1.School of Precision Instrument and Optoelectronics Engineering, Tianjin University, Tianjin 300072, China 2.College of Science, Langfang Normal University, Langfang 065000, China 3.College of Physics, Guizhou University, Guiyang 550025, China
Fund Project:Project supported by the National Key Research and Development Program of China (Grant Nos. 2014YQ060773, 2017YFC0803600).
Received Date:21 May 2019
Accepted Date:18 July 2019
Available Online:01 September 2019
Published Online:05 September 2019
Abstract:A variety of new materials have improved the production and life of human beings. Two-dimensional nano materials have become a research hotspot due to their unique physical and chemical properties. Molybdenum disulfide (MoS2) is representative of transition metal sulfide, with excellent mechanical properties and chemical stability. In order to study the influence of external electrical field on the molecular structure and spectrum, here in this work, the density functional theory with the hybrid B3LYP at Def2-TZVP level is employed to calculate the geometrical parameters of the ground state of MoS2 molecule under external electric fields ranging from 0 to 0.1 a.u. (0?5.1423 × 1010 V/m). Based on the optimized structures, the time-dependent density functional theory at the same level as the above is adopted to calculate the absorption wavelengths and the molar absorption coefficients for the first ten excited states of MoS2 molecule under external electric fields. The results show that the most strongest absorption band is located at 483 nm with a molar absorption coefficient of 461 L·mol–1·cm–1 in the UV-Vis absorption spectrum. The intramolecular charge transfers as a whole with the enhancement of the external electric field. The electric dipole moment increases with the external electric field rising, while the total molecular energy decreases with external electric field increasing. With the enhancement of the external electric field, the absorption peaks show a significant redshift. When the electric field increases to 0.1 a.u., the redshift is obvious. This can be explained as follows. When the external electric field is weaker, the electron transfer in the molecule is not significant. However, with the augment of the external electric field, the electron transfer in the molecule occurs as a whole. This makes the electron interaction between Mo and S weaker, thus the electron transition is more likely to occur. The energy required for excitation is reduced, and the wavelength of the excited state becomes longer, that is, the absorption peak takes a redshift. With the enhancement of the external electric field, the molar absorption coefficient increases obviously. This is because the overall transfer of the external electric field to the electron makes the electron cloud density of the MoS2 molecule increase and the number of electrons in transition augment. This work provides a theoretical basis for the utilization and improvement of MoS2 photoelectric properties, and also enlightens the application research of other photoelectric materials. Keywords:MoS2/ external electric field/ density functional theory/ ultraviolet-visible spectrum
表1不同电场下原子Mo和S的电荷Q/a.u. Table1.The charges of Mo and S (unit: a.u.) of the MoS2 molecule under different external electric fields.
图 2 MoS2分子占据轨道侧视图 Figure2. The side view of MoS2 occupied molecular orbital.
MoS2的电偶极矩随着外电场的增强而增大, 如图3所示, 这是由于外电场诱导出分子的感生偶极矩[15], 同时伴随着电子云相对分子骨架的移动和分子骨架的变形, 即分子的极化, 两者的叠加导致了分子偶极矩的变化. 图 3 分子电偶极矩u随外电场的变化 Figure3. The relation of dipole moment and electric field intensity of MoS2.
MoS2分子的基态总能量与外电场的关系见图4, F = 0 a.u.时, 基态总能量E = –864.7004 a.u., 但当F增大到0.10 a.u.时, 基态总能量E减小为–865.0555 a.u., 且减小的趋势加剧. 这是由于内外电场的叠加使得分子几何结构发生了变化: 无外电场时, 分子结构松弛, 分子总能量较大; 但伴随着外电场的增强, 分子结构变得更加紧致、稳固, 分子总能量也随之降低, 计算结果与(1), (2)式也是相符的. 图 4 外电场下分子总能量E的变化 Figure4. The relationship between total energy of MoS2 molecule and electric field intensity.
4.不同电场下MoS2分子的紫外-可见吸收光谱24.1.无外场时MoS2的激发态波长 -->
4.1.无外场时MoS2的激发态波长
在优化分子基态几何结构的基础上, 采用TD-DFT计算了MoS2分子的前10个激发态, 得到了分子UV-Vis光谱, 如图5所示. Jelena Pe?i?等[33]利用Perkin-Elmer Lambda 4B UV-Vis光谱仪, 测量了以N-Methyl-2-pyrrolidone为溶剂的MoS2在350—750 nm波长范围的紫外-可见光谱, 结果显示最强吸收出现在451 nm处. 对比文献中的实验数据, 我们的计算与之符合较好, 这也说明了我们的方法是可信的, 并且我们的工作也是对文献很好的补充. 图 5 不同外电场下的MoS2分子UV-Vis吸收光谱(Epsilon: 摩尔吸收系数) Figure5. UV-Vis absorption spectra of MoS2 molecule under different external electric fields (Epsilon: molar absorption coefficient).
