 ,2,*, 杜政霖,1,*
,2,*, 杜政霖,1,*Challenges of High-Throughput Computing in Genomic Data Analysis for Large-Scale Cohort Studies
Zeng Jingyao1, Yuan Na1, Wei Wenjuan2, Li Gen,2,*, Du Zhenglin,1,*通讯作者: * 李根(E-mail:gen.li@genetalks.com)杜政霖(E-mail:duzhl@big.ac.cn)
收稿日期:2019-11-29网络出版日期:2020-02-20
| 基金资助: |
Received:2019-11-29Online:2020-02-20
作者简介 About authors

曾瀞瑶,中国科学院北京基因组研究所国家基因组科学数据中心,博士,助理研究员,主要从事大规模人群队列基因组研究与肿瘤多组学数据整合和挖掘,并主导了中国人遗传变异数据库的设计与开发。
在本文贡献了第一章的写作并修改全文。
Zeng Jingyao, PhD, is a research assistant in National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences. The research area of her focuses mainly on genomic analysis of large-scale cohort studies and neoplastic omics data integration and mining. And she was also responsible for the design and development of Chinese Genomic Variation Database.
She contributed to section 1 and the paper modification.
E-mail: zengjy@big.ac.cn

苑娜,中国科学院北京基因组研究所生命与健康大数据中心,计算机硕士,工程师,主要从事大规模人群队列遗传变异解析及数据库开发。
本文贡献了第二章的写作。
Yuan Na, is currently an engineer in Big Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences, mainly engaged in large-scale cohort studies and database website development.
She contributed to section 2.
E-mail:yuann@big.ac.cn

魏文娟,博士,在圣路易斯华盛顿大学和中国科学院上海生化所联合培养硕博连读(2008-2014),后在阿肯色儿童医院及北卡州立大学进行临床及科研工作(2014-2015)。回国后加入人和未来参与基因检测与基因数据相关工作(2016至今)。在分子生物学、生物统计、生物信息学、精准医疗方面具有丰富的经验。
在本文中与李根共同负责第三与第四章的写作。
Wei Wenjuan was a joint Ph.D. student of Washington University in St. Louis, School of Medicine and Chinese academy of science, Shanghai institute for biological science (2008-2014). After graduation, she worked in Arkansas Children’s Hospital and North Carolina State University on biostatistics (2014-2015). Later joined Genetalks and worked on genetic test and genetic data solutions (2016-now). Dr. Wei has wide experience in molecular biology, biostatistics, bioinformatics, and precision medicine.
She and Li Gen co-contributed to the writing of sections 3 and 4.
E-mail:weiwenjuan@sibcb.ac.cn

李根,目前任人和未来生物科技(长沙)有限责任公司首席信息官,博士毕业于国防科技大学,主要研究领域包括基因分析高性能计算、动态编译优化、基因数据压缩。
与魏文娟共同完成了本文第三与第四章的写作。
Li Gen, is currently CIO of Genetalks Inc., He received Ph.D. degree in Computer Science and Technology at National University of Defense Technology. His research interest includes high-performance computing for genomic analysis, dynamic compilation optimization, and genomic data compression.
He and Wei Wenjuan co-contributed to the writing of sections 3 and 4.E-mail:gen.li@genetalks.com

杜政霖,中国科学院北京基因组研究所生命与健康大数据中心,博士,高级工程师,主要研究方向是基因组学和生物信息学。
负责文章整体架构设计与摘要撰写。
Du Zhenglin, is currently a senior engineer in National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences. His research interest includes genomics and bioinformatics.
