1.Schools of Materials Science and Engineering, China University of Petroleum (East China), Qingdao 266580, China 2.Schools of Mechanical and Electronic Engineering, Qingdao Binhai University, Qingdao 266555, China
Fund Project:Project supported by the Natural Science Foundation of Shandong Province, China (Grant No. ZR2017MEE005)
Received Date:03 May 2019
Accepted Date:21 August 2019
Available Online:01 November 2019
Published Online:05 November 2019
Abstract:In this work, the competitive adsorption behavior of H2 and CO on strained Fe(110) are investigated by the first-principles method based on the spin-polarized density functional theory to study the hydrogen embrittlement of steels. The results show that the most stable adsorption site for CO is top site, and the orbital of CO molecule hybridizing with Fe 3p and 4s states illustrates a strong electronic interaction between them. The adsorption energy values of CO at the four calculated adsorption sites are more negative than those of H2, which favors the binding with Fe(110) surface. The potential energy variations for CO and H2 molecules close to the surface are calculated. The attractive force of the Fe(110) surface acting on CO in 1.5–3 ? is greater than that acting on H2. The pre-adsorbed CO increases the dissociation energy barrier of H2 from 0.08 eV to 0.13 eV but reduces the force between H2 and surface. The surface tensile strain enhances the interaction between hydrogen and Fe(110), which, however, is reduced by the compressive strain. The opposite tendency is found in the adsorption of CO. The binding strength of CO is stronger than that of H2 on the strained Fe(110) surface. The difference in adsorption energy between CO and H2 decreases with tensile strain increasing. The effect of surface strain and partial pressure of CO gas phase on the surface coverage ratio of H atom are also calculated quantitatively based on thermodynamics at 298 K, with the partial pressure of H2 set to be 10 MPa. The surface ratio of the H atom decreases with partial pressure of CO increasing. The hydrogen coverage drops nearly to zero when the partial pressure of CO reaches a certain value. This result reveals that CO can inhibit hydrogen adsorption on Fe surface. In the case where the surface ratio of hydrogen decreases to 1%, the corresponding CO partial pressures are 105 Pa, 1.1 × 103 Pa, 2.4 × 105 Pa on –2%, 0, 2% strained Fe(110) surface, respectively. High CO partial pressure is needed to suppress the hydrogen adsorption since the binding strength of CO is close to that of H2 on the expanded surface. Keywords:hydrogen embrittlement/ competitive adsorption/ adsorption of hydrogen
表1CO与H2在Fe(110)表面不同位置的吸附能(eV) Table1.Adsorption energies of CO and H2 on high symmetry sites of Fe(110).
图2为CO分子吸附在top位前后的分波态密度(PDOS)图. 可以看出吸附前, 孤立的CO分子中C原子和O原子有强烈键合作用, 从低能到高能的分子轨道分别是3σ, 4σ, 1π, 5σ, 2π. 其中5σ是最高占据分子轨道, 2π是最低未占据分子轨道. 吸附前, Fe原子的态密度分布在–8—22 eV能量区间. CO吸附使得Fe原子自旋向上态密度和自旋向下态密度差异减小. 吸附后, Fe原子的4s, 3p在–22.6 eV, –8.9 eV处, 3d在–5.7 eV出现较大的共轭峰. 表明表面Fe原子与C原子存在强烈的杂化耦合作用. 该杂化耦合作用使得C原子的2p态展宽, 形成成键态和反键态. 成键态使得C原子与Fe原子的作用增强, 是CO在Fe表面吸附能较大的原因. 图 2 CO, Fe(110)面及其top位吸附前后分波态密度图 (a)吸附前; (b)吸附后 Figure2. Projected local density of states from Fe on clean Fe surface and a CO molecular in vacuum (a) and with a CO adsorbed surface (b).