24.2.外电场对激发态波长和摩尔吸收系数的影响 -->
4.2.外电场对激发态波长和摩尔吸收系数的影响
在优化的MoS2分子基态几何结构的基础上, 采用TD-DFT方法计算了了0—0.10 a.u.的外电场对分子前10个激发态的UV-Vis光谱产生的影响, 如图5所示. 可以看出, 当外电场较弱时, 谱峰的波长变化并不明显, 但随着外电场的增强, 吸收峰显著红移, 当外电场增大到0.1 a.u.时, 最强吸收峰波长红移到753 nm, 相比无电场时的吸收峰向长波方向移动了270 nm, 红移非常明显, 这是由于当外电场较弱时, 分子中的电子云主要集中在Mo和S的周围, 如图6占据轨道所示; 但随着外电场的增强, Mo和S周围的电子云沿电场方向发生了整体转移, 这使得Mo和S之间的电子云密度变小, Mo和S之间的相互作用随之变弱, 外层电子更易跃迁到高能级, 也就是跃迁时所需的能量减小, 吸收峰波长变长, 即吸收峰红移. 当外电场增大到0.1 a.u., 分子占据轨道的电子云整体转移非常显著, 因此, 此时的红移也就相当明显了. 图 6 不同外电场下MoS2激发态的分子轨道(占据轨道, 填充了电子的分子轨道; 跃迁轨道, 分子被激发时, 电子从占据轨道跃迁到的空轨道; 轨道权重, 分子某一激发态, 构成轨道的各个组成部分的贡献, 图中给出的是所占比例最大的部分) Figure6. Excited state orbital