 ,*中国科学院宁波材料技术与工程研究所,浙江 宁波 315201
,*中国科学院宁波材料技术与工程研究所,浙江 宁波 315201The Application of Materials Genome Approach in Materials Design
Qian Xu, Tian Ziqi,*Ningbo Institute of Materials Technology & Engineering, Chinese Academy of Sciences, Ningbo, Zhejiang 315201, China通讯作者: * 田子奇(E-mail:tianziqi@nimte.ac.cn)
收稿日期:2019-11-29网络出版日期:2020-02-20
| 基金资助: |
Received:2019-11-29Online:2020-02-20
作者简介 About authors

钱旭,中国科学院宁波材料技术与工程研究所,硕士在读,就读于中国科学技术大学纳米科学技术学院,现在中国科学院宁波材料技术与工程研究所联合培养。目前主要从事催化模拟方面的高通量计算研究工作。
本文负责检索文献并完成文章写作。
Qian Xu, is a master candidate studying at the School of Nanoscience and Technology of the University of Science and Technology of China. He is currently co-cultivated by the Ningbo Institute of Materials Technology and Engineering of the Chinese Academy of Sciences. He is mainly engaged in high-throughput calculation research in catalytic simulation.
In this paper, he is responsible for completing the analysis of both domestic and international research review and finishing the manuscript.
E-mail: qianxu@nimte.ac.cn

田子奇,中国科学院宁波材料技术与工程研究所,副研究员,分别于2009年和2014年在南京大学获得物理化学学士和博士学位。之后在加州大学河滨分校从事了三年博士后研究。现在在中国科学院宁波材料技术与工程研究所任副研究员。主要从事气体分离与转化材料的理论研究工作。
本文指导钱旭完成了论文的写作。
Tian Ziqi received his B. S. and Ph. D. in physical chemistry from Nanjing University in 2009 and 2014, respectively. Then he worked as a postdoctoral researcher at University of California, Riverside for three years. Currently he is an associate professor in Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Science. His research mainly focuses on theoretical investigation of novel material for gas separation and conversion.
In this paper, he is responsible for the manuscript writing instruction.E-mail:tianziqi@nimte.ac.cn

摘要
【目的】本文主要介绍材料基因方法在一系列材料设计中的应用,如开发高性能催化材料、热电材料、金属有机框架(MOFs)材料、锂电池材料以及钙钛矿型光伏材料。【方法】将高通量计算与机器学习等数据挖掘技术结合,通过高通量计算产生一定规模的数据库,进而对材料数据库进行数据挖掘和分析。【结果】利用数据内在规律发现并筛选出潜在的新材料。【局限】目前,很多理论预测的材料在实验中合成制备还比较困难,因此理论与实验还需要更加深入地结合。【结论】随着计算机数据技术以及实验合成方法的进一步发展,材料基因方法将会在材料开发方面展现出更显著的作用。
关键词:
Abstract
[Objective] In this paper, we introduce the application of materials genome approach on materials design, including explorations of catalytic materials, thermoelectric materials, metal organic framework (MOF) materials, lithium battery materials and perovskite photovoltaic materials. [Methods] High-throughput computing is combined with data mining techniques, machine learning for instance. Database is generated from the high-throughput computing, and then data mining and deep analysis are performed. [Results] Potential novel materials are screened and discovered based on the data analysis. [Limitations] Currently, some hypothetical materials are hardly realized in experiments. Thus, the theoretical predictions and experiments need to be integrated more deeply. [Conclusion] With the further development of computational and experimental technology, materials genetic approach will perform a more significant role in materials development.
Keywords:
PDF (11648KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
钱旭, 田子奇. 材料基因方法在材料设计中的应用. 数据与计算发展前沿[J], 2020, 2(1): 128-141 doi:10.11871/jfdc.issn.2096-742X.2020.01.011
Qian Xu.
