全文HTML
--> --> -->对于含有TPE基团的TPA和AIE材料, 实验者通常在TPE上添加给电子基团以增强TPA强度, 而给体的取代位置和数目存在多种可能. 不同的取代方式可以产生不同的多枝结构, 分枝之间的相互作用对非线性光学响应具有重要影响[17,18]. 因此, 需要研究给体的位置和数目对TPA性质的影响. 虽然给体或受体强度对TPA性质的影响已经被广泛研究, 但是关于给体或受体位置和数目对TPA性质影响的研究还很少[19]. Chattopadhyaya等[19]研究了一系列给体/受体取代的双蔥分子的TPA性质, 发现添加三对给体/受体的双蔥分子的TPA强度比具有特定排列的一对给体/受体的双蔥分子小. 他们的结果清楚地表明, 增加给体/受体基团的数目不一定增强体系的TPA强度. 据我们所知, TPE基团上给体位置和数目对TPA性质影响的研究还未见报道.
最近, Gu等[10]合成了一种新的光敏剂TPEDC分子. 该分子具有强TPA和荧光, 可以用于光动力学治疗. TPEDC分子含甲氧基、TPE和氰基基团, 具有D-共轭桥(π)-A型分子结构. 以TPEDC分子为模型, 我们研究了给体位置和数目对TPA性质的影响. 并且, 在研究结果的基础上, 根据具有AIE特性的分子结构特点, 综合考虑了结构与TPA性质之间的关系, 理论设计了具有更大TPA截面的分子结构, 为实验者合成具有TPA和AIE特性的功能材料提供了理论指导.











采用响应函数方法[21]来计算双光子跃迁矩阵元. 响应函数方法是一种含时微扰方法. 在该方法中, 双光子跃迁矩阵元可以通过二次响应函数的留数得到


当单色偏振光激发分子材料时, 微观TPA截面






应用Gaussian16[23]程序包, 在杂化密度泛函B3LYP的水平上, 采用6-31g(d, p)基矢, 对所有分子结构进行了几何优化. 在CAM-B3LYP泛函的水平上, 采用6-31g(d)基矢, 在Dalton2013[24]程序包中计算了分子的TPA截面. 并且, 通过极化连续模型考虑了水溶剂的影响. 我们曾应用B3LYP泛函计算过各种类型分子的TPA性质[15,16,25-29], 因此, 也采用B3LYP泛函计算了TPA截面. 发现两种泛函的计算结果相对趋势是一致的, 可以得到基本一致的结论. 对于电荷转移分子, CAM-B3LYP泛函由于考虑了长程修正, 原理上计算结果更准确, 近年来被广泛使用. 因此, 本文均采用CAM-B3LYP泛函计算TPA性质.
3.1.给体位置效应
TPEDC分子简称T分子, 其化学结构式见图1. 通过观察T分子的结构式, 可以想到, 在TPE单元上的两个甲氧基可能存在几种不同的取代方式. 并且, 两个乙烯基桥存在顺式和反式异构体结构. 考虑这两方面, 得到了T分子的几种异构体结构T1—T5. 它们的化学结构式如图1所示. T, T2和T4分子的结构差异是TPE单元上的两个甲氧基取代位置不同. T1, T3和T5分子具有顺式构型, 相应的反式构型分别是T, T2和T4分子. 优化的几何结构如图2所示. 优化结构显示, 所有分子的TPE部分均成螺旋桨结构. 分子中心的两个苯环不在一个平面上, 有一定角度的扭转, 二面角约为33°. 菁基两侧的苯环也不在一个平面上, 二面角约为72°. 此外, 计算结果显示, 这些分子异构体的能量从小到大依次为T, T1, T4, T5, T3, T2, 能量差异非常小. T与T1分子的能量差只有0.002 kcal/mol, T2与T3能量差为0.003 kcal/mol, T4与T5能量差为0.001 kcal/mol, T与T2的能量差也只有0.08 kcal/mol. 图 1 T和T1?T5分子的化学结构式
图 1 T和T1?T5分子的化学结构式Figure1. Chemical structures of T and T1?T5 molecules.
 图 2 优化的T和T1?T5分子的结构
图 2 优化的T和T1?T5分子的结构Figure2. Optimized geometries of the T and T1?T5 molecules
通过响应函数方法计算了水溶液下所有异构体的TPA性质. 六个最低激发态的TPA波长