He contributed to the organization of the paper and the writing of abstract.E-mail:duzhl@big.ac.cn

摘要
【目的】为推动精准医学研究的发展,世界各国相继开展大规模人群队列基因组测序计划,通过对数以万计个体进行全基因组测序,构建人群特异的基因组变异图谱。这些海量基因组数据产出,对计算速度和计算通量提出了新的要求,迫切需要速度更快、通量更高的计算平台来处理与解读这些生物序列信息。由于基因组数据自身的特点、数据解析过程的多样性和复杂性,致使在大规模人群基因组变异解析中高通量计算资源的使用效率低、计算速度慢、耗时长,服务器与本地数据交换不便,因此需要针对基因组变异解析进行多方面优化,通过软硬件开发来解决应用中存在的多种问题。本文拟对这些优化方法进行分析和综述。【方法】在高通量计算系统中,系统IO瓶颈问题是基因组变异解析并行化效率低的主要原因,通常采用基于分布式非结构化存储数据库以及对象存储系统,以提升IO的大规模可扩展能力,解决分析流程中存在的IO问题;同时通过基因组数据的高效压缩算法,可减少数据IO和传输压力。为了加快基因组数据解析速度,可在软件上采用神经网络等算法优化基因组解析方法,在硬件上使用FPGA(现场可编程逻辑门阵列)或GPU异构计算,以提高数据处理速度。【结果】综合来看,以上多方面的优化可以大幅提升基因组数据分析中高通量计算的性能,解决基因组数据处理中的存储墙问题,提高高通量计算资源的使用效率,大大减少全基因组变异解析的计算时间。【结论】高通量计算在基因组数据解析应用中存在的多种问题,可通过软硬件开发和优化得以解决,从而显著改进高通量计算在大规模人群队列变异解析应用中的计算效率,促进今后人群队列基因组研究与应用的广泛开展。
关键词:
Abstract
[Objective] In order to promote the precision medicine research, large-scale population genomic studies have been carried out globally, and population-specific genome variation maps have been built by whole genome sequencing of thousands of individuals. These projects output massive genomic data, which needs high-throughput computing (HTC) to process. However, due to the characteristics of genomic data and the diversity and complexity of process workflows, HTC computing resources are not fully utilized in genomic data analysis tasks, so that the computing speed is slow and the data exchange over servers is inconvenient. Therefore, it is necessary to optimize HTC platforms for genomic data analysis from software and hardware aspects. This paper analyzes and summarizes these optimization methods. [Methods] In an HTC system, the bottleneck of system IO is the main cause for the low parallel efficiency in genomic data processing. Generally, distributed unstructured storage database and object storage system are used to improve the scalability of large-scale IO and solve the IO problems in data processing. Meanwhile, the IO load can be reduced by using the efficient compression algorithms of genomic data. In order to accelerate genomic data processing, algorithms such as neural networks can be used to optimize genome analysis methods, and FPGA or GPU heterogeneous computing can be used to improve the speed of data analysis. [Results] In brief, the above optimization can greatly improve HTC performance by solving the IO wall problem in genomic data analysis and improving the efficiency of HTC resources, which greatly reduces the computing time of genome-wide variation analysis. [Conclusions] The software and hardware improvements can significantly increase the HTC efficiency and speed in genomic data analysis, and can promote the application of high-throughput computing on large-scale cohort studies in the future.
Keywords:
PDF (9319KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
曾瀞瑶, 苑娜, 魏文娟, 李根, 杜政霖. 高通量计算在大规模人群队列基因组数据解析应用中的挑战. 数据与计算发展前沿[J], 2020, 2(1): 117-127 doi:10.11871/jfdc.issn.2096-742X.2020.01.010
Zeng Jingyao.