引言
在人类历史中,人类文明的每一次变革往往都对应着材料科学的每一次突破性进展。传统的材料科学作为一门以实验为基础的系统科学,主要依赖“试错”的研究方法,从提出假设到实验验证,不断迭代从而不断接近目标材料。这种研究方式耗时耗力,往往一种新材料从原型被发现,到发展成熟投入应用都需要很长的周期。进入20世纪,以有机氟聚合物、先进半导体等为代表的一系列材料,开发周期一般在二十年左右。随着工业水平和社会需求的快速发展,传统研究方法已经无法满足新材料开发的需求,迫切需要新的研究方法来快速搜索新材料,推进材料科学快速发展[1]。如何突破传统实验试错法耗时长、成本高的束缚,是研究者们主要关注的问题。随着计算技术的快速发展,“材料基因方法”成为了材料科学的新兴领域。在真实实验前,可以根据体系特征和材料的组成基元,自动化快速生成海量可能的结构,集中实施大规模计算——即“高通量计算”,以筛选可能的材料。同时也可以将已有的实验数据结合,建立结构与性质之间的联系,为新材料的设计提供大量的信息,预测可能的高性能材料,突破传统的试错过程。[2]。数据挖掘能够通过调用常见的分类回归算法,例如逻辑回归算法、K近邻算法、最小二乘法以及支持向量机等方法对目标体系进行快速分类预测。此外,对于体系构效关系特征值的选取和描述,还可以使用皮尔逊相关系数来进行分析。将高通量计算与机器学习等数据分析挖掘技术紧密结合,统称为“材料基因方法”。
在过去的十年里,材料基因方法在材料科学领域得以广泛应用,逐渐成为解决材料设计的可行途径。材料基因方法一般可分为三个步骤:(1) 通过对广泛材料结构的电子结构和性质进行计算,实现虚拟材料的增加[3];(2) 将合理的材料信息储存在数据库中[4];(3) 借助机器学习和理论推演等数据分析手段进行数据分析,并对材料性质和功能信息分类、整合和预测[5]。此外,材料的性质信息也会反馈到数据库,从而不断改进和完善整个数据驱动的材料设计流程。这三步工作相互独立又紧密耦合,对大数据时代科学研究具有很高的参考价值[1]。
催化材料、金属有机框架材料、热电材料、锂离子电池材料以及钙钛矿光伏材料各自具有相对有规律的结构组成方式和丰富的结构组分/结构组成基元,便于程序化大规模生成海量潜在结构,并实施高通量计算以及进一步进行数据处理,是材料基因方法合适的实施领域,本文将着重就这五类材料基于高通量计算的优化设计进行简要介绍,作为材料基因方法的应用范例。除此以外,其他材料领域也已有材料基因方法的应用,但限于篇幅和笔者的知识水平有限,暂不在本文涉及。
1 催化材料的设计
得益于表面反应理论与密度泛函方法的发展,通过理论计算能够越发深入地描述表面反应过程,对于催化剂的构效关系理解也更加深刻,因此采用理论计算来指导设计新催化剂得到了越来越广泛的关注[6]。近年来,通过使用密度泛函理论(Density functional theory, DFT)对催化材料表面进行电子结构探究,极大地促进了对固体表面化学性质的认识,为非均相催化和电催化领域反应机理提供了更深入的理解,从而对新型催化材料的探索提供了指导。从DFT计算中获得的活性物种的表面吸附能可以有效描述表面催化活性,因此被作为预测材料催化活性的一种描述符,被广泛运用到机器学习中[7]。Li等[8]基于吸附能的预测开发了一个机器学习框架,用于加速发现双金属催化剂。这种方法基于不同活性中间体与催化材料之间吸附能存在的线性关系,将复杂反应的动力学参数简化,通过高通量计算获得的大量数据,筛选出最优指纹特征,结合一部分吸附物种在金属表面的吸附能来训练人工神经网络,从而估算复杂非线性的吸附物与催化位点之间的相互作用,最终准确预测潜在催化剂。相比于案例式(case by case)DFT计算的高计算成本,此工作提供了一种替代方法,通过考察指纹描述符与已知催化剂吸附性能之间的相关性,快速预测新催化剂反应活性。
