 , 李孟1, 唐建伟1,2, 张倩1
, 李孟1, 唐建伟1,2, 张倩1
1. 武汉理工大学市政工程系, 武汉 430070;
2. 中国人民解放军 95338部队, 广州 510405
收稿日期: 2019-03-13; 修回日期: 2019-05-02; 录用日期: 2019-05-02
基金项目: 中央高校基本科研业务费专项资金资助(No.2019-zy-130,2019Ⅲ105CG)
作者简介: 许入义(1995-), 女, E-mail:m13855148713@163.com
通讯作者(责任作者): 张倩, E-mail:qianzhang@whut.edu.cn
摘要: 以3,4-二甲基苯胺(3,4-dimethylaniline,3,4-DMA)为苯胺类污染物的代表,通过降解贡献度分析、不同影响因素下动力学公式的建立及中间产物分析来探究其在高盐光电催化氧化体系下的同步耦合反应机制.结果表明,氯类活性物质与羟基自由基对3,4-DMA 10 min降解的贡献率分别为87.55%和7.9%,UV直接氧化、阳极直接氧化、光生空穴直接氧化的作用均可忽略.在不同的初始pH值(A)、初始氯化钠浓度(B)、电流密度(C)、光照强度(D)条件下,光电体系中的光催化与电催化反应均表现出协同效应,且电催化反应占主导.4种影响因素可通过影响氯类活性物质与羟基自由基的产生来对耦合机制产生作用.根据所得动力学公式ln(c0/ct)=0.0042A-0.2422B0.3378C1.4742D0.2300t,4种因素对反应体系的作用强弱顺序为:电流密度(C)>初始氯化钠浓度(B)>初始pH值(A)>光照强度(D).基于GC/MS对中间产物的检测结果,提出3,4-DMA的两种可能降解路径:①3,4-DMA依次降解为3,4-二甲基苯酚、邻二甲苯、邻甲基苯甲酸、甲苯、苯甲醛、烷烃类物质、CO2和H2O;②3,4-DMA依次降解为含N联苯类物质、脂肪酸类物质、CO2和H2O.
关键词:3, 4-二甲基苯胺(3, 4-DMA)光电催化氧化耦合机制动力学降解路径
Study on the reaction mechanism of photoelectrocatalytic oxidation of aniline contaminants
XU Ruyi1
, LI Meng1, TANG Jianwei1,2, ZHANG Qian1 1. Department of Civil Engineering, Wuhan University of Technology, Wuhan 430070;
2. 95338 Troops of the Chinese People's Liberation Army, Guangzhou 510405
Received 13 March 2019; received in revised from 2 May 2019; accepted 2 May 2019
Abstract: On the basis of the analysis of degradation contribution, establishment of kinetic formulas under different influencing factors, and analysis of intermediates, the synchronous coupling degradation mechanism of 3, 4-dimethylaniline (3, 4-DMA), a representative aniline pollutant, in high salt photoelectrocatalytic system was investigated. Results indicated that chlorine active substances and hydroxyl radical respectively contributed 85% and 11% to the degradation rate of 3, 4-DMA in 10 minutes, and the degradation of 3, 4-DMA by the direct oxidation of UV, anode and photogenerated holes was negligible. Under different conditions of initial pH values (A), initial sodium chloride concentration (B), current density (C), and light intensity (D), both photocatalysis and electrocatalysis posed a synergistic effect on the degradation of 3, 4-DMA and the contribution of electrocatalysis was dominant. The four factors could have certain impact on the coupling mechanism through affecting the production of chlorine active substances and hydroxyl radical. According to the obtained kinetic formula ln(c0/ct)=0.0042A-0.2422B0.3378C1.4742D0.2300t, the influence of the four factors followed the order:current density (C) > initial sodium chloride concentration (B) > initial pH (A) > light intensity (D). The degradation intermediates were detected via GC-MS analysis, and two possible 3, 4-DMA degradation pathways were proposed as follows:① 3, 4-DMA being sequentially degraded to 3, 4-dimethylphenol, o-xylene, o-methylbenzoic acid, toluene, benzaldehyde, alkane, CO2 and H2O; ②3, 4-DMA being sequentially degraded to N-containing biphenyls, fatty acids, CO2 and H2O.
Keywords: 3, 4-dimethylaniline (3, 4-DMA)photoelectrocatalytic oxidationcoupling mechanismkineticsdegradation pathway
1 引言(Introduction)3, 4-二甲基苯胺(3, 4-dimethylaniline, 3, 4-DMA)是一种苯胺类污染物, 含3, 4-DMA的废水往往具有高盐、难降解和致癌等特点.因此, 3, 4-DMA被美国EPA列为优先控制的129种污染物, 也被列入“中国环境优先污染物黑名单”中(周文敏等, 1990).目前, 关于3, 4-DMA废水处理工艺的相关报道十分少见(杨唯艺等, 2018), 而同类型苯胺类污染物的常见处理方法有吸附、膜分离、生物降解及絮凝沉降等(Gómez et al., 2009), 但这些方法因为处理能力有限或投资成本过高而在实际应用时受到限制.