| T | T1 | T2 | T3 | T4 | T5 | ||||||||||||
| ${\lambda _{{\rm{tp}}}}$/nm | $\sigma $/GM | ${\lambda _{{\rm{tp}}}}$/nm | $\sigma $/GM | ${\lambda _{{\rm{tp}}}}$/nm | $\sigma $/GM | ${\lambda _{{\rm{tp}}}}$/nm | $\sigma $/GM | ${\lambda _{{\rm{tp}}}}$/nm | $\sigma $/GM | ${\lambda _{{\rm{tp}}}}$/nm | $\sigma $/GM | ||||||
| S1 | 743 | 719 | 743 | 696 | 740 | 707 | 738 | 672 | 743 | 638 | 743 | 629 | |||||
| S2 | 650 | 4 | 649 | 2 | 651 | 10 | 651 | 6 | 652 | 8 | 653 | 4 | |||||
| S3 | 612 | 944 | 612 | 858 | 614 | 893 | 612 | 873 | 615 | 880 | 615 | 827 | |||||
| S4 | 572 | 305 | 572 | 334 | 578 | 212 | 575 | 259 | 578 | 136 | 580 | 175 | |||||
| S5 | 556 | 3 | 556 | 5 | 557 | 4 | 556 | 5 | 557 | 4 | 557 | 5 | |||||
| S6 | 540 | 15 | 540 | 9 | 542 | 2 | 540 | 5 | 542 | 3 | 541 | 2 | |||||
表1分子六个最低激发态的TPA波长


Table1.The TPA wavelength


2
3.2.给体数目效应
可以看出, 这些分子体系具有多枝结构, 分枝之间的复杂耦合决定了分子的TPA性质. 已有研究表明, 对于多枝分子, 分枝间的耦合对分子的光学性质有重要影响[17,18]. 根据耦合作用的强弱, TPA强度相对于分枝单元会表现出协同加强、简单加和、甚至削弱的不同趋势[30-32]. 人们通常采用简单的弗兰克激子模型来研究分枝间的耦合作用[17,18]. 在该模型中, 分枝间的相互作用主要考虑偶极-偶极相互作用. 在TPE各分枝上是否添加给体, 会影响TPE各分枝的偶极矩, 进而影响分枝间的相互作用. 因此, 在TPE上添加更多的给体取代基能否产生更强的TPA, 还需要进一步仔细研究. 为此, 我们考虑了R, S和U几种分子结构, 它们的化学结构式和相应的优化几何结构如图3所示. 对于R, S, T和U分子, TPE部分的给体数目不同. 结构R中的TPE部分没有连接甲氧基, 结构S中只有一个末端甲氧基, 结构U包括三个甲氧基, 其中两个添加在TPE基团的侧位上, 一个位于分子的末端. 图 3 R, S和U分子的化学结构式和优化的几何结构
图 3 R, S和U分子的化学结构式和优化的几何结构Figure3. Chemical structures and optimized geometries of the R, S and U molecules.
R, S和U分子的TPA性质的计算结果列于表2中. 与T分子类似, 这些分子仍然具有三个较强的TPA态. 对于S和U分子, 第三激发态具有最大的TPA截面, 而R分子TPA截面最大的态是第五激发态. R分子的第四和第五激发态的能量几乎是简并的. 可以发现, 随着取代基数目的增加, 三个TPA吸收峰的位置依次发生红移. 例如, 第三激发态的