除了上述神经网络方法预测吸附能外,Jinnouchi等[9,10]使用贝叶斯线性回归方案,基于MAE为0.1 eV的单晶平板表面的DFT计算数据,预测了RhAu纳米颗粒NO分解关键中间体的吸附能。并基于具有相似原子构型的位点有望具有相似活性的事实,使用平滑重叠原子位置(Smooth Overlap Atomic Position , SOAP)相似性内核作为描述符。最终,通过这些描述符了解了表面分离,催化速率以及组成的依赖性,并获得了NO分解过程中RhAu纳米颗粒上活性位的详细结构。与此同时,Toyao等[11]发现,将树性回归与十二个数据库中的可用描述符相结合,能够预测甲烷相关的中间体在Cu基合金上的吸附能且其RMSE为0.26 eV,并运用此发现优化甲烷利用率来进行甲醇合成以及甲烷的氧化偶联。Ulissi等[12]以及Tran等[13]利用神经网络电位来预测金属间化合物的吸附能。以上工作说明,高通量计算与机器学习相结合的材料基因方法在研究设计催化材料方面展现出巨大的潜力,并且能够有效地在多金属合金以及化合物的广阔材料空间中有效快速地确定合适的候选催化剂。
为了能更加准确地通过数据技术预测催化材料表面的吸附能,以设计潜在的优异催化材料,需要考虑不同催化表面以及不同吸附位点。目前多数基于DFT计算的表面科学研究很大程度上依赖于研究人员的直觉,往往仅考虑了部分代表性的表面,吸附物种的吸附构象也源自研究人员主观的初始猜测。这就阻碍了不同表面吸附性质计算的自动化、高通量实施。尽管采用贝叶斯优化等方法能够对吸附物结构进行全局优化,甚至对给定系统的势能面进行无穷采样,但计算成本过高。为了解决上述问题,Joseph H. Montoya等[14]提出了一种高通量计算工作流程,以自动化计算固体表面的吸附能(工作流程如图1所示)。这一系统能够为任意固体结构构造对称的吸附界面模型,引入了能够根据界面结构特性自动确定吸附结构的算法,并能生成不同的吸附构型,计算得到的数据可实现高通量获取。整个工作流程实现了灵活处理各种吸附物、体相材料以及不同表面模型,无需手动调节单个作业吸附态的几何结构和计算参数,显著降低了计算管理量。为通过高通量计算快速形成表面吸附性质计算数据库、应用数据挖掘方法、发现用于催化的先进材料提供了强有力的工具。最近,Boes等[15]在这个方向上进一步进行了详细说明,报告了基于图论的方法来枚举表面和独特的化学吸附结构,对任何可能的吸附结构创建独特的系统表面表示,并为这些结构生成了三维初始猜测。
图1
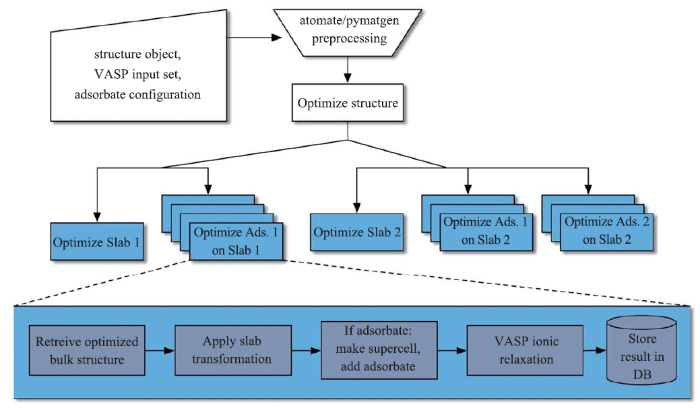 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1用于计算表面物种吸附能的自动化工作流程
Fig.1Automated workflow for calculating surface species adsorption energy
在实施高通量计算构造催化材料数据库方面,Osman Mamun等[16] 在双金属合金材料方面开展了大量工作。双金属合金主要由37种选定的金属组合构成,他们通过高通量DFT计算,考察了1998种双金属合金以及37种纯金属表面的H、C、N、O和S的原子吸附,考虑了不同的吸附位点,生成了包含90000多个系统计算结果的数据集,并将数据集在www.Catalysis-hub.org平台上公开[17](数据库平台示意图如图2所示)。与表面吸附和催化过程相关的理论计算数据库建设,对于后续开展数据挖掘发现新材料有很大的促进作用。
图2
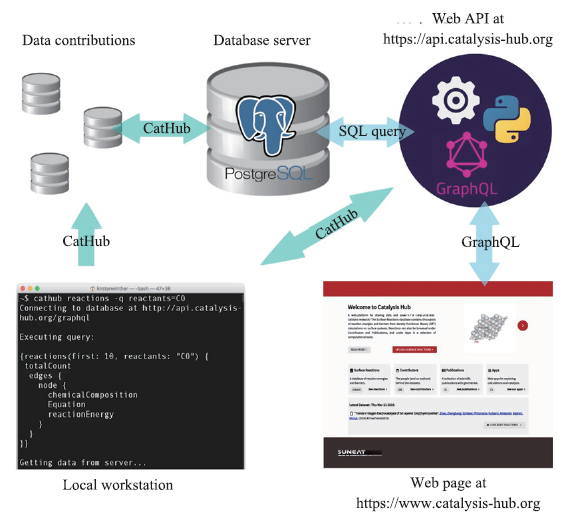 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2Catalysis-hub数据库平台示意图