式中Eslab, Eslab+H分别是表面吸附氢原子前后体系的能量; $ E_{\rm H_2} $表示氢分子的能量. 表1列出了氢分子的吸附能. 氢分子在tf位置的吸附能最负, 为–1.33 eV, 表明tf位的吸附最稳定, 与Wang等[20]的计算数据–1.36 eV基本一致. 比较CO和H2的吸附能数据可以看出, 4个吸附位CO的吸附能均比H2的吸附能更负, 表明CO与Fe(110)表面的结合强度大于H2与Fe(110)面的结合强度. 其中CO吸附在sb位与H2吸附在tf之间结合强度差值最小, 为0.31 eV. 计算了H原子在表面tf位吸附前后的态密度, 如图3所示. 从图3可以看出, 吸附后H的1s电子向左移动到–7.3— –5.2 eV, 存在明显的波峰. Fe原子的4s电子在–7.8— –5.8 eV之间出现新的峰、3p电子与3d电子在–6.2 eV出现弱小的峰, 表明Fe的4s, 3p, 3d电子与H的1s电子存在共轭杂化作用, 共轭峰值较小, 说明杂化作用较弱. H原子的吸附对Fe原子自旋向上和自旋向下的态密度差异影响较小. 图 3 H与Fe(110)面tf位吸附前后分波态密度图 (a)吸附前; (b)吸附后 Figure3. Projected local density of states from Fe on clean Fe surface and a H atom in vacuum (a) and with a H adsorbed surface (b).
23.3.势能随距离的变化 -->
3.3.势能随距离的变化
