre P, et al. 2010. Structure and infrared spectrum of the Ag+-Phenol ionic complex[J]. Journal of Physical Chemistry A, 114(42): 11053-11059. DOI:10.1021/jp100853m |
| Lyu L, Han M, Cao W, et al. 2019. Efficient fenton-like process for organic pollutant degradation on cu-doped mesoporous polyimide nanocomposites[J]. Environmental Science, Nano, 6: 798-808. DOI:10.1039/C8EN01365A |
| Lyu L, Zhang L, He G, et al. 2017. Selective H2O2 conversion to hydroxyl radicals in the electron-rich area of hydroxylated C-g-C3N4/CuCo-Al2O3[J]. Journal of Materials Chemistry A, 5: 7153-7164. DOI:10.1039/C7TA01583F |
| Lyu L, Zhang L, Hu C. 2016. Galvanic-like cells produced by negative charge nonuniformity of lattice oxygen on d-TiCuAl-SiO2 nanospheres for enhancement of Fenton-catalytic efficiency[J]. Environmental Science:Nano, 3: 1483-1492. DOI:10.1039/C6EN00290K |
| Lyu L, Zhang L, Wang Q, et al. 2015. Enhanced Fenton catalytic efficiency of γ-Cu-Al2O3 by σ-Cu2+-ligand complexes from aromatic pollutant degradation[J]. Environmental Science &Technology, 49: 8639-8647. |
| Ma J, Yang Q, Wen Y, et al. 2017. Fe-g-C3N4/graphitized mesoporous carbon composite as an effective Fenton-like catalyst in a wide pH range[J]. Applied Catalysis B:Environmental, 201: 232-240. DOI:10.1016/j.apcatb.2016.08.048 |
| Miti Z, Nikoli G S, Caki M, et al. 2009. FTIR spectroscopic characterization of Cu(Ⅱ) coordination compounds with exopolysaccharide pullulan and its derivatives[J]. Journal of Molecular Structure, 924-926: 264-273. DOI:10.1016/j.molstruc.2009.01.019 |
| Nichela D A, Berkovic A M, Costante M R, et al. 2013. Nitrobenzene degradation in Fenton-like systems using Cu(Ⅱ) as catalyst.Comparison between Cu(Ⅱ)- and Fe(Ⅲ)-based systems[J]. Chemical Engineering Journal, 228: 1148-1157. DOI:10.1016/j.cej.2013.05.002 |
| Sakaki T, Shibata M, Miki T, et al. 1996. Reaction model of cellulose decomposition in near-critical water and fermentation of products[J]. Bioresource Technology, 58(2): 197-202. DOI:10.1016/S0960-8524(96)00099-5 |
| Schick R, Strasser I, Stabel H H J W R. 1997. Fluorometric determination of low concentrations of H2O2 in water:Comparison with two other methods and application to environmental samples and drinking-water treatment[J]. Water Research, 31(6): 1371-1378. DOI:10.1016/S0043-1354(96)00410-1 |
| Silvia B, Alessandro D, Francesca B, et al. 2002. The structure of the peroxo species in the TS-1 catalyst as investigated by Resonant Raman Spectroscopy[J]. Angewandte Chemie International Edition, 41(24): 4734-4737. DOI:10.1002/anie.200290032 |
| Song W, Cheng M, Ma J, et al. 2006. Decomposition of hydrogen peroxide driven by photochemical cycling of iron species in clay[J]. Environmental Science & Technology, 40(15): 4782-4787. |
| Wang W, An T, Li G, et al. 2017. Earth-abundant Ni2P/g-C3N4 lamellar nanohydrids for enhanced photocatalytic hydrogen evolution and bacterial inactivation under visible light irradiation[J]. Applied Catalysis B-Environmental, 217: 570-580. DOI:10.1016/j.apcatb.2017.06.027 |
| Yuan Z, Xiao L. 2012. Heterogeneous UV/Fenton degradation of TBBPA catalyzed by titanomagnetite:Catalyst characterization, performance and degradation product[J]. Water Research, 46: 4633-4644. DOI:10.1016/j.watres.2012.06.025 |
| Zhang P, Song T, Wang T, et al. 2017. In-situ synthesis of Cu nanoparticles hybridized with carbon quantum dots as a broad spectrum photocatalyst for improvement of photocatalytic H2 evolution[J]. Applied Catalysis B:Environmental, 206: 328-335. DOI:10.1016/j.apcatb.2017.01.051 |
| Zhang Y, Pan Q, Chai G, et al. 2013. Synthesis and luminescence mechanism of multicolor-emitting g-C3N4 nanopowders by low temperature thermal condensation of melamine[J]. Scientific Reports, 3: 1943. DOI:10.1038/srep01943 |