Fig.2Catalysis-hub database platform diagram
当前的多数表面高通量计算仅考虑了反应的热力学可能性,形成的数据库包含了大量表面吸附能数据[18]。而由于确定表面反应动力学能垒的过渡态计算需要很高的计算成本,数据库极少涉及动力学能垒信息。随着机器学习等数据处理技术的发展,能垒的计算将可能显著加快[19]。另一方面,表面性质计算数据库的不断丰富,也为以机器学习为代表的替代预测模型提供了针对性的数据集。未来巨大组成和自由度的化学空间探索,将更多依靠高通量计算与替代模型的组合使用。各种新数据方法的应用将大大降低计算成本,缩短研究周期,为新材料的快速探索和准确预测做出更大贡献。
2 金属有机框架的筛选
近年来金属有机框架(Metal-Organic Frameworks,MOFs)作为一种新型多孔材料日益受到关注。MOFs主要是由无机金属离子与有机配体自组装形成的具有周期性网络结构的晶体材料。由于密度低、比表面积大、结构功能具有广阔的设计调控空间、孔道尺寸可调控等特点,MOFs在气体吸附分离以及储能等领域具有很好的应用前景[20]。随着对MOFs研究的不断深入,已合成的MOFs数量呈指数增长,由于存在大量可能的金属节点和连接配体结构以及丰富的组合方式,MOFs的结构数量几乎可以认为是无限的,如何在如此众多的候选材料里,快速为给定的应用找到最佳的MOFs结构呢?随着计算技术的发展和高通量计算筛选技术的出现,设计识别潜在的高性能MOFs材料并揭示其内在构效关系成为可能[21]。
目前实验室新合成的MOFs晶体结构会存储在Cambridge Structural Database(CSD)数据库[22]中,但不会贴上MOFs标签。数据库中许多MOFs结构中包含溶剂分子,结构信息存在不同程度的无序、氢原子的缺失、原子重叠等问题[21],对于基于已有实验结构的高通量计算筛选,首先要获得合理的结构。Watanabe等[23]为了筛选用于气体吸附分离的MOFs,在CSD数据库中识别提取出30 000种可能的MOFs材料,检查这些材料是否存在骨架特征结构。对含有溶剂分子的结构,根据电中性原则自动剔除溶剂分子;对难以纠正的无序原子进行标记并剔除。对于合理的结构采用力场方法(Universal Force Field,UFF)计算吸附剂与吸附质的相互作用,使用高通量巨正则蒙特卡洛方法(Grand Canonical Monte Carlo)以及分子动力学(Molecular Dynamics)模拟气体在材料中的吸附和扩散性能,从而探索最有希望用于分离CO2/N2的膜材料。之后,Goldsmith等[24]围绕气体捕获和存储应用,使用结构合理化算法,从CSD数据库中高效识别清理MOFs结构,并对筛选出的约22 700种化合物进行表面积、孔体积以及氢气吸收性能表征,最终预测了4 000多种化合物的理论储氢量(结构处理流程图如图3所示)。Gokay Avci等[25]运用高通量计算对MOFs材料进行了筛选,以此来评估其吸附剂和膜对CO2/H2分离的性能。最终发现孔径狭窄、孔隙率低的MOFs是从H2中分离CO2的最佳吸附材料,相反,孔径大、孔隙率高的MOFs是对H2进行选择性分离的最佳膜材料。Cigdem Altintas等[26]筛选了最新的MOFs数据库来显示MOFs对CH4/H2分离的最终极限性能,研究了4 350种MOFs的结构特性及其气体分离性能之间的关系,这也将触发后续的实验性工作,加快具有更好分离性能的新型MOFs的设计。Seda Keskin等[27]在无限稀释下模拟了3765种MOFs中单组分H2及N2的吸附和扩散,并计算了所有组分的H2及N2渗透率以及H2/N2的选择性MOFs膜。