基于光电催化的高级氧化技术因具有适用范围广、处理迅速彻底且不产生二次污染物等优点, 近年来一直被广泛关注(范崇政等, 2001).Ronaldo等(1999)采用DSA电极(Ti/Ru0.3Ti0.7O2)电化学辅助的光催化电极降解活性蓝19(C. I. Reactive Blue 19), 发现120 min时活性蓝19几乎全部降解.Wen等(2013)在研究微囊藻毒素(MC-LR)光电催化降解实验中发现, 起氧化作用的活性物质主要是光激发二氧化钛产生的空穴, 即羟基自由基和氧负离子.Yang等(2013)采用同位素H2O18证实在光电过程中产生的羟基自由基主要通过光生空穴吸附氢氧根或水产生的.传统的光电催化工艺是指将半导体光催化剂粉末固定在导电的金属上, 同时将固定后的催化剂作为工作电极, 在一定的光源照射下, 采用外加恒电流或恒电压的方法迫使光生电子对向电极方向移动, 从而与光生空穴发生分离的一种技术(Ma et al., 2014).该方法可有效抑制电子与空穴的复合, 提高光催化的量子化效率.但由于光电极极板面积与厚度有限, 产生的空间电荷层数量与光生量子的数量十分有限, 无法对高浓度、大流量的有机物进行有效降解(Waldner et al., 2003).有****(刘艳彪, 2011)设计了基于TiO2纳米管阵列电极的光催化燃料电池体系, 并将其用于对0.22 mmol·L-1四环素的降解研究, 结果表明, 在反应8 h时几乎实现了四环素的全部降解.但该体系仅能针对低浓度、小体积和间歇式的有机物进行降解, 且反应耗时长.为克服传统光电催化工艺的弊端, 本文研发了一种新型的光电耦合催化氧化工艺, 将悬浮态的纳米二氧化钛(TiO2)光催化剂和紫外光作为光催化子系统, 钛基二氧化铱阳极(Ti/IrO2)和石墨阴极作为电催化子系统, 两个子系统在同一个混沌体系中进行催化氧化反应.电催化析氧副反应产生的氧气可为光催化反应提供电子受体, 电极仍可促进光生电子与空穴的分离, TiO2由于悬浮在溶液中, 光生量子的产生数量不再受限, 且悬浮的TiO2不受传质控制, 极大地提高了污染物的降解速率.而紫外光照不仅能促进羟基自由基的产生, 且会激发3, 4-DMA及其中间产物, 使得它们更容易被氧化降解, 同时提高了光催化与电催化的反应速率.
因此, 本文通过降解贡献度分析、各因素对耦合机制的影响及中间产物的检测与分析来重点研究所建立的光电催化体系降解3, 4-DMA时的耦合机制.以反应速率常数kobs值与氯类活性物质的浓度变化量为参考, 对在不同反应条件下析氧副反应对光电体系中光催化与电催化反应的不同作用、外加电流对羟基自由基产量的影响、紫外光照对电催化反应的影响进行详细讨论.以期找到在不同初始pH值、初始氯化钠浓度、电流密度、光照强度作用下光电体系中光催化与电催化不同的耦合机制, 并对4种因素作用的强弱进行排序.最后, 结合3, 4-DMA降解中间产物的分析, 对光电体系降解苯胺类污染物的耦合机制进行详细的解释说明.
2 材料与方法(Materials and methods)2.1 仪器与试剂仪器:直流稳压电源(RXN305D, 深圳市兆信电子科技有限公司); 分光光度计(UV-1100, 上海美谱达仪器有限公司); 旋转蒸发仪(RV10, 艾卡仪器设备有限公司); GC-MS分析仪(Agilent, 安捷伦科技有限公司); 精密pH计(PB-10, 北京赛多利斯仪器系统有限公司).
试剂:3, 4-二甲基苯胺(3, 4-DMA)、氯化钠、硫酸氢钾、亚硝酸钠、硫酸钠、氨基磺酸铵、95%乙醇、氢氧化钠、盐酸、叔丁醇均为分析纯, 二氯甲烷为色谱纯, 均购自国药集团化学试剂有限公司; 愈创木酚为分析纯, 购自麦克林生化科技有限公司; P25纳米二氧化钛(TiO2)购自广州合仟科技有限公司; Ti/IrO2阳极和石墨阴极购自苏州舒尔泰工业科技有限公司.
2.2 实验方法2.2.1 3, 4-DMA降解实验所有反应溶液的3, 4-DMA浓度均为5 mg·L-1, TiO2投加量为300 mg·L-1, 实验温度为室温.按光催化实验要求加入TiO2光催化剂暗室搅拌30 min, 用去离子水清洗阳极板和阴极板2遍, 开启紫外灯, 开启直流电源开始光电耦合催化氧化实验.实验时间10 min, 间隔2 min, 用注射器取样, 再将水样通过0.22 μm聚醚矾膜(PES, 津腾)过滤, 以去除水体中的TiO2颗粒.