引言
高通量测序技术和信息技术推动着生命科学进入了大数据时代,基因组大数据已经成为推动生物学和医学发展的关键,对于提升生命科学原始创新能力、破解人类健康难题起到重要的支撑作用。借助高通量计算、人工智能、深度学习等一系列新型信息技术,拓展基因组大数据研究的新理论和新方法,必将促使生物医学研究向“数据密集型科学”的新范式转变,让精准医学从观念走向实践。本文将针对高通量计算在生物学研究领域的应用进行深入探讨,以大规模人群基因组变异解析应用为例,详细分析高通量计算在整个研究体系中如何发挥作用及存在的挑战,并针对发现的问题综述了基因组数据分析中改进高通量计算性能的方法与实际效果。最后,本文对高通量计算未来在基因组数据计算应用方面的发展提出思考与展望。1 研究背景
自2001年人类基因组计划完成至今,高通量测序技术发生了翻天覆地的发展和变革[1,2]。通量更高、耗时更短、测序成本大幅度降低,这些因素都促使该领域每年以指数级的增长速度产出基因组、转录组、蛋白组、表观组及代谢组等多组学数据,生物信息在数年内已迅速膨胀成数据的海洋[3]。与此同时,这些海量数据也将推动我们从一个积累数据向解析数据的时代转变,数据量的巨大积累往往蕴含着潜在突破性发现的可能,并最终揭示由量变到质变而凸显出来的生物学奥秘。为推动精准医学的发展,多个国家均已相继开展大规模人群队列基因组测序计划,旨在构建人群特异的基因组变异图谱,并为逐步实现对癌症等特定疾病的临床早期诊断与治疗提供强有力支撑和指导。从最初的千人基因组计划与HapMap计划开始,世界各国先后开展了本民族的大规模人群队列基因组研究,例如英国的万人(UK10K)[4]和十万人基因组计划[5]、冰岛人基因组[6]、美国万人基因组项目[7]、日本千人基因组计划(1KJPN)[8]。这些大规模人群队列研究通过对数以万计的个体进行全基因组重测序,鉴定并获得了千万数量级的单核苷酸多态性(SNP)、插入缺失(indel)以及大片段结构变异等多种类型的基因组变异数据。作为拥有全世界五分之一人口的中华民族,同样开展了包括中国人抑郁症人群队列项目(CONVERGE)[9]、90个汉族人基因组深度测序(90Han)[10]及中国科学院精准人群队列研究项目(CASPMI)[11]在内的多个中国人群队列研究。其中CONVERGE项目的平均测序深度仅为1.7X,而90Han虽具有较高测序深度(80X),但包含较小的样本量。中国科学院精准人群队列项目取样覆盖中国10个民族、32个省市自治区,完成了约一千个样本的高深度全基因组测序(~30X)。人类基因组数据十分庞大,其参考序列长度为3Gb,而一个人的高深度测序数据往往高达100Gb,这无疑对运行分析任务的计算机集群系统性能有着极高的要求,尤其是进行大规模人群队列的全基因组变异解析研究。挑战往往伴随机遇而来,庞大的数据产出对计算速度和通量出了新的要求,我们迫切需要一些速度更快、通量更高的计算平台来处理与解读这些海量生物序列信息。
随着“大数据”时代的到来,高性能计算(HPC)的主要应用从传统的科学与工程计算逐步演变为以数据处理为核心,这给传统高性能计算机体系结构带来了巨大挑战,促使高通量计算(HTC)应运而生[12,13]。高通量计算正是顺应时代需求,针对各领域对快速精准处理大数据的需求,应运而生的一种新兴计算模式。与追求高性能和低功耗的传统计算系统不同,高通量计算主要致力于在单位时间处理更多的任务,强调高通量、强实时和低延迟。服务器处理器芯片也相应地从传统追求单个任务的“快”,变为单位时间处理任务数量的“多”。目前许多新兴计算应用如云计算、人工智能及物联网均依赖于高通量计算系统来实现,高通量计算也已在材料学[14]、芯片结构设计[15]等领域取得一些卓有成效的应用成果。面向精准医学研究领域的海量基因组测序数据,同样需要一批具有足够存储能力、足够计算资源及并行计算能力的大型高通量计算机,来有效保障本领域对大规模人群队列基因组变异解析应用的计算需求。