Hilal Daglar等[28]使用高通量计算对MOFs进行了筛选,评估其在混合基质膜(Mixed Matrix Membranes , MMMs)中作为填料的性能并对此进行了经典的蒙特卡洛和分子动力学模拟,计算了7822种合成MOFs的CO2和N2的渗透率,最终,评估了109 508种不同类型的基于MOFs的MMMs的CO2渗透率和CO2/N2的选择性。Qiao等[29]结合蒙特卡洛及分子动力学模拟,在MOFs的几何特征(密度、孔隙率及表面积等)与膜性能的评估标准(渗透率和渗透选择性)之间建立了结构性能关系,并运用主成分分析法(Principal Component Analysis , PCA)评估描述符之间的相互关系,应用多元线性回归及决策树模型进一步处理,最终确定了7种最佳的MOFs膜,用于从CH4中一步分离CO2和N2。
图3
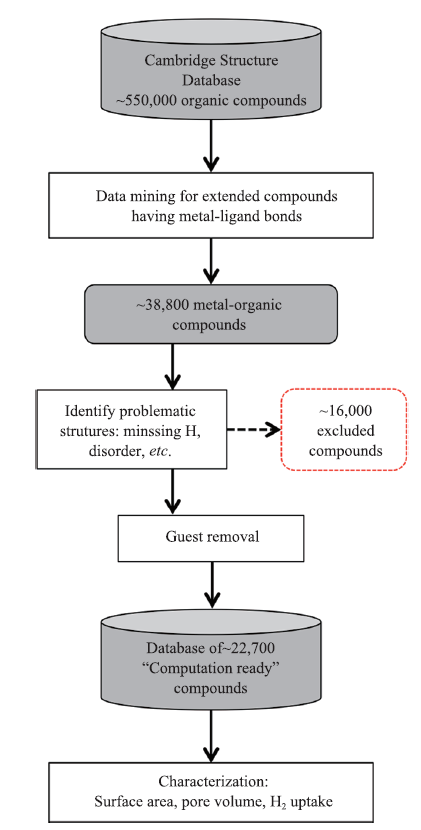 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3晶体结构数据处理分析流程图
Fig.3Process flow of analysis on crystal structure data
除了从现有数据库中筛选有特定功能的MOFs材料,还可以通过高通量计算预测实验中未制备但可望具有极佳性能的结构,从而指导实验合成制备。Wilmer等[30]从晶体学数据库中得出MOFs的常见构造块,经自动重组生成了137 953种假想的MOFs,采用高通量巨正则蒙特卡洛模拟甲烷的吸附等温线,最终确定了300多种可用于高压、室温下甲烷存储的潜在材料,并通过实验证实了其中一种结构的预测甲烷吸附量。类似的,Diego等[31]借助高通量巨正则蒙特卡洛模拟,研究了122 835种假设的MOFs以及39种理想的碳基结构在室温不同压力下的甲烷的吸附量,从而归纳出增加强作用位点的体积密度,同时提高输运温度,可能是一种提高多孔材料的甲烷输送量的合适方法。
随着合成和预测的MOFs结构数量不断增加,高通量计算筛选技术有望在特定应用的新MOFs开发中扮演更重要的角色。
3 热电材料的筛选
热电材料是一种新型的能量转换材料,能够实现电热之间的直接能量转换。热电优值(ZT值)常被用来衡量热电材料能量转换效率,为了达到较高的ZT值,热电材料应具有优异的电运输性能以及较低的热导率[32]。传统的热电材料研究一般关注少量样本的电热输运性质理解和优化,难以发现系统性规律,对新体系设计优化提供的帮助有限。随着材料基因方法的发展,研究者们将理论计算与热电材料设计相结合,取得了一系列突破性的进展。Madsen[33]在国际晶体结构数据库(ICSD)中用高通量方法自动搜索了570种含锑化合物中的新热电材料。Wang等[5]通过使用AFLOW考察了2500多种新化合物,从中得出功率因素与材料固有参数之间存在很强相关性的结论。Xi等[34]使用基于材料信息平台的高通量工作流程,运用三个筛选标准:(1)IB,IIB,IIIA和IVA为阳离子,S,Se和Te为阴离子;(2)FCC阴离子亚晶格;(3)阳离子配位数= 4。从82 412种材料中筛选出214种候选材料并对它们进行了自动计算。最终,根据其半导体材料的性质,对214种候选材料依据带隙标准(> 0.1eV)再次进行筛选,进而减少到161种候选材料,结合计算出的功率因数,成功预测了新型高性能热电材料。Zhang等[35]运用高通量计算基于热电材料的晶体结构特征以及三个筛选标准对无机晶体结构数据库的材料进行筛选,最终确定13种化合物作为潜在的基于Cu-S的热电材料。Robert等[36]对洛伦兹数极低且热电品质因数极高的材料进行了高通量计算搜索,最终确定了潜在的具有高品质因数以及低洛伦兹数的热电材料。Guo等[37]对具有高电性能的热电材料Half-Heusler化合物使用经过实验研究的NbFeSb及ZrNiSn化合物的热电性质作为筛选标准进行了筛选。Chen等[38]使用Materials Project里收录的48 000多种材料体系作为研究对象,对其中大约25 000种半导体材料进行电、热传输性质的理论研究,运用数据技术对热电材料进行探索,在多样的数据中找到预测相关热电材料性能的特征描述符,例如电负性、带隙等。在此基础上使用密度聚类算法将5 431种材料的数据分成六类,分别分析了对热电材料性质影响很大的关键描述符。