2.2.2 中间产物分析实验按照与降解实验相同的方法与步骤, 用二氯甲烷(DCM)有机溶剂对样品萃取3次, 每次50 mL, 再合并有机相, 用无水硫酸钠干燥过夜.过滤后用德国艾卡RV10旋蒸仪进行旋蒸浓缩, 蒸馏浓缩至5 mL后, 依次关闭冷凝回流装置、旋转马达和加热器, 最后减压, 打开蒸馏瓶取样, 用GC/MS气相质谱联用仪对降解中间产物进行分析.
2.3 分析方法3, 4-DMA测定采用甲氧基苯酚分光光度法(杨晓芬等, 2002), 在456 nm处测量.氯离子浓度采用离子色谱仪测定, 用AgNO3滴定法(GB11896-89)校核.自由氯浓度采用国标法《水质游离氯和总氯的测定N, N-二乙基-1, 4-苯二胺分光光度法》(HJ586- 2010)测定.3, 4-DMA的降解中间产物分析在美国Entech公司-美国Agilent公司的GC/MS气质联用仪上进行, 分析条件如下:气相色谱条件色谱柱为Agilent 222-5532LTM DB-5ms (G3900-63004), 载气为高纯He, 柱流量为1.0 mL·min-1, 模式为分流, 初始温度为250 ℃(打开), 压力为7.63 psi(打开); 柱温为280 ℃, 质谱离子源温度为230 ℃, 四极杆温度为150 ℃, 采用正离子化采集模式, 30 psi.
3 结果与讨论(Results and discussion)3.1 3, 4-DMA降解贡献度分析在光电反应体系中, 3, 4-DMA可通过UV直接氧化、空穴直接氧化、羟基自由基氧化、阳极直接氧化、氯类活性物质氧化5个途径被降解.为了进一步讨论各反应途径对3, 4-DMA降解的贡献度, 进行了如图 1所示的各组实验.
图 1(Fig. 1)
 |
| 图 1 各降解途径贡献度分析 Fig. 1Analysis of the contribution of each degradation pathway |
由图 1可知, 仅含TiO2催化剂的反应体系中3, 4-DMA降解率小于0.2%, TiO2的吸附作用可忽略.对以硫酸钠支持的单光和单电系统均不投加TiO2粉末, 其最大降解率分别为1.1%和2.5%, 10 min时降解率贡献度分别为0.75%与1.6%.由于硫酸根离子的惰性, 其在光催化与电催化系统中不参与氧化反应.因此, 通过降解率可说明UV直接氧化与阳极直接氧化作用效果微弱, 可忽略.叔丁醇(Tert-butyl alcohol, TBA)是一种小分子的醇类物质, 可以与羟基自由基发生反应(反应速率常数K=1.9×109 L·mol-1·s-1), 虽然从热力学上看, 其与光生空穴也存在反应的可能性, 但由于醇类水溶性极强, 很难与催化剂表面的光生空穴直接反应(Miguel et al., 2016), 故以50 mL·L-1 TBA作为羟基自由基的抑制剂.比较加入TBA与未加入TBA的光电耦合系统发现, 20 min之前降解率差值小于5%, 10 min时降解率贡献度为7.9%, 说明在本反应体系中通过羟基自由基氧化而降解的3, 4-DMA所占比重较小.同时, 在加入50 mL·L-1 TBA的基础上, 将电解质替换为硫酸钠, 3, 4-DMA在10 min时的降解率为4.55%.此部分降解率为UV直接氧化、阳极直接氧化、空穴直接氧化的共同作用, 除掉前两者的贡献度, 空穴直接氧化在10 min时降解率的占比为2.2%, 其作用微弱, 可忽略.当仅将氯化钠电解质变为硫酸钠时, 降解率下降十分明显, 10 min时由100%降为12.45%, 氯类活性物质的贡献度为87.55%, 说明氯类活性物质的氧化作用在3, 4-DMA降解过程中占据绝对的主导地位.因此, 在光电耦合催化氧化高盐3, 4-DMA降解过程中, 氯类活性物质发挥了最大的作用, 羟基自由基氧化作用贡献较小, 而TiO2吸附、UV直接氧化、阳极直接氧化、空穴直接氧化作用均可忽略.
3.2 各影响因素对耦合机制的作用3.2.1 反应动力学方程的建立3, 4-DMA的降解过程符合准一级模型, 则其反应速率可表示为式(1).对等式两边同时积分, 并将t=0时, C=C0代入, 则可得式(2).