2 人群队列基因组变异解析的主要步骤
大规模人群队列研究是对特定人群的表型进行观察和随访,对个体进行全基因组测序,并在此基础上对人群进行基因组变异解析。通过基因组测序数据与参考基因组进行序列比对,可以鉴定大量的单核苷酸多态性(SNP)、拷贝数变异(CNV)、插入缺失(indel)、结构变异(SV)等变异信息,结合个体的相关临床表型信息,可以找出与疾病、性状相关的基因位点。人群队列全基因组数据的遗传变异位点解析主要包括以下五个步骤(图1):图1
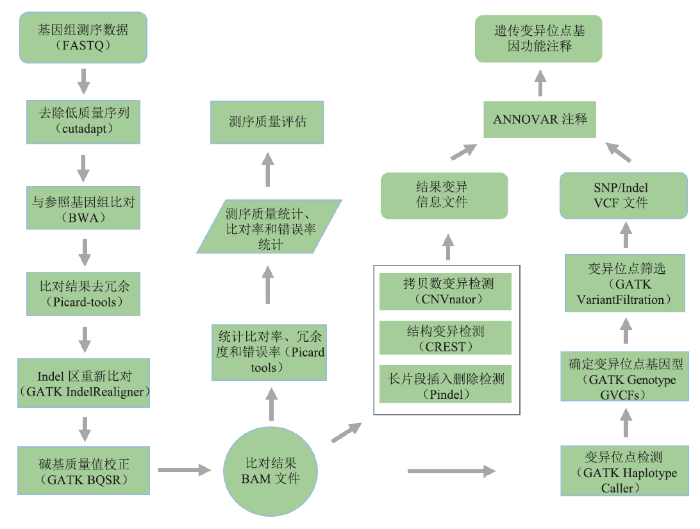 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1基因组变异解析的分析流程
Fig.1The workflow of genomic variant analysis
(1)基因组测序原始数据的质控和处理
使用 FastQC [16]软件做质控,对测序数据进行质量控制,统计测序长度、质量值、数量以及Q20、Q30比率、总数据量等信息,判断测序数据情况是否符合后续分析要求。对于可用的数据使用Cutadapt [17]软件进行去接头及低质量序列过滤。
(2)与参考基因组比对
根据测序序列与参考基因组的序列相似性,将测序序列定位到染色体上,该分析使用BWA [18]的MEM算法完成。
(3)比对结果排序和去冗余
将BAM文件中同一染色体上的测序数据按照坐标顺序从小到大进行排序,使用Picard-tools(http://broadinstitute.github.io/picar)的SortSam完成,然后使用Picard-tools的MarkDuplicates去除由PCR扩增所形成的冗余数据。
(4)基因组变异位点及结构变异检测
对于SNP和小Indel(< 50bp)使用GATK [19]完成变异检测。在变异检测前,需要重新校正碱基质量值,然后使用HaplotypeCaller进行变异检测,使用GenotypeGVCFs确定每个样本的基因型。对于检测出的变异位点,使用VariantFiltration模块通过特定指标进行过滤,结合质量值、测序深度、重复性等因素做进一步的过滤筛选,最终得到可信度高的变异位点数据。对于SV的检测,主要鉴定基因组上发生的大片断插入、缺失、倒位、易位、拷贝数变异等类型的变异(长度50bp以上)。
(5)位点注释
根据现有的多种数据库,使用ANNOVAR [20]软件,对检测到的多种类型的基因组变异进行功能注释,注释变异对应的基因,评估变异对基因功能的影响程度以及与特定疾病的关联性。
3 应用中存在的主要问题
目前,在一般配置的集群服务器上,计算一个测序深度30X的人全基因组数据,通常需要30~40个小时,远跟不上测序数据产生的速度,因此需要提高全基因组数据分析效率。影响基因组数据分析效率的主要因素有两方面:一是变异解析流程在高通量计算系统中并行化效率低;二是基因组变异解析的流程复杂,处理速度慢、耗时长。以下针对这两个方面分别讨论可行的解决方法。3.1 人群队列基因组变异解析的并行化效率低
高通量计算系统的节点之间的连接通常存在高通信延迟,由此产生的“存储墙”问题尤为明显。围绕同一计算应用的计算节点数量越多,整个计算集群在通信IO上损耗的性能越大,当节点数量达到一定量时,计算性能反而负向增长,严重降低了对大型数据集的计算效能。以一个样品全基因组深度测序数据分析为例,全部计算需要经历序列比对、排序去重、变异检测三个主要步骤。在这三个步骤里,存在两个存储墙:第一,在比对步骤里, 若以1对N分发模式将数据切分发进300个计算节点,即便节点通过万兆网络直连也需要超过7分钟。如果采用M对N模式,IO系统的平衡设计又是一个巨大挑战。