| R | S | U | ||||||
| ${\lambda _{{\rm{tp}}}}$/nm | $\sigma $/GM | ${\lambda _{{\rm{tp}}}}$/nm | $\sigma $/GM | ${\lambda _{{\rm{tp}}}}$/nm | $\sigma $/GM | |||
| S1 | 719 | 500 | 729 | 646 | 754 | 769 | ||
| S2 | 631 | 27 | 642 | 19 | 658 | 7 | ||
| S3 | 595 | 597 | 603 | 785 | 621 | 931 | ||
| S4 | 557 | 4 | 567 | 640 | 580 | 118 | ||
| S5 | 556 | 853 | 557 | 4 | 557 | 4 | ||
| S6 | 540 | 99 | 541 | 29 | 541 | 11 | ||
表2分子六个最低激发态的TPA波长


Table2.The TPA wavelength


2
3.3.电荷转移
TPA性质与分子内电荷转移密切相关. 当分子从基态跃迁到激发态时, 分子内的电荷会重新分布. 为了更好地理解给体位置和给体数目对分子内电荷转移的影响, 计算了T, T2和T4以及R, S和U在基态和第一激发态的自然键轨道电荷分布情况. 为了便于分析, 将所研究的分子分成两部分, 四苯基乙烯基团连同给体甲氧基一起作为A部分, 剩余部分为B部分. 以T分子为例, 如图4所示. 图 4 T分子的A和B两部分
图 4 T分子的A和B两部分Figure4. The A and B parts of the T molecule.
表3列出了分子基态和激发态A和B两部分的电荷计算结果. 其中QA0和QA1分别代表A部分基态和第一激发态的电荷, QA表示A部分基态和第一激发态的电荷差. 同样, QB0和QB1分别代表B部分基态和第一激发态的电荷, QB表示B部分基态和第一激发态的电荷差. 所有电荷单位为e, 1e = 1.602 × 10–19 C. 分子T的A部分第一激发态电荷是0.3978e, 比基态电荷0.0298e具有更强的电正性. 相应地, B部分的第一激发态相比基态表现出更强的电负性. 这说明分子T的A部分是电子给体, 而B部分为电子受体. 对于其他分子, 情况亦如此. 可以看到, T给体电荷差QA为0.3680e, T2和T4的QA分别为0.3609e和0.3772e, 电荷差相差不大. 这说明给体位置对分子内电荷转移有影响, 但影响并不明显. 比较R, S, T和U分子的电荷差, 发现它们的电荷差依次增大. S比R的电荷差增加了0.0377e, T比S的电荷差增加了0.0438e, U比T又增加了0.0625e. 因此, 随着给体数目的增加, 从基态到第一激发态的分子内电荷转移明显增强.
| QA0/e | QA1/e | QA/e | QB0/e | QB1/e | QB/e | |