热电材料的热导率与材料的晶体结构和组成原子质量等因素有着密不可分的关系。随着热电材料领域数据量的快速增长,基于大量数据,生成热导率预测模型成为一种快捷有效的材料预测手段[39]。Carrete等[39]对元素周期表里所有非放射性元素进行排列组合,从中得到大约79 000种半哈斯勒合金结构的材料,随后通过形成能的计算以及热力学分析等方法,得到了75种稳定的半哈斯勒合金,最后结合机器学习模型来估算热导率。相比限制在半哈斯勒区域进行探索,Atsuto Seko等[40]不通过经验知识来限制探索空间。他们通过第一性原理近似求解玻尔兹曼输运方程,来评估岩盐、闪锌矿和纤锌矿三种原始结构的化合物的低晶格热导率。将计算结果用来构建模型,随后使用贝叶斯优化,对具有多种结构和化学组成的数据库进行“虚拟筛选”,最终对排名靠前的化合物进行第一性原理的低晶格热导率计算,以验证筛选结果。
虽然高通量计算指导的热电材料筛选已经取得了一定的成果,但仍然存在很多挑战。首先,预测的材料经过实验合成表征,分析实验数据与理论预测之间的差异根源极具挑战性。另一方面,筛选出的热电材料在实验中也许没有可行的合成方法,预测的材料难以得到验证,预测的可靠性时常停留在理论阶段。总之材料基因方法在热电材料领域还有很大发展空间,理论计算预测应该与实验方面进一步结合,准确预测高性能热电材料仍然是一个挑战,为未来的研究提供了大量机会[41]。
4 锂电池材料筛选
锂电池因其高能量密度的优点,已成为便携式电子设备不可或缺的组件,且广泛应用到生活的各个领域中,例如混合动力汽车等。为了进一步提高锂离子电池性能,满足应用中不断增长的需求,研究者们进行了广泛的研究,以期实现高电压、高能量密度、低成本、高安全性、长寿命和环境友好等一系列目标[42]。近年来,高通量计算大大加快了对新型锂电池电极材料的筛选。相较于较为成熟的碳阳极材料,寻找满足需求的阴极材料是发展高性能锂离子电极的一大难点。Geoffroy Hautier等[43]通过高通量计算考察了不同含锂化合物和含钠化合物的稳定性以及可能的充放电电压和能量密度,发现将一种已知材料Na3Mn(CO3)(PO4)中的Na替换为Li,可能实现高理论容量(>200mAh/g),同时也提出了若干新颖的碳磷酸盐和碳硅酸盐可作为候选阴极材料。为了兼顾高工作电压和高安全性,Anubhav Jain等[44]使用高通量密度泛函计算评估了1 400多种阴极材料的工作电压与热力学氧气释放温度的关系,研究了聚阴离子基团、氧化还原能力等对材料性能的影响,筛选出几种兼具高工作电压和高安全性的阴极材料。基于尖晶石结构的锂锰氧化物被认为是一种典型的锂离子电池的阴极材料候选体系。Zhang等[45]开发了基于从头算和相图计算(CALPHAD)方法的高通量计算框架,系统描述了成分-结构-性能关系,并评估尖晶石化合物在电池应用中的性能[46],继而通过实验对整体性能进行评估,得到在正八面体位置掺杂的出色阴极材料。高通量计算的流程图如图4所示。
图4
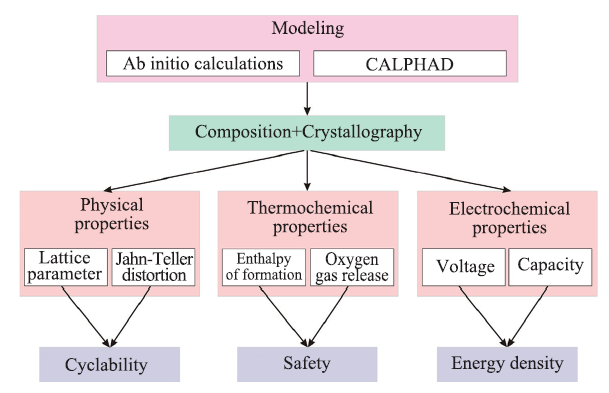 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4锂电池材料高通量计算框架的流程图
Fig. 4Flowchart of high-throughput calculation framework for lithium battery materials