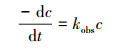 | (1) |
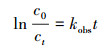 | (2) |
将初始pH值(A)、初始氯化钠浓度(B)、电流密度(C)、光照强度(D)这4个主要影响因素作为自变量, 建立kobs与4种影响因素的关系式:
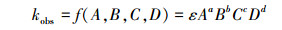 | (3) |
3.2.2 初始pH值的影响当其它试验条件保持不变时, 设置溶液初始pH=3.0、6.4和9.0, 分别建立单光、单电与光电系统的动力学公式来探讨不同初始pH值对耦合机制的影响.从图 2可以看出, 随着pH值的改变, 单电与单光系统的速率常数变化呈现完全相反的趋势, 单光系统在碱性条件下反应速率最快, 而单电系统则是酸性条件下反应速率最快.对于光电系统, 其kobs值均大于同条件下单光与单电体系之和, 说明在不同pH值作用下光电系统中的光催化与电催化反应存在协同效应.光电系统反应速率变化趋势与单电系统保持一致, 说明电催化反应在光电系统中占主导, 但由于光电系统中电催化反应抵消了一部分光催化反应的反向作用, 反应速率总体变化幅度小于单电系统.
图 2(Fig. 2)
 |
| 图 2 初始pH值对反应速率的影响 Fig. 2The effect of initial pH on reaction rate |
进一步分析光催化反应发现, 在酸性条件下3, 4-DMA以质子化存在, TiO2表面带正电, 同性相斥(迪安, 2003), 3, 4-DMA难以吸附到TiO2表面, 酸性条件对光催化反应不利.为了更好地讨论pH值对耦合机制的影响, 分别对单光、单电及光电体系中的溶解氧进行了测定.对于存在电催化反应的体系, 析氧副反应4OH--4e→O2+H2O是影响反应速率的一个重要因素(Waldner et al., 2003), 而溶液中的溶解氧含量可用来间接判断析氧副反应进行的程度.由图 3可知, 单电与光电体系的溶解氧含量在碱性条件最大, 中性条件次之, 酸性条件最小.原因为酸性条件下Ti/IrO2电极的析氧电位更高, 析氧副反应受到抑制, 溶液中由析氧副反应产生的溶解氧含量也随之下降(Cui et al., 2012); 碱性条件则恰好相反.总的来看, 光电体系的溶解氧含量低于单电体系, 原因可能为在光电系统中析氧副反应产生的氧气被光催化反应利用产生羟基自由基(张丽, 2014), 使得体系中溶解氧值有一定的下降.
图 3(Fig. 3)
 |
| 图 3 不同反应体系中的溶解氧值 Fig. 3Dissolved oxygen values of different reaction systems |
根据3.1节所得结论, 氯类活性物质对3, 4-DMA降解的贡献度高达87.55%, 其中, 氯类活性物质的种类主要为自由氯、氯自由基及氯类氧化物, 且在以氯化钠为电解质的光电催化反应中氯离子变为氯类活性物质是其主要的转化途径(秦微等, 2015).若能对各类氯类活性物质的浓度进行定量测定, 则能更好地反映各类影响因素对耦合机制的作用.但氯自由基寿命极短, 难以准确对其进行定量测定(Varanasi et al., 2018), 而氯类氧化物种类复杂, 要准确进行定性与定量更是难以实现.基于以上原因, 本文参照国标法对溶液中的氯离子与自由氯浓度进行了测定, 希望通过氯离子浓度的减少量来间接说明氯类活性物质的生成量, 并通过自由氯含量来判断其是否为氯类活性物质的主要成分.由表 1数据可知, 在不同pH条件下, 光电系统中氯离子减少量与自由氯增加量的变化趋势都和速率常数保持一致, 说明在pH=3下析氧副反应受到抑制的同时, 溶液中产生了更多的氯类活性物质, 加快了反应的进行.相反, 在pH=9时, 氯类活性物质产量减少, 反应变慢.同时, 自由氯增加量在氯离子减少量中占比小于2%, 说明自由氯含量极少, 对3, 4-DMA降解的贡献不大.
表 1(Table 1)
| 表 1 不同初始pH值条件下光电体系中氯类活性物质浓度的变化量 Table 1 Changes in the concentration of chlorine active substances in photoelectrocatalytic system under different initial pH values | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
表 1 不同初始pH值条件下光电体系中氯类活性物质浓度的变化量 Table 1 Changes in the concentration of chlorine active substances in photoelectrocatalytic system under different initial pH values
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
考虑到调为酸性条件使实验操作变得复杂, 且对于光电体系来说, pH=6.4时的降解速率常数与pH=3时仅相差0.0274 min-1, 因此, 确定最佳pH条件为6.4.为了进一步讨论初始pH值与光电系统表观速率常数kobs之间的关系, 对pH值与kobs进行趋势分析, 得出kobs=0.4217A-0.8634.
3.2.3 初始氯化钠浓度的影响由于不同氯化钠浓度对单光体系反应速率影响很小, 且单光体系的反应速率常数在光电系统中占比小于10%, 因此, 主要针对单电与光电系统的动力学拟合结果进行讨论.如图 4所示, 当氯化钠浓度范围为1000~6000 mg·L-1时, 单电与光电体系的kobs值均持续上升, 分别从0.1239、0.1630 min-1增至0.2201、0.2954 min-1, 且增加速率相近.在不同氯化钠浓度作用下, 光催化与电催化仍存在协同效应.结合表 2分析可得, 当氯化钠浓度小于8000 mg·L-1时, 氯化钠浓度的增加可促进氯离子向氯类活性物质转化.但在不同氯化钠浓度条件下, 自由氯含量仍较少, 最大仅为2.5 mg·L-1.