为分析CO对H2的吸附以及解离过程的影响, 计算H2, CO分子逐渐靠近Fe(110)表面的势能变化. 计算过程中限定吸附物的Z向坐标, 仅对X, Y坐标进行弛豫. Wang 等[33]研究发现垂直构型靠近铁表面的H2难以解离. 且前面分析表明CO垂直吸附于Fe(110)表面. 因此本文分别计算与表面垂直的CO、平行H2以及预先吸附CO平行的H2在靠近表面过程中的势能变化, 计算结果如图4所示. 图中横坐标各距离分别为CO中的C原子到表面的距离、H2两个H原子的中心到表面的距离以及预先吸附CO时H2的中心到表面的距离. H2在靠近表面的过程中, 能量先降低, 后又逐渐升高, 到2.4 ?能量到达最高点. 结构优化中发现距离小于2.4 ?时, 如不固定H2的Z坐标, H2直接解离为H原子并吸附在tf位. 距离减小过程中, CO的吸附能不断降低, 到达1.5 ?时, 能量最低, 形成CO的分子吸附态. 能量随距离变化的斜率表示作用力的大小. 1.5—3 ?范围内, 表面对CO的吸引力明显大于H2, 这是表面优先吸附CO的原因之一. 图 4 CO与H2吸附过程中的势能变化 Figure4. Potential energy variations of CO and H2 moving towards Fe(110).
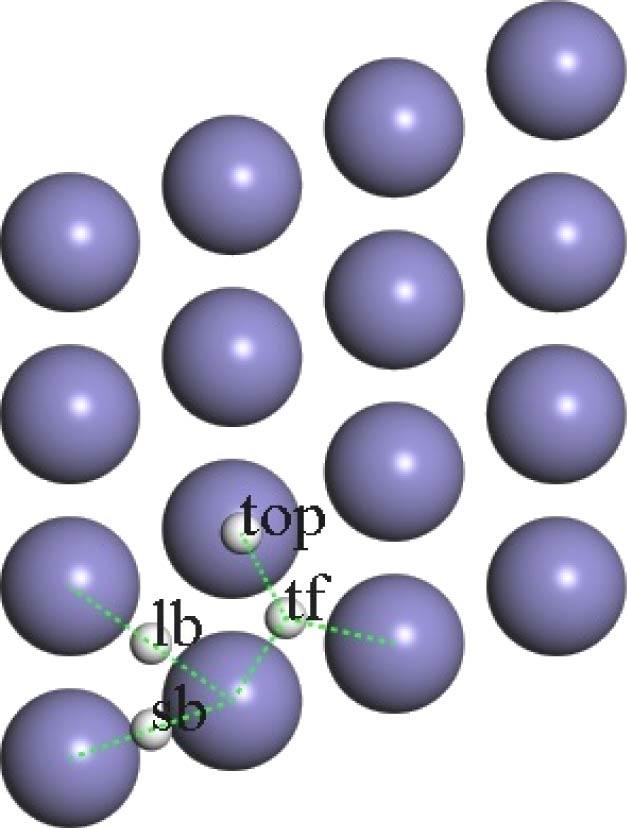 图 1 Fe(110)面及其对称点
图 1 Fe(110)面及其对称点








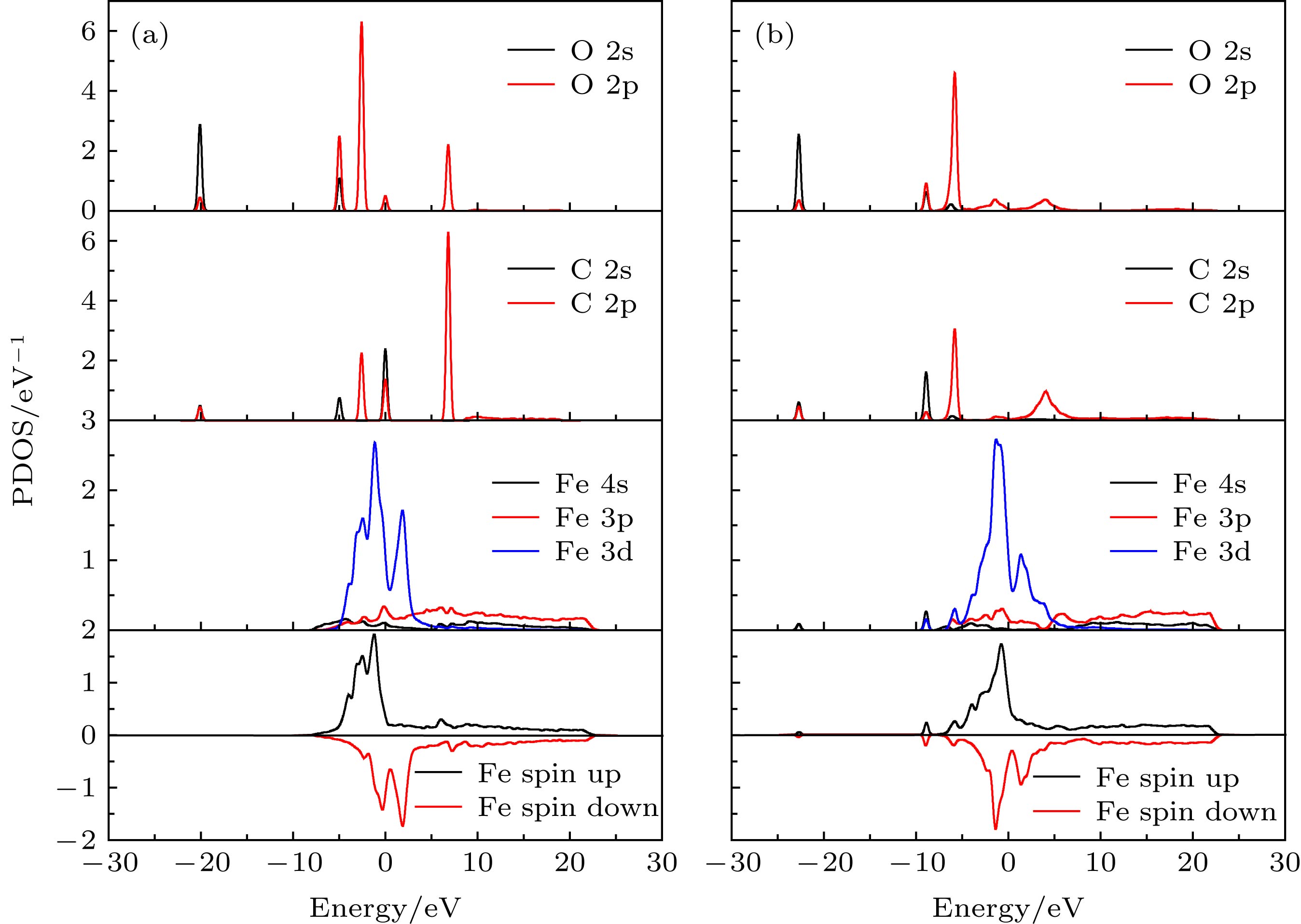 图 2 CO, Fe(110)面及其top位吸附前后分波态密度图 (a)吸附前; (b)吸附后
图 2 CO, Fe(110)面及其top位吸附前后分波态密度图 (a)吸附前; (b)吸附后