| T | 0.0298 | 0.3978 | 0.3680 | – 0.0298 | – 0.3978 | – 0.3680 |
| T2 | 0.0292 | 0.3901 | 0.3609 | – 0.0292 | – 0.3901 | – 0.3609 |
| T4 | 0.0294 | 0.4066 | 0.3772 | – 0.0294 | – 0.4066 | – 0.3772 |
| R | 0.0271 | 0.3136 | 0.2865 | – 0.0271 | – 0.3136 | – 0.2865 |
| S | 0.0284 | 0.3526 | 0.3242 | – 0.0284 | – 0.3526 | – 0.3242 |
| U | 0.0305 | 0.4340 | 0.4305 | – 0.0305 | – 0.4340 | – 0.4305 |
表3分子各部分的基态电荷和第一激发态电荷(单位: e)
Table3.Net charges (unit: e) for divided parts of the molecules in the ground states and in the first excited states.
2
3.4.分子设计
理论设计出同时具有较强TPA和AIE特性的材料对双光子荧光显微成像技术的应用具有重要意义. 在分子设计中, 需要考虑已经确定的结构与性质关系. 已有研究表明, 许多结构因素在增强有机分子的TPA中起到了重要作用, 如给电子基团和吸电子基团的强度, π共轭中心特性和分子平面性, 以及分子的共轭长度和聚集方式等. 我们在TPEDC体系的基础上, 考虑了一些结构因素, 理论设计了同时具有TPA和AIE特性的分子, 并计算了设计分子的TPA性质.设计分子的化学结构式和相应的优化几何结构如图5所示. 为了提高分子的平面性, 增加分子的共轭长度, 设计了T分子的同分异构体X分子. 考虑到三苯胺基团也具有螺旋桨结构, 并且常用于AIE材料设计中, 在T分子的基础上, 用三苯胺基团代替四苯基乙烯基团, 设计了Y分子. 最后将Y做进一步改进, 设计了Z分子. 优化结构显示, X分子中心的三个苯环相互之间扭转了约33°, 均位于分子的主干方向上, 相比于T分子, 增加了分子的平面性和共轭长度. 与T分子类似, Y分子中菁基两侧的苯环扭转角约为70°左右. 含有三苯胺基团的Z分子与Y分子相比, 平面性更好, 共轭长度也有所增加.
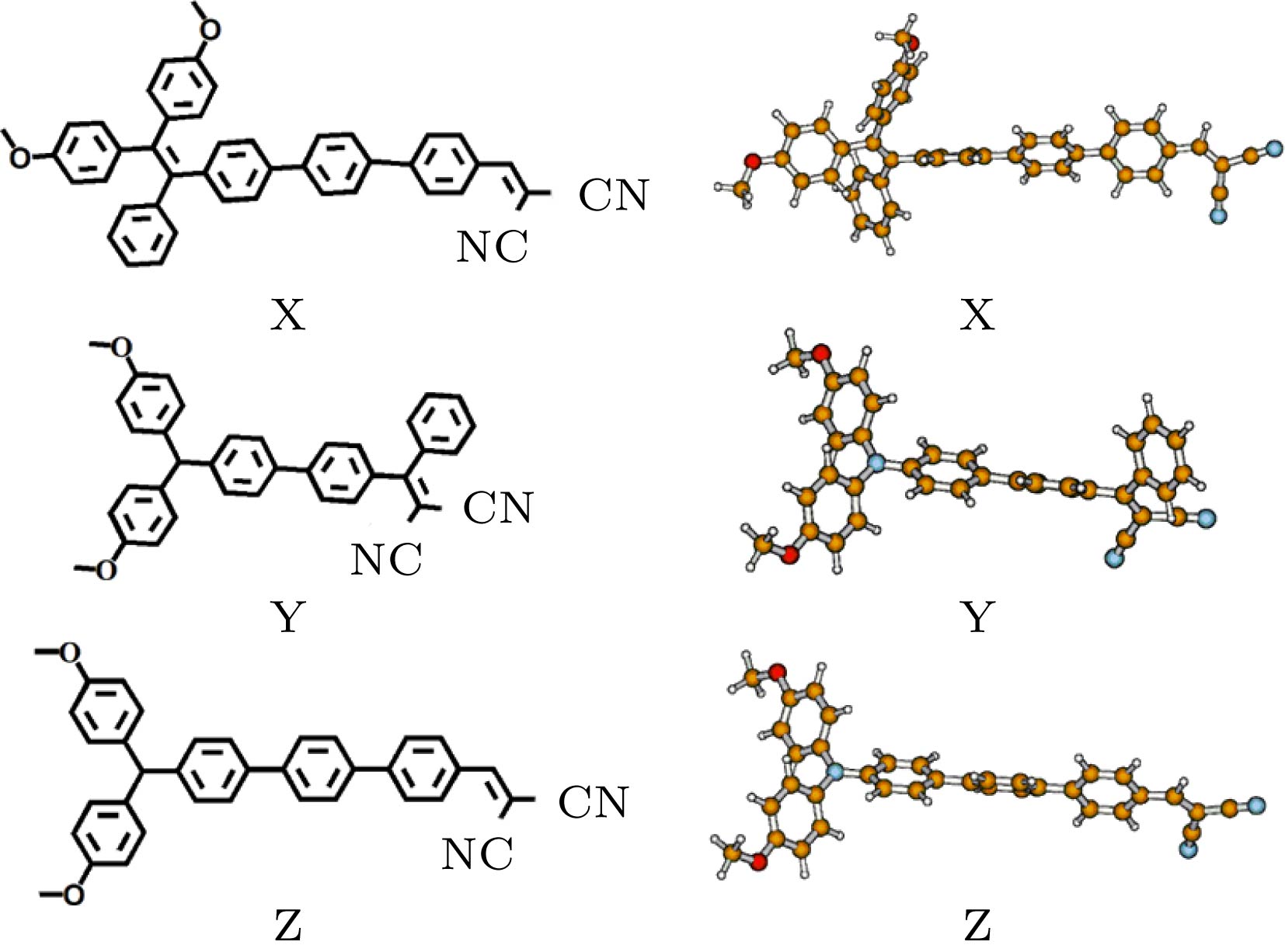 图 5 X, Y和Z分子的化学结构式和优化几何结构
图 5 X, Y和Z分子的化学结构式和优化几何结构Figure5. Chemical structures and optimized geometries of the X, Y and Z molecules.
X, Y和Z分子TPA性质的计算结果列于表4. 相对于实验中的T分子, 三个设计分子的TPA截面均有明显提高. X和Y分子的最大TPA态仍在第三激发态. X分子的最大TPA截面