图 4(Fig. 4)
 |
| 图 4 初始氯化钠浓度对反应速率的影响 Fig. 4The effect of NaCl concentration on reaction rate |
表 2(Table 2)
| 表 2 不同初始氯化钠浓度条件下光电体系中氯类活性物质浓度的变化量 Table 2 Changes in the concentration of chlorine active substances in photoelectrocatalytic system under different initial sodium chloride concentration | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
表 2 不同初始氯化钠浓度条件下光电体系中氯类活性物质浓度的变化量 Table 2 Changes in the concentration of chlorine active substances in photoelectrocatalytic system under different initial sodium chloride concentration
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
而当氯化钠浓度进一步增加至8000 mg·L-1时, 单电与光电系统的反应速率均开始下降.其原因可能有以下3个方面:①溶液中的高浓度氯离子可与光催化过程产生的羟基自由基快速反应, 生成了氧化活性远不及羟基自由基的活性物质ClOH·-:HO·+Cl-→ClOH·-(Kormann et al., 1999); ②氯离子可能在光催化剂的羟基自由基表面形成表面复合物TiCl4(Jia et al., 2012), 或在光催化剂颗粒表面产生一种强极性的环境(刘仁龙, 2004), 使有机物向光催化剂活性位的扩散与迁移阻断; ③高氯离子强度会导致催化剂TiO2团聚, 降低其量子小尺寸效应(王阿楠等, 2014).为了进一步探究光电耦合体系氯化钠初始浓度与表观速率常数kobs之间的关系, 对氯化钠初始浓度与kobs进行趋势分析, 得出kobs=0.0157B0.3378.
3.2.4 电流密度的影响其它影响因素保持不变的情况下, 考察电流密度分别为1.5、2.5、3.5、5.0 mA·cm-2时的耦合机制.如图 5所示, 电流密度的增加对单电与光电系统的反应速率均有促进作用, 但对光电体系的促进作用更强.因此, 不同电流密度影响下, 光催化与电催化反应表现出较强的协同效应, 在I=5.0 mA·cm-2时, 协同度高达41.66%.其原因为对于光电耦合体系, 电流密度的加大对光催化与电催化反应均有利.对于光催化反应而言, 电流密度的增加可减少光生电子-空穴对的复合, 使得体系产生了更多的羟基自由基, 从而提高光催化速率.而对于电催化反应, 结合表 3数据可得, 电流密度的增加进一步促使氯离子转化为氯自由基、自由氯等氯类活性氧化物质, 加快了电催化速率.当光催化反应与电催化反应速率同时提高时, 体系的反应速率可得到极大提高.
图 5(Fig. 5)
 |
| 图 5 电流密度对反应速率的影响 Fig. 5The effect of current density on reaction rate |
表 3(Table 3)
| 表 3 不同电流密度条件下光电体系中氯类活性物质浓度的变化量 Table 3 Changes in the concentration of chlorine active substances in photoelectrocatalytic system under different current densities | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
表 3 不同电流密度条件下光电体系中氯类活性物质浓度的变化量 Table 3 Changes in the concentration of chlorine active substances in photoelectrocatalytic system under different current densities
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
进一步分析发现, 对于光电耦合系统, 当电流密度从1.5 mA·cm-2变为2.5 mA·cm-2时, kobs增幅最大, 从0.1229 min-1增至0.2954 min-1.而当电流密度从2.5 mA·cm-2变为5.0 mA·cm-2时, kobs仅增加了19.57%.因此确定2.5 mA·cm-2为最佳电流密度值.为了进一步讨论光电耦合体系电流密度C与表观速率常数kobs之间的关系, 对电流密度与kobs进行趋势分析, 得出kobs=0.0520C1.4742.
3.2.5 光照强度的影响保持其它试验条件不变, 考察当紫外灯功率分别为6、12和18 W时, 光照强度对耦合机制的影响, 单光与光电耦合系统的动力学拟合结果如图 6所示.
图 6(Fig. 6)
 |
| 图 6 光照强度对反应速率的影响 Fig. 6The effect of light intensity on reaction rate |
从图 6可以看出, 随着光照强度的增加, 单光与光电体系的kobs值均逐渐增大, 其中, 光电体系增加更为明显, 在不同光照强度作用下光催化与电催化反应仍具有协同效应.同时分析表 4数据发现, 光照强度的增加对溶液中氯离子的减少量与自由氯的增加量并无太大影响, 说明光照强度的增加并不能通过促进氯类活性物质的产生来提高反应速率.而是促进了羟基自由基的产生来直接提高光催化反应的速率, 也可能是通过紫外光的光量子来激发3, 4-DMA及其中间产物, 使得有机物更容易被氧化, 同时提高光催化与电催化的反应速率.但由于本反应中所用紫外灯瓦数较低, 影响较弱, 紫外灯的作用并不明显, 考虑能耗使用6 W紫外灯即可.为了进一步探究反应速率常数kobs与光照强度的关系, 对光照强度与kobs进行趋势分析, 得出kobs=0.1936D0.2300.