第二,在排序与去重步骤,涉及对数十亿条序列进行分布式混洗、去重、切割,这些序列粒度非常细,细粒度数据的交换是延迟敏感的,大量的网络握手导致网络交换带宽将无法有效发挥交换能力,集群计算能力大部分时间处于等待序列数据交换状态。解决数据IO问题通常采用分布式文件系统。制约文件系统的可扩展性(容量和性能)的主要因素包括两个主要方面:一是容量和IO瓶颈;二是元数据读写瓶颈问题,主要包括文件元数据服务带来的性能瓶颈等问题。面向对象存储技术用网络替代总线有效地解决了容量和IO瓶颈的问题,但是对于元数据读写瓶颈,依然没有较好的解决方案。著名的面向对象存储的分布式文件系统包括Lustre、CEPH以及GLuster等。Lustre和CEPH都存在元数据服务器,因而存在单点故障问题。Lustre和GLuster系统适合进行大文件读写,而对于小文件的读写,由于Lustre存在元数据服务器,进行小文件读写时会带来巨大的元数据服务器访问压力,Gluster虽然没有元数据服务器,但是IO方面优化不足,本地文件系统的元数据访问成为了系统瓶颈。在全基因组数据计算过程中,对大型文件数据交换与海量小粒度文件数据交换并存,使得已有的分布式文件系统难以处理。
为解决以上问题,提升IO的大规模可扩展能力,主要有两种方法:一是利用基于分布式非结构化数据库存储,定制成块存储与分发服务集群,以N:M方式聚合带宽进行瞬间高并发的数据分发;二是利用对象存储系统进行数百至数千台机器间的大规模数据混洗和分发。此外,对于数据的分割和计算必须在生物学意义下确保计算准确性,同时调节系统负载均衡。比如在变异检测前,数据切分按照染色体区域等生物学依据进行有效切割,调节计算负载的均衡程度,并合理设置重叠距离,以确保数据切割不会导致信息丢失。通过在高通量计算环境中进行合理的数据切割,以此来提高数据传输效率(图2)。在系统构建上,首先构造一个分层分布式数据库StageDB,作为数据分发系统,能够支持随机数据块同时发送到数百个计算节点上。该系统将FASTQ 文件切割为多个数据块,并将这些数据块存储在一个主机节点和多个备份节点中,这样的情况下,即使多台机器同时读取同一个数据块也能保证读取速度。采用多层级多节点分配和排序的方法,并调整数据切割和分配的顺序。首先,将比对输出的结果划分到多个区域,并将它们发送到相应的计算节点,接着在各个节点上完成排序;然后,将各个节点排序的结果切割为数百个小文件上传到对象存储上,由对象存储传输到后续计算节点。通过分层排序和切割提供足够的带宽,以及对象存储提供的小文件并行存储,从而打破第二个IO墙,实现近线性的性能加速[21]。基于这种思路建立的高通量计算系统,具有良好的扩展性和吞吐能力,从而最大限度地解决基因组变异解析中存在的IO问题。
图2
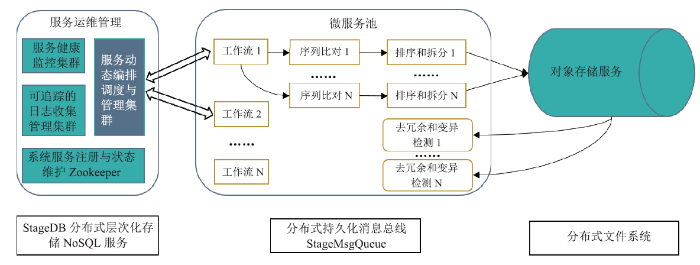 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2面向基因组变异解析的高通量计算集群系统结构
Fig.2System structure of the HTC system for genomic variant analysis
另一方面,大规模人群队列研究的开展越来越广泛,由此而产生的数据量庞大,给存储和传输带来严峻的挑战,因此需要开发专用的压缩工具以将测序数据进行高效压缩。QUIP [22]利用与 FASTQ 数据的所有三个部分中的order-3和高阶马尔可夫链模型相关的算术编码;LW FQZip [23]分别使用增量和运行长度有限编码方案来压缩元数据和质量分数,读取由轻量级映射模型进行预处理, 然后将三个组件组合在一起, 由通用工具 (如LZMA) 进行压缩;Fqzcomp [24]通过order-k上下文建模估计字符概率, 并借助算术编码器以FASTQ格式压缩测序数据。但如果目标和参考的相似性较低, 基于引用的算法可能是不确定的,因此提出了无参考的方法来解决这一问题。