此外,对于阳极材料的研究近些年也逐渐受到关注。Scott Kirklin等[47]研究了过渡金属硅化物、磷化物以及锡化物这三类活性-惰性化合物的阳极材料,运用高通量计算得到了几种潜在的阳极材料,包括CoSi2、TiP和NiSi2,它们的储放锂可能显著优于商业化的石墨碳电极。
除了寻找容量更高的电极材料来提高锂电池性能外,如何防止阴极降解也是提高锂电池寿命需要解决的问题。通常电极材料的降解原因主要包括电极材料活性表面积减少以及过渡金属离子溶解到电解质中等变化[48],这些过程会导致锂电池循环性能下降。常用的抑制阴极材料退化的策略是在阴极施加保护层,常见的稳定氧化物,如Al2O3、MgO、ZnO等,在抑制过渡金属流失和容量衰减过程中性状可能会发生很大的变化,因此寻找稳定涂层材料也是新的研究热点。Muratahan Aykol等[49]提出了一种高通量密度泛函理论计算框架,该框架包括描述候选氧化物以及含氧阴离子化合物的热力学稳定性、电化学稳定性以及材料除酸能力的反应模型。通过筛选130 000种含氧材料,发现了潜在的优异涂层材料,如WO3、LiBO2、Li2SrSiO4、Li2CaSiO4、LiMn2O4等。这些涂层材料相比常规涂层,在氢氟酸清除以及热力学性能方面表现更优。
5 钙钛矿型光伏材料的优化设计
随着光伏产业的迅速发展,钙钛矿型光伏材料,尤其是混合有机-无机卤化物钙钛矿作为一种新一代太阳能电池材料,凭借其优异的光电性能成为当下的研究热点[50]。目前研究最多的混合卤化物钙钛矿是铅基卤化物,它们易于合成且成本低廉,但化学稳定性差、有毒的缺点也限制了它们的应用,因此快速寻找无毒且稳定的新型杂化无铅卤化物钙钛矿材料成为当下需要解决的问题[51]。Sudip Chakraborty等[52]介绍了一种基于密度泛函理论的高通量计算筛选以及向下选择策略来加速无铅钙钛矿型太阳能电池材料的发现。根据多种可能的钙钛矿结构如ABX3、A2BX6、A3B2X9等,进行能量和能量稳定性的筛选;随后基于带隙等基本描述符进行光学响应的筛选和过滤;接着对载流子的有效质量以及迁移率进行附加筛选并在此之后进行更高级的电子结构计算,得出准确的能带结构和光学性质;最终,进行对最终得到结构的实验验证。Takahito Nakajima等[53]使用密度泛函理论对11 025种ABX3与A2BBX6形式的有机-无机卤化物钙钛矿进行了高通量计算,发现了51种低毒卤化物钙钛矿材料。Li等[54]展示一种无铅混合卤化物半导体的高通量计算和筛选方法:在基于钙钛矿和非钙钛矿结构以及几种典型有机阳离子的24种原型结构上,建立了一个包含4 507种设想的杂化化合物材料库,随之采用密度泛函计算对结果数据库进行高通量筛选,最终确定了23种可用于发光二极管以及13种可用于太阳能转换的潜在材料,展示了设计新颖的有机-无机功能材料的新途径。
机器学习技术的应用,绕开了部分复杂的量子力学计算,极大加速了材料设计的进程。最近的研究和应用也表明,通过机器学习可以更高效、更精确地执行钙钛矿稳定性与组成离子半径等特征之间的映射关系。Balachandran 等[55]基于390个实验上已经报告的ABO3化合物的数据集应用了两个机器学习模型,以此预测了235个新的潜在的ABO3钙钛矿且置信度高于50%。Li等[56]利用Materials Project[57]提供的1 929种钙钛矿氧化物的热力学数据,对机器学习模型进行训练,并打算基于此模型预测更多的钙钛矿氧化物。Xie和Grossman等[58]根据Materials Project中DFT计算出的数据建立了卷积神经网络的智能晶体图,便于直接从晶体结构预测材料性能。Li等[59]提出了一种将机器学习与DFT计算相结合的策略来预测稳定的卤化物钙钛矿。