表 4(Table 4)
| 表 4 不同光照强度条件下光电体系中氯类活性物质浓度的变化量 Table 4 Changes in the concentration of chlorine active substances in photoelectrocatalytic system under different light intensities | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
表 4 不同光照强度条件下光电体系中氯类活性物质浓度的变化量 Table 4 Changes in the concentration of chlorine active substances in photoelectrocatalytic system under different light intensities
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.2.6 3, 4-DMA反应动力学模型的建立根据以上分析结果, 3, 4-DMA光电耦合催化反应的速率常数kobs与初始pH值(A)、初始氯化钠浓度(B)、电流密度(C)、光照强度(D)的关系式为:
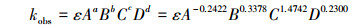 | (4) |
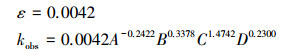 | (5) |
 | (6) |
为了验证该模型的准确性, 选用温度20 ℃, 3, 4-DMA初始浓度5 mg·L-1, NaCl初始浓度6000 mg·L-1, TiO2浓度300 mg·L-1, 电流密度2.5 mA·cm-2, 光照强度6 W, 初始pH值分别为3.0、6.4、9.0的3组实验数据进行检验, 将实验值与理论计算值进行对比, 结果如图 7所示.从图 7可以看出, 实验值与模型计算值较接近, 说明通过分析得出的准一级模型对光电耦合催化条件下3, 4-DMA的降解过程具有较高的拟合度, 该模型可用来准确描述3, 4-DMA的降解动力学过程.
图 7(Fig. 7)
 |
| 图 7 在确定试验条件下试验值与计算值的比较 Fig. 7Comparison of test values and calculated values under defined test conditions |
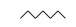
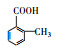
3.3 3, 4-DMA降解路径分析根据GC/MS检测结果, 3, 4-DMA降解过程中发现了3, 4-二甲基苯酚、邻二甲苯、甲基苯、苯甲醛、烷烃类物质等小分子, 也同时发现N-苄烯丁胺等联苯类大分子物质.共检测到8种中间产物, GC/MS图谱见图 8, 具体出峰时间、名称与结构式见表 5.
图 8(Fig. 8)
 |
| 图 8 3, 4-DMA降解中间产物的GC/MS图谱 Fig. 8GC/MS spectrum of 3, 4-DMA degradation intermediates |
表 5(Table 5)
| 表 5 3, 4-DMA降解中间产物 Table 5 3, 4-DMA degradation intermediates | |||||||||||||||||||||||||||||||||||||||||||||
表 5 3, 4-DMA降解中间产物 Table 5 3, 4-DMA degradation intermediates
| |||||||||||||||||||||||||||||||||||||||||||||

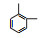
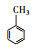
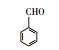


根据检测到的中间产物, 通过合理的推断与分析, 提出如图 9所示的两种可能降解途径.对于路径一, t=3.507 min时出现了3, 4-二甲基苯酚, 推测反应初始时3, 4-DMA被氯类活性物质、光生空穴、羟基自由基等氧化生成3, 4-二甲基苯酚.接着在保留时间t=4.453 min时发现了邻二甲苯, 推测3, 4-二甲基苯酚中—OH官能团继续被氧化性物质攻击, 进而将羟基脱去, 生成水和氧气, 变成邻二甲苯.在t=9.703 min时检测出了邻甲基苯甲酸, 说明邻二甲苯上的甲基很有可能发生了甲基→醛基→羧基的一个过程.因此, 推测邻二甲苯上的甲基先进行单电子转移反应脱氢生成了甲基自由基(—CH2·), 接着由于电催化系统析氧副反应的不断进行, 溶液中氧含量增加, —CH2·接触氧气生成过氧自由基—CH2OO·, —CH2OO·容易得到电子生成醛基—CH2O.—CH2O可以发生单电子转移反应脱去H原子生成羰基自由基, 羰基自由基再与氧气反应生成羰基过氧自由基(COOO·)继而生成COOOH, COOOH具有强氧化性, 极易与电子和氢氧根离子反应生成羧酸.在t=4.932和6.024 min时分别发现了甲基苯和苯甲醛, 说明羧基在氯类活性物质和羟基自由基等氧化性物质的攻击下可以发生脱羧反应进而生成甲基苯, 甲基苯的甲基继续发生醛基化的反应, 即在单电子转移反应脱氢后生成苄基自由基, 然后被氧气氧化为苄基过氧自由基, 最后再被电子还原成苯甲醛(Epstein et al., 1987; Augugliaro et al., 2000; Fan et al., 2004).t=8.203和8.389 min时都检测到烷烃类物质, 推测苯甲醛的醛基再次经过醛基→羧基→脱羧化的过程进而生成苯环, 然后被氯类活性物质和羟基自由基开环生成正烷烃(Marci et al., 2003), 最后进一步被矿化.