| X | Y | Z | ||||||
| ${\lambda _{{\rm{tp}}}}$/nm | $\sigma $/GM | ${\lambda _{{\rm{tp}}}}$/nm | $\sigma $/GM | ${\lambda _{{\rm{tp}}}}$/nm | $\sigma $/GM | |||
| S1 | 747 | 947 | 824 | 1543 | 802 | 2259 | ||
| S2 | 663 | 40 | 634 | 737 | 602 | 1362 | ||
| S3 | 615 | 1906 | 579 | 1763 | 582 | 1 | ||
| S4 | 560 | 1 | 576 | 91 | 540 | 57 | ||
| S5 | 542 | 65 | 570 | 606 | 527 | 738 | ||
| S6 | 525 | 502 | 560 | 50 | 546 | 4 | ||
表4分子六个最低激发态的TPA波长


Table4.The TPA wavelength


为了便于比较, 还模拟了X, Y, Z和T分子的TPA光谱, 如图6所示. 可以看到, 这些分子在600—850 nm之间均有多个较强的TPA吸收峰. 设计分子的吸收峰强度均比实验中的T分子强. X, Y和Z在长波范围内均有较强的吸收峰, 这些吸收峰相对T分子有不同程度的红移. 当三苯胺基代替四苯基乙烯基之后, Y和Z的吸收峰都发生了较大红移. 根据(1)式, 双光子跃迁矩阵元与分子的激发能和偶极矩有直接关系. 相关的激发能越低, 越有利于增大TPA截面. 与X和T相比, Y和Z的吸收波长有较大的红移, 说明它们的激发能较低, 因此Y和Z具有更强的TPA.
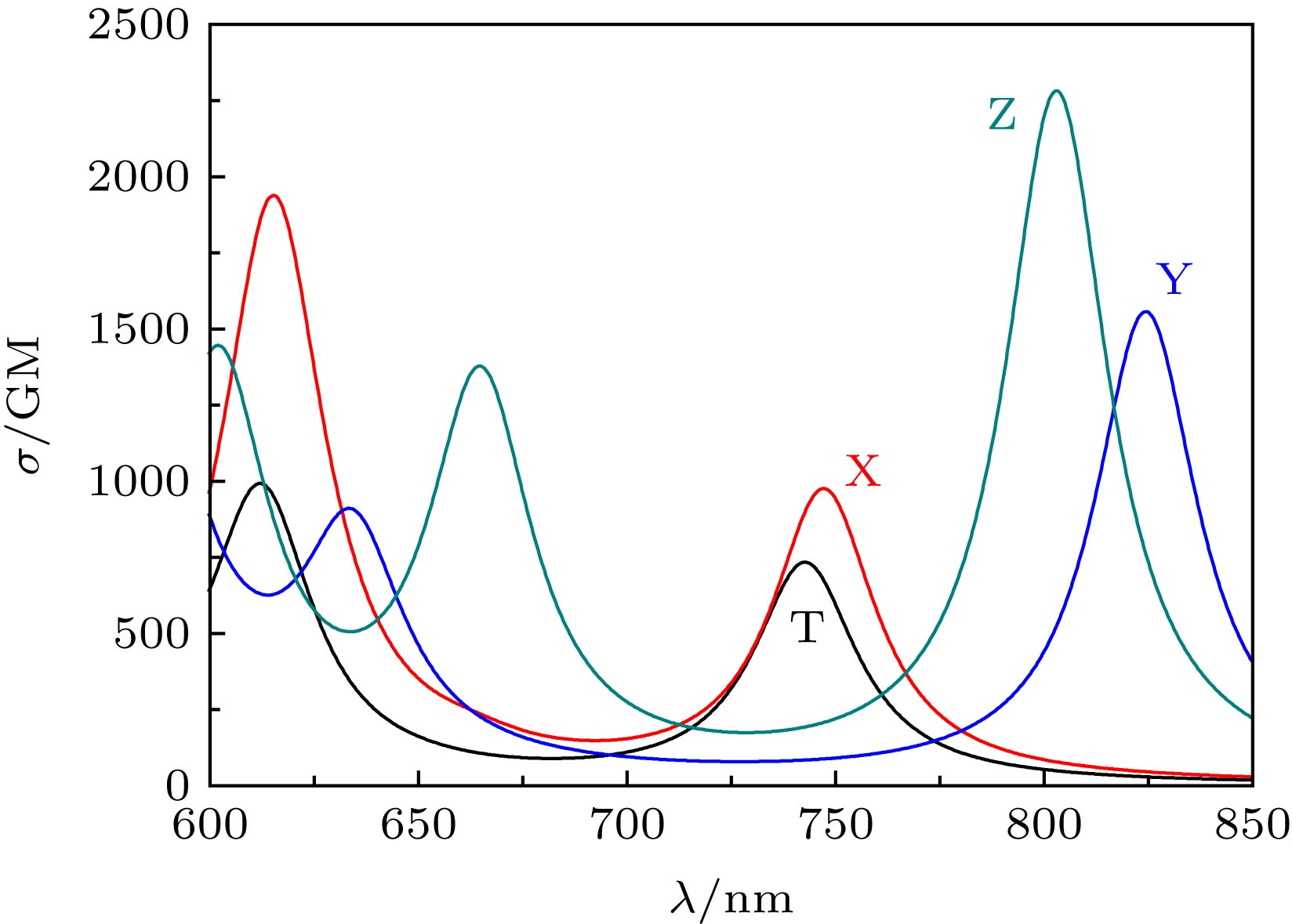 图 6 X, Y, Z和T分子的TPA谱
图 6 X, Y, Z和T分子的TPA谱Figure6. TPA spectra of the X, Y, Z and T molecules.
对于设计TPA和AIE材料, 不仅要求分子材料具有大的TPA截面, 还要求在高浓度下具有较高的荧光量子产率. 例如, 双光子荧光探针的性能通常采用TPA活性截面来表征, 即TPA截面与荧光量子产率的乘积. 因此, 在提高TPA截面的同时, 还应该考虑对荧光量子产率的影响. 比如, 增加分子平面性可以提高TPA截面, 但可能会降低分子的发光效率. 我们理论设计的分子采用了具有螺旋桨结构的四苯基乙烯基和三苯胺基, 可以有效避免紧密堆积和荧光猝灭, 因此可能具有AIE特性[3-6], 从而具有较好的应用前景.