将已计算出354种钙钛矿的DFT结果作为训练集,在钙钛矿稳定性和组成离子半径之间建立性质-特征关系并通过与354种DFT计算的246种钙钛矿的实验可成型性进行比较,验证此模型的有效性。最终得出机器学习模型预测的稳定性趋势与可用的实验数据是高度吻合的,以此证明机器学习模型在设计稳定钙钛矿方向上有着应用潜力。相关原理如图5所示。Lu等[60]开发了一种基于高通量计算数据与机器学习相结合的方法来预测混合有机无机钙钛矿光伏材料,整个工作流程如图6所示。运用如图7所示的皮尔逊关系图筛选出14个最佳特征值,成功地从5 158个未发现的结构中筛选出六种具有适当带隙的斜方形无铅钙钛矿。
图5
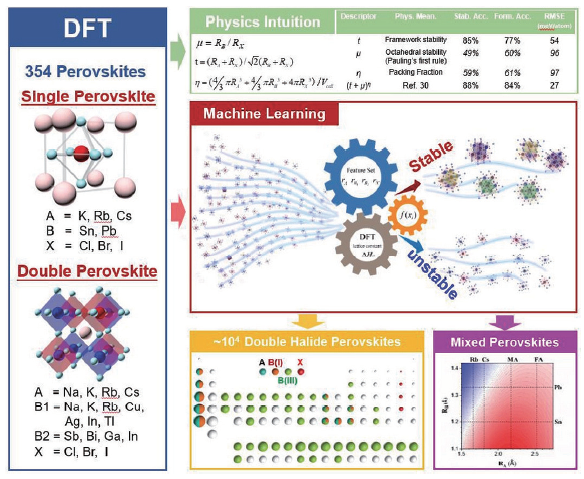 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5基于DFT计算的机器学习模型在钙钛矿卤化物高通量稳定性工程上的工作原理图
Fig. 5Working principle diagram of machine learning based on DFT calculation in high-flux stability engineering
图6
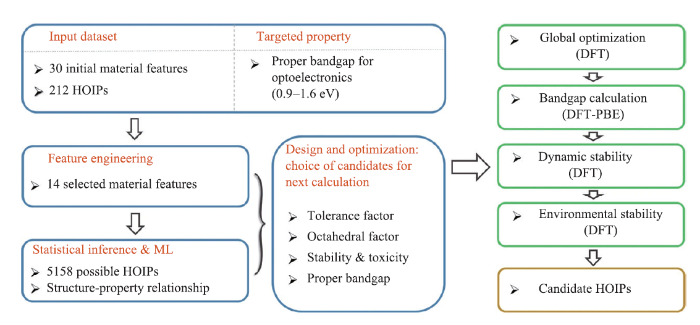 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6混合有机无机无铅钙钛矿设计框架
Fig. 6Framework for designing organic-inorganic lead-free perovskite
图7
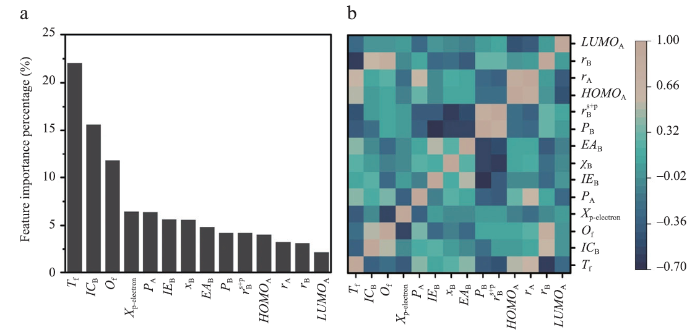 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7所选特征的重要性和相关性
Fig.7Importance and relevance of selected features
此外合适描述符的探索也扮演着非常重要的角色。Lu等[61]在先前的工作基础上,通过结合高通量计算与机器学习技术进行多步筛选,提出了两个新的描述符,用来描述实现混合无机钙钛矿的可形成性,为构建描述符开辟了一条新的道路。未来,高通量计算与机器学习等技术在光伏材料领域的应用将会更加广泛,同时对有效特征描述符的开发也将提出更高的要求。
6 总结与展望
本文从催化材料的设计、金属有机框架材料的筛选、热电材料的筛选、锂电池材料的筛选以及钙钛矿型光伏材料的设计方面介绍了高通量计算的典型应用。高通量计算在材料研究领域逐渐兴起,并且随着计算技术的迅猛发展,高通量计算与机器学习等数据挖掘技术结合的材料基因方法将为材料科学发展注入新的活力。但理论模拟与设计还应与实验更加紧密地结合,期待计算筛选出来的新材料结构能够在实验中实现合成和验证,这也将是未来数据驱动的材料设计领域发展所需要面临的挑战。利益冲突声明
所有作者声明不存在利益冲突关系。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[J].
[本文引用: 2]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 2]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 2]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 2]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]
[J].
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}