图 9(Fig. 9)
 |
| 图 9 3, 4-DMA总降解路径图 Fig. 9The total network of 3, 4-DMA degradation |
对于路径二, 在t=13.124 min时发现生成联苯类大分子的物质.这一方面可能是阴极极板和TiO2电子还原半反应使得3, 4-DMA还原, 也很有可能是芳香自由基间的重组.因此, 推断在羟基自由基的氧化下脱去3, 4-DMA的氨基形成氨基自由基(—NH2·), 氨基自由基电负性很高(Bustillo et al., 2018), 容易与苄基自由基反应生成苯甲氨, 苯甲氨与羟基自由基和二氧化氯发生电子转移反应生成苯甲胺自由基(Xu et al., 2010), 最后再和苄基自由基结合生成氮联苯类物质.根据相关****的研究, 联苯类物质可以被羟基自由基和二氧化氯进一步氧化开环生成脂肪酸类物质(Chen et al., 2012), 最后被矿化生成二氧化碳、水.
因此, 3, 4-DMA的可能降解路径为:①3, 4-DMA依次降解为3, 4-二甲基苯酚、邻二甲苯、邻甲基苯甲酸、甲苯、苯甲醛、烷烃类物质、CO2和H2O; ②3, 4-DMA依次降解为含N联苯类物质、脂肪酸类物质、CO2和H2O.
4 结论(Conclusions)1) 贡献度分析结果表明, 以10 min时3, 4-DMA降解率来看, 氯类活性物质的贡献度为87.55%, 羟基自由基为7.9%, 空穴直接氧化占2.2%, UV直接氧化与阳极直接氧化分别为0.75%与1.6%, 氯类化性物质氧化占绝对的主导地位.
2) 不同初始pH值(A)、初始氯化钠浓度(B)、电流密度(C)与光照强度(D)条件下, 光电体系的光催化与电催化反应均表现出协同效应, 4种因素可通过影响氯类活性物质与羟基自由的产生来影响光催化与电催化的耦合效果.
3) 4种因素对光电耦合机制产生的影响各有不同, 根据所得动力学方程ln(c0/ct)=0.0042A-0.2422B0.3378C1.4742D0.2300t, 各因素影响的强弱顺序为:电流密度(C)>初始氯化钠浓度(B)>初始pH值(A)>光照强度(D).
4) 基于GC-MS检测结果, 推测3, 4-DMA的降解路径分为由3, 4-DMA→3, 4-二甲基苯酚→邻二甲苯→邻甲基苯甲酸→甲苯→苯甲醛→烷烃类物质→CO2、H2O的第一类路径和由3, 4-DMA→含N联苯类物质→脂肪酸类物质→CO2、H2O的第二类路径.
参考文献
| 迪安J A. 2003. 兰氏化学手册[M]. 北京: 科学出版社. |
| Augugliaro V, Loddo V, Marci G, et al. 2000. Photocatalytic degradation of toluene in aqueous suspensions of polycrystalline TiO2 in the presence of the surfactant tetradecyldimethylamino-oxide[J]. Studies in Surface Science & Catalysis, 130(14): 1973–1978. |
| Bustillo-Lecompte C F, Kakar D, Mehrvar M. 2018. Photochemical treatment of benzene, toluene, ethylbenzene, and xylenes (BTEX) in aqueous solutions using advanced oxidation processes:Towards a cleaner production in the petroleum refining and petrochemical industries[J]. .Journal of Cleaner Production, 186: 609–617.DOI:10.1016/j.jclepro.2018.03.135 |
| Cui Y H, Feng Y J, Liu J, et al. 2012. Comparison of various organic compounds destruction on rare earths doped Ti/Sb-SnO2 electrodes[J]. J Hazard Mater, 239-240(4): 225–232. |
| Doudrick K, Yang T, Hristovski K, et al. 2013. Photocatalytic nitrate reduction in water:Managing the hole scavenger and reaction by-product selectivity[J]. Applied Catalysis B:Environmental, 136(1): 40–47. |
| Epstein A J, Ginder J M, Zuo F, et al. 1987. Insulator-to-metal transition in polyaniline:Effect of protonation in emeraldine[J]. Synthetic Metals, 21(1): 63–70. |
| Chen F, Yu S, Dong X, et al. 2012. High-efficient treatment of wastewater contained the carcinogen naphthylamine by electrochemical oxidation with γ-Al2O3 supported MnO2 and Sb-doped SnO2 catalyst[J]. Journal of Hazardous Materials, 227-228: 474–479.DOI:10.1016/j.jhazmat.2012.05.024 |
| Fan Z Y, Huang J L, Wang P, et al. 2004. Kinetics of aniline oxidation with chlorine dioxide[J]. Journal of Environmental Sciences, 16(2): 238–241. |
| 范崇政, 肖建平, 丁延伟. 2001. 纳米TiO2的制备与光催化反应研究进展[J]. 科学通报, 2001, 46(4): 265.DOI:10.3321/j.issn:0023-074X.2001.04.001 |
| 冯炜.2010.纳米晶体TiO2制备、活性及光催化降解1-萘酚废水路径研究[D].天津: 天津大学http://d.wanfangdata.com.cn/Thesis/Y1874448 |
| Gómez J L, León G, Hidalgo A M, et al. 2009. Application of reverse osmosis to remove aniline from wastewater[J]. Desalination, 245(1/3): 687–693. |