 图 3 H与Fe(110)面tf位吸附前后分波态密度图 (a)吸附前; (b)吸附后
图 3 H与Fe(110)面tf位吸附前后分波态密度图 (a)吸附前; (b)吸附后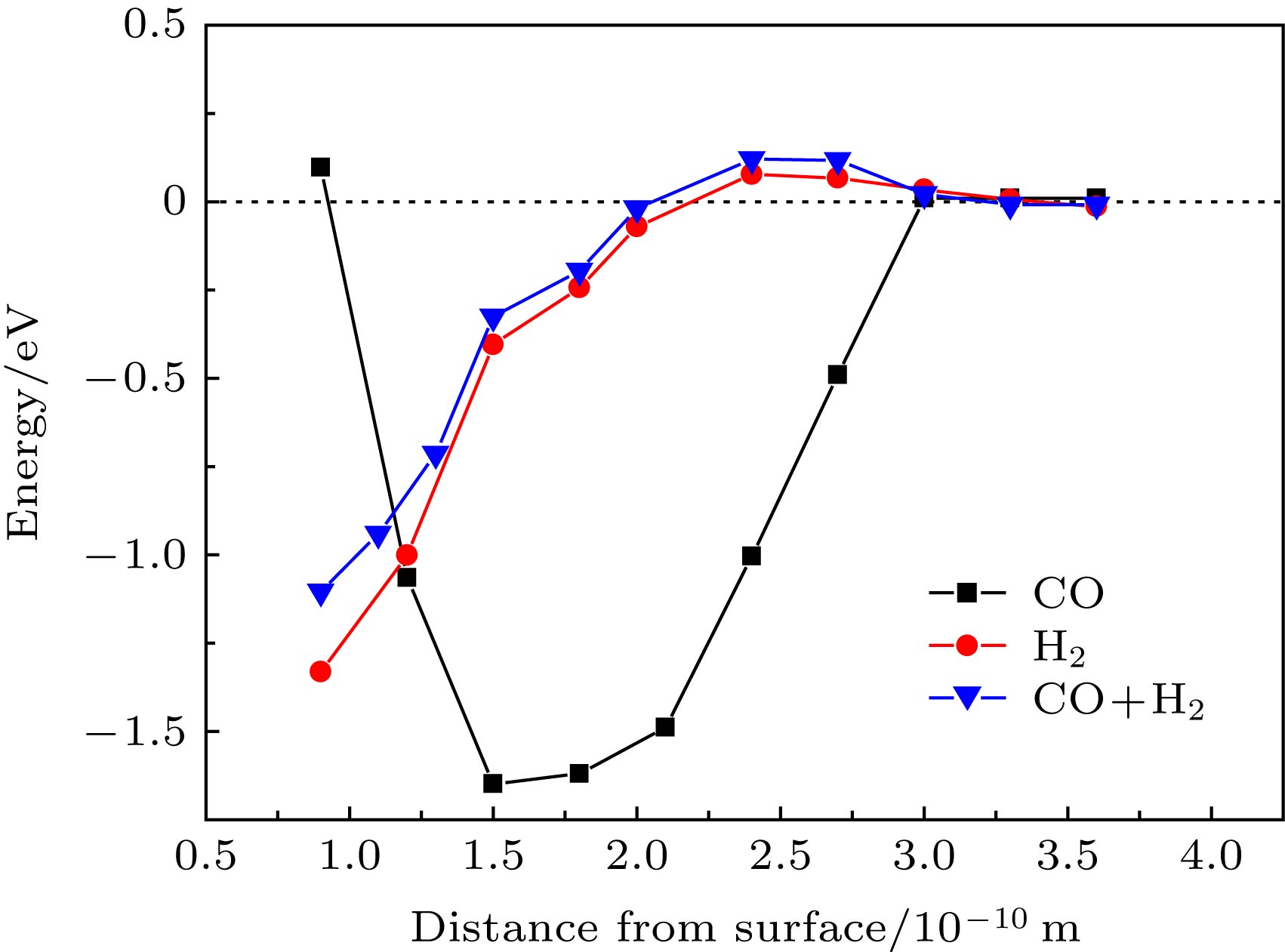 图 4 CO与H2吸附过程中的势能变化
图 4 CO与H2吸附过程中的势能变化 图 5 CO吸附能、H2吸附能和应变之间的关系
图 5 CO吸附能、H2吸附能和应变之间的关系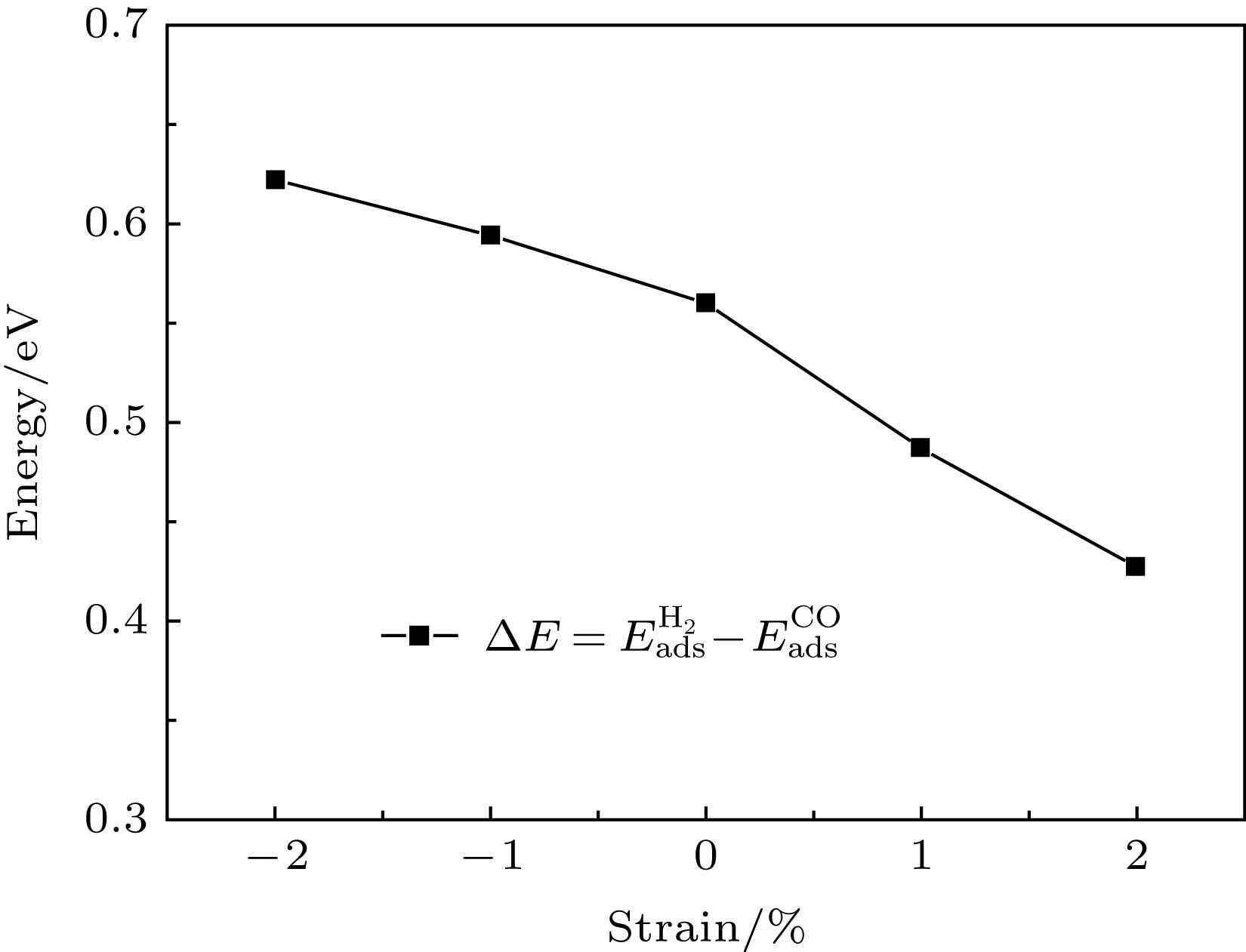 图 6 H2和CO吸附能的差与应变之间的关系
图 6 H2和CO吸附能的差与应变之间的关系
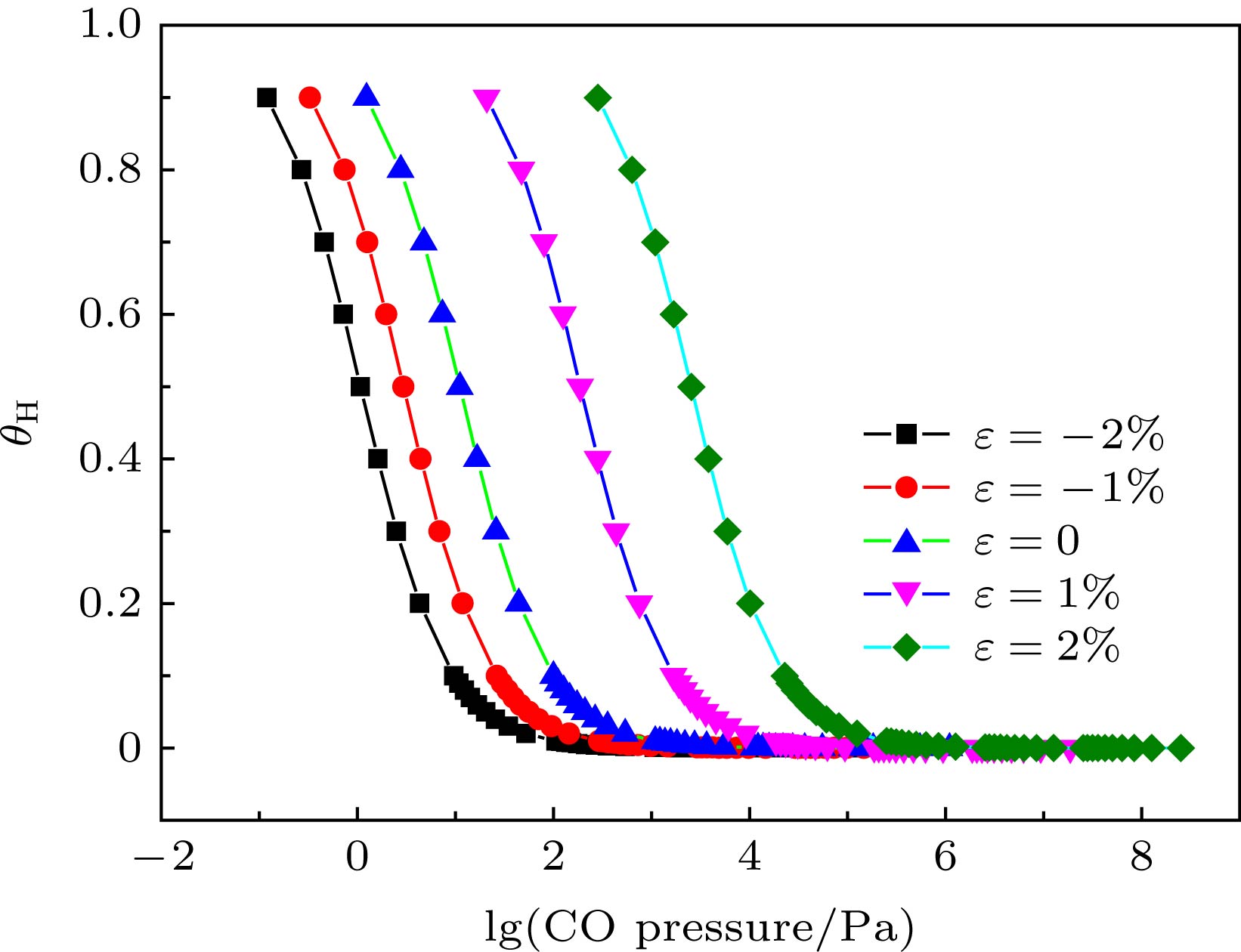 图 7 CO的分压与θH之间的关系
图 7 CO的分压与θH之间的关系