Grumbac提出一种专用于基因组序列的压缩方法[25],它的主要思想是基于经典的字典压缩方法Lempel-Ziv 和 Leme-pel [26]压缩算法,重复和回文是用最重要的事件的长度和位置进行编码。作为生物压缩的延伸[25], Biocompress [27]利用相同的方案, 并使用在不存在显著重复的情况下, 对order-2进行算术编码。DSRC [28]算法将序列拆分为多个块, 并用 LZ77 [26]独立压缩和赫夫曼[29] 编码。它在压缩和解压缩速度上都比 QUIP 快, 但在压缩比方面低于后者。DSRC2 [30]是DSRC的多线程版本, 将输入拆分为三个流进行预处理,预处理后, 元数据、读序和质量分数将在 DRSC 中单独压缩。重新组织读序的提升算法 SCALCE [31]在压缩比和压缩速度方面都可以优于大多数其他算法。GTZ压缩算法是基于Context Model压缩技术,采用混合多阶压缩编码器。多阶压缩编码流程主要分为预测模型和编码两个部分。首先,使用1、2、4、6阶预测模型,混合使用匹配模型,减少预测错误偏差,使用加权平均器对预测概率值进行加权平均,然后对该概率值进行量化,并增加插值以减少预测偏差、提高压缩率,进而使用位算数编码器对概率值进行编码压缩(图3)。GTZ是能够确保数据解压100%一致还原的无损压缩,可将FASTQ文件最低压至原大小的2.53%[32]。
图3
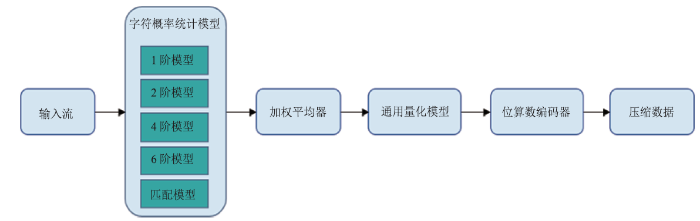 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3GTZ测序数据压缩算法
Fig.3The GTZ compression algorithm for sequencing data
综上所述,基因大数据高通量计算系统的构建为生物技术行业带来新的机会,科研工作者可以在短时间内分析大量基因组测序数据,有利于大规模人群队列基因组研究的开展。
3.2 全基因组变异解析速度慢、耗时长
基因组变异解析中的某些步骤并行化效率低,难以通过增加CPU计算节点来缩短数据分析时间。为了缩短计算时间、降低计算成本,可从软硬件两方面提高数据处理速度,在软件上改进和优化基因组解析算法,在硬件上使用高通量计算系统中的FPGA(现场可编程门阵列)或GPU异构计算节点。在硬件方面,FPGA不仅有软件的可编程性和灵活性,同时又有ASIC电路级并行的能力,FPGA提供的强大的可编程硬件计算能力可以满足基因组数据快速计算的需求。FPGA计算加速系统主要由三大部分组成(图4):一是PCI-E接口模块以及对应的DMA数据传输模块,用于与CPU进行高速数据交换;二是硬件计算加速核,这部分硬件电路可以专门针对特定计算需要进行电路定制;三是板载DDR存储控制模块,这部分模块被用于为计算核心提供临时数据存储的高速板载内存通道。针对全基因组变异解析可建立基于FPGA的异构计算系统,采用FPGA和CPU协同工作的异构加速技术,利用各自的特性加速基因数据的高通量计算。通过对全基因组分析中的热点计算核Smith-Waterman算法和pairHMM算法进行FPGA硬件重编程,优化原算法的体系结构层面以实现最大的加速效能;而其他功能则在CPU上优化的多线程软件中运行,以实现最大的灵活性。不同计算核的协同并发使整个系统的计算功能得到最佳呈现。
图4
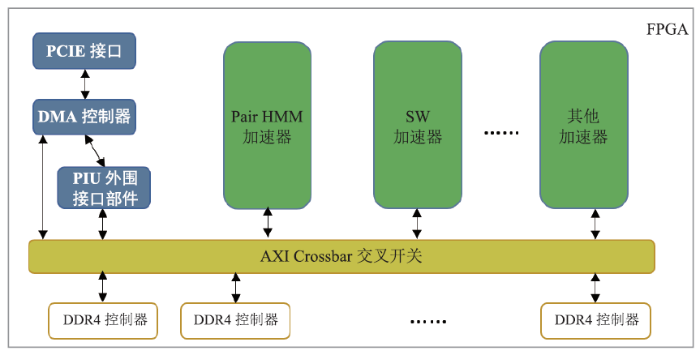 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4用于基因组变异解析的FPGA内部体系结构
Fig.4FPGA internal architecture diagram for genomic variant analysis