| Jia H, Zhao J, Fan X, et al. 2012. Photodegradation of phenanthrene on cation-modified clays under visible light[J]. Applied Catalysis B:Environmental, 123-124(4): 43–51. |
| Kormann C. 1991. Photolysis of chloroform and other organic molecules in aqueous TiO2 suspensions[J]. Environmental Science and Technology, 25(3): 494–500.DOI:10.1021/es00015a018 |
| Liao W, Zhang Y, Zhang M, et al. 2013. Photoelectrocatalytic degradation of microcystin-LR using Ag/AgCl/TiO2 nanotube arrays electrode under visible light irradiation[J]. Chemical Engineering Journal, 231(6501): 455–463. |
| 刘仁龙.2004.纳米TiO2光催化氧化处理苯酚水溶液的研究[D].重庆: 重庆大学http://cdmd.cnki.com.cn/Article/CDMD-10611-2004127213.htm |
| 刘艳彪.2011新型电极材料光/电催化降解有机污染物及污染物化学能的综合利用[D].上海: 上海交通大学http://cdmd.cnki.com.cn/Article/CDMD-10248-1012016680.htm |
| Ma J, Yang M, Sun Y, et al. 2014. Fabrication of Ag/TiO2 nanotube array with enhanced photo-catalytic degradation of aqueous organic pollutant[J]. Physica E:Low-dimensional Systems and Nanostructures, 58(1): 24–29. |
| Marci G, Addamo M, Augugliaro V, et al. 2003. Photocatalytic oxidation of toluene on irradiated TiO2:comparison of degradation performance in humidified air, in water and in water containing a zwitterionic surfactant[J]. Journal of Photochemistry and Photobiology A:Chemistry, 160(1/2): 105–114. |
| Pelaez M, Falaras P, Likodimos V, et al. 2016. Use of selected scavengers for the determination of NF-TiO2 reactive oxygen species during the degradation of microcystin-LR under visible light irradiation[J]. Journal of Molecular Catalysis A:Chemical, 425(4): 183–189. |
| Pelegrini R, Peralta-Zamora P, Andrade A R D, et al. 1999. Electrochemically assisted photocatalytic degradation of reactive dyes[J]. Applied Catalysis B:Environmental, 22(2): 83–90.DOI:10.1016/S0926-3373(99)00037-5 |
| 秦微, 谢陈鑫, 王震, 等. 2015. 光电催化氧化处理高氨氮废水技术研究[J]. 广东化工, 2015, 42(5): 90–91.DOI:10.3969/j.issn.1007-1865.2015.05.048 |
| Varanasi L, Coscarelli E, Khaksari M, et al. 2018. Transformations of dissolved organic matter induced by UV photolysis, Hydroxyl radicals, chlorine radicals, and sulfate radicals in aqueous-phase UV-Based advanced oxidation processes[J]. Water Research, 135: 22–30.DOI:10.1016/j.watres.2018.02.015 |
| Waldner G, Pourmodjib M, Bauer R, et al. 2003. Photoelectrocatalytic degradation of 4-chlorophenol and oxalic acid on titanium dioxide electrodes[J]. Chemosphere, 50(8): 989–998.DOI:10.1016/S0045-6535(02)00612-4 |
| 王阿楠, 袁滕, 骆永明. 2014. 二氧化钛(P25)光催化降解二苯砷酸的研究[J]. 环境科学, 2014, 35(10): 3800–3806. |
| Xu R, Li J, Wang J, et al. 2010. Photocatalytic degradation of organic dyes under solar light irradiation combined with Er3+:YAlO3/Fe- and co-doped TiO2 coated composites[J]. Solar Energy Materials and Solar Cells, 94(6): 1157–1165.DOI:10.1016/j.solmat.2010.03.003 |
| 杨唯艺, 李孟, 张倩, 等. 2018. K2FeO4氧化降解3, 4-二甲基苯胺的机理研究[J]. 中国环境科学, 2018, 38(5): 1744–1751.DOI:10.3969/j.issn.1000-6923.2018.05.017 |
| 杨晓芬, 赵美萍, 李元宗, 等. 2002. 水中苯胺类化合物的分光光度法测定[J]. 分析化学, 2002, 30(5): 540–543.DOI:10.3321/j.issn:0253-3820.2002.05.007 |
| 张丽.2014.可见光响应型LaTaON2光阳极及其光电化学分解水性能研究[D].南京: 南京大学http://d.wanfangdata.com.cn/Thesis/Y2941969 |
| 周文敏, 傅德黔, 孙宗光. 1990. 水中优先控制污染物黑名单[J]. 中国环境监测, 1990(4): 3–5. |