基因组数据分析中涉及大量测序序列和参考基因组之间的比对计算,在序列比对阶段,需要利用Smith-Waterman算法对序列进行打分来实现测序序列与参考基因组的精细比对;在突变检测阶段,需要利用PairHMM算法评估测序序列和组装的单倍型序列的相似度进而获得可能基因分型的概率。通过合理的设计,将这些高度密集的动态规划类计算移植到FPGA上进行大批量数据并发处理,减少CPU计算压力。利用这种设计思路构建的典型FPGA计算加速系统,一个测序深度30X人类全基因组数据解析仅需0.5小时,一天内可以完成超过5T数据量的基因组数据分析任务,处理能力与传统CPU服务器节点处理能力比达到 1:60。
在软件算法方面,随着以深度学习为代表的人工智能技术的发展,基于深度神经网络技术在各个领域都有广泛的应用,Google的DeepVariant项目即是基因组变异解析方面的一个典型实例。由于神经网络模型在推理方面数据计算的依赖关系简单、计算并行性高、计算模式相对统一,因此基于深度神经网络的DeepVariant算法,在已有通用计算框架支持下(如Tensorflow等)易于通过GPU进行加速。DeepVariant中卷积神经网络(Convolution Neural Network,CNN)的输入是参考基因组及覆盖该候选变异位点的测序数据,根据位点的基因型可分为三种类别:与参考基因组相同的纯合子、杂合子、与参考基因组不同的纯合子。CNN通过对大量已标记的数据进行学习,然后可以用于判定未标记数据的变异属于哪一类基因型。DeepVariant采用的卷积神经网络是Inception-v3 [33],这一网络结构是对GoogleLeNet网络(也称为Inception-v1)[34]的优化。DeepVariant和GoogleLeNet网络共同的一个重要特点是通过堆叠多个的Inception模块,构建层次更深的网络结构,提高网络的判定能力,并且由于采用了较小尺寸的卷积核,从而降低了网络的参数数目,加快了网络的训练速度。与传统的GATK变异检测方法相比,二者检出的突变数目相当但差异较大,两种方法的一致性仅为85%。这一方面是由于DeepVariant与GATK的数学模型不同,另一个是神经网络本身的计算结果具有不可解释性,其结果的准确性与其训练集有很大关系。在计算时间上,AI算法天然具有计算数据并行性,在GPU异构服务器上(32 core CPU, 2 NVIDIA Tesla M60 GPU),DeepVariant获得了相比单纯CPU计算约4.7x的提速。
综上所述,通过神经网络等算法及GPU或FPGA异构计算的使用,可大大减少全基因组变异解析的计算时间,加快人群队列基因组研究的数据分析速度。
4 未来趋势与展望
随着测序技术的发展和测序成本的进一步降低,测序技术的应用必将越来越广泛,海量基因组数据应运而生,由此带来的生物信息计算需求也呈指数增长,无论是计算的多样性以及计算密度均是如此。面对繁杂且大量的计算需求,只有借助高通量计算才能应对大规模人群队列基因组数据解析的重任。从高通量计算在基因组学应用的发展方向来看,一方面FPGA、GPU与CPU进行混合异构计算将成为未来生物信息计算加速的常态化手段,另一方面进行更多加速软件的开发势在必行。基于数据的计算特性选择适合的异构硬件进行计算,可以有效提高高通量计算的效能,让计算能够适应数据量的爆炸性增长。除了大规模人群队列基因组研究外,有关肿瘤、微生物、第三代数据组装的研究也越来越广泛的开展,相信提高相关计算效率、降低计算成本的加速软件也将越来越多的开发与应用。利益冲突声明
所有作者声明不存在利益冲突关系参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[C].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[C].
[本文引用: 2]
[J].
[本文引用: 2]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}