diagram of MoS2 molecule under different external electric fields (occupied orbitals, molecular orbitals filled with electrons; a transition orbital is a vacant orbital that an electron jumps from an occupied orbital to when a molecule is excited; orbital weight, the contribution of each component of an excited state of a molecule to the composition of the orbital, shown in the figure is the one with the largest proportion).














 图 1 MoS2的分子结构
图 1 MoS2的分子结构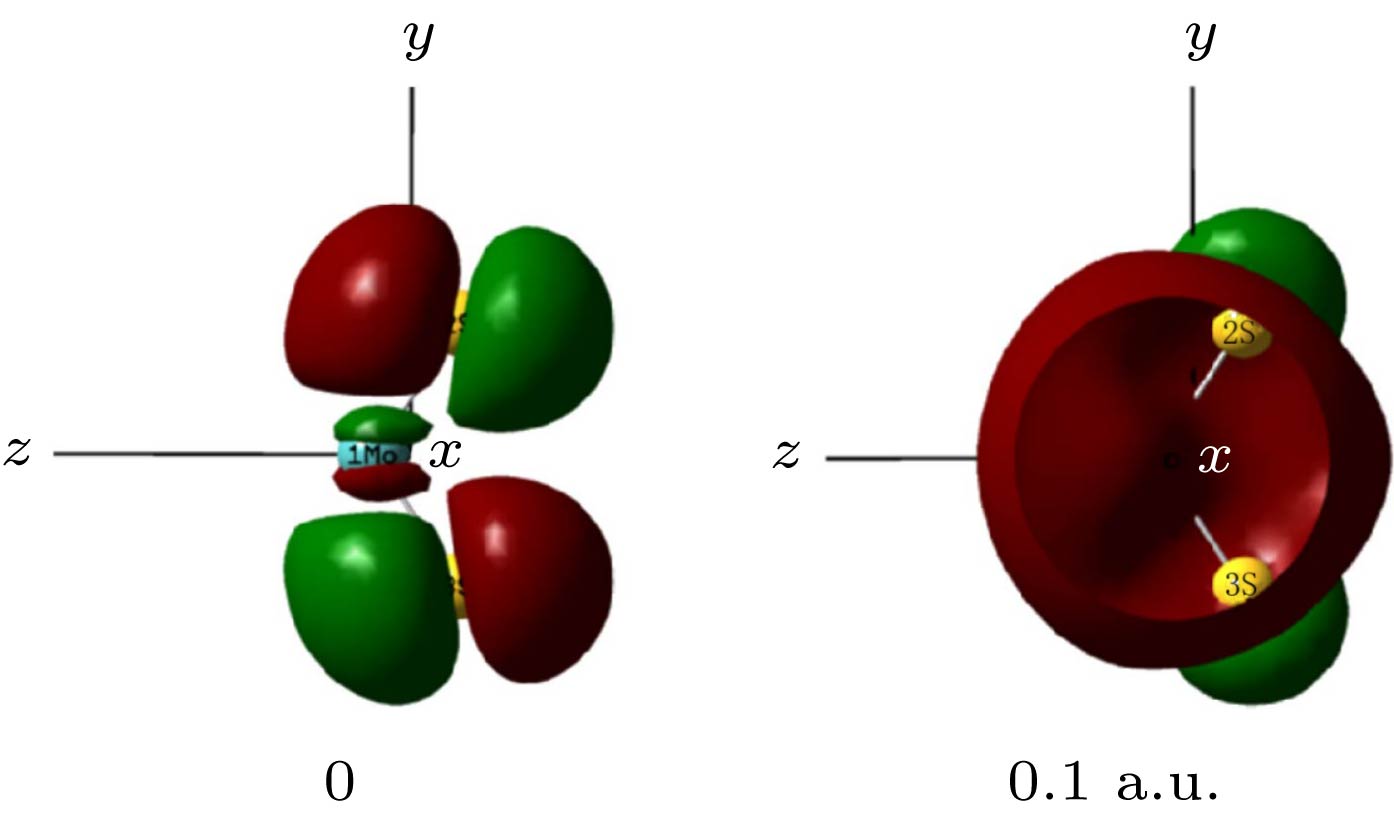 图 2 MoS2分子占据轨道侧视图
图 2 MoS2分子占据轨道侧视图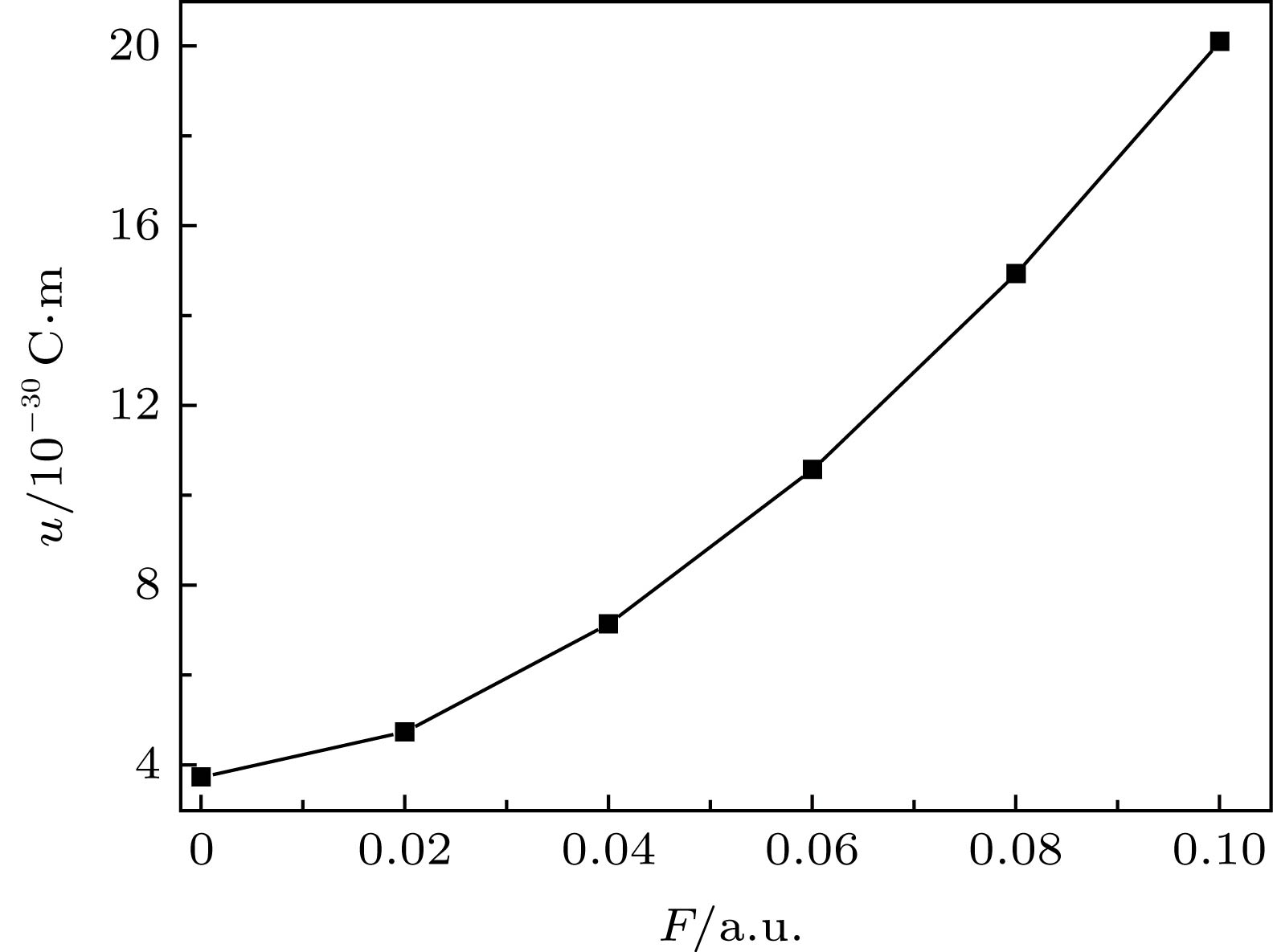 图 3 分子电偶极矩u随外电场的变化
图 3 分子电偶极矩u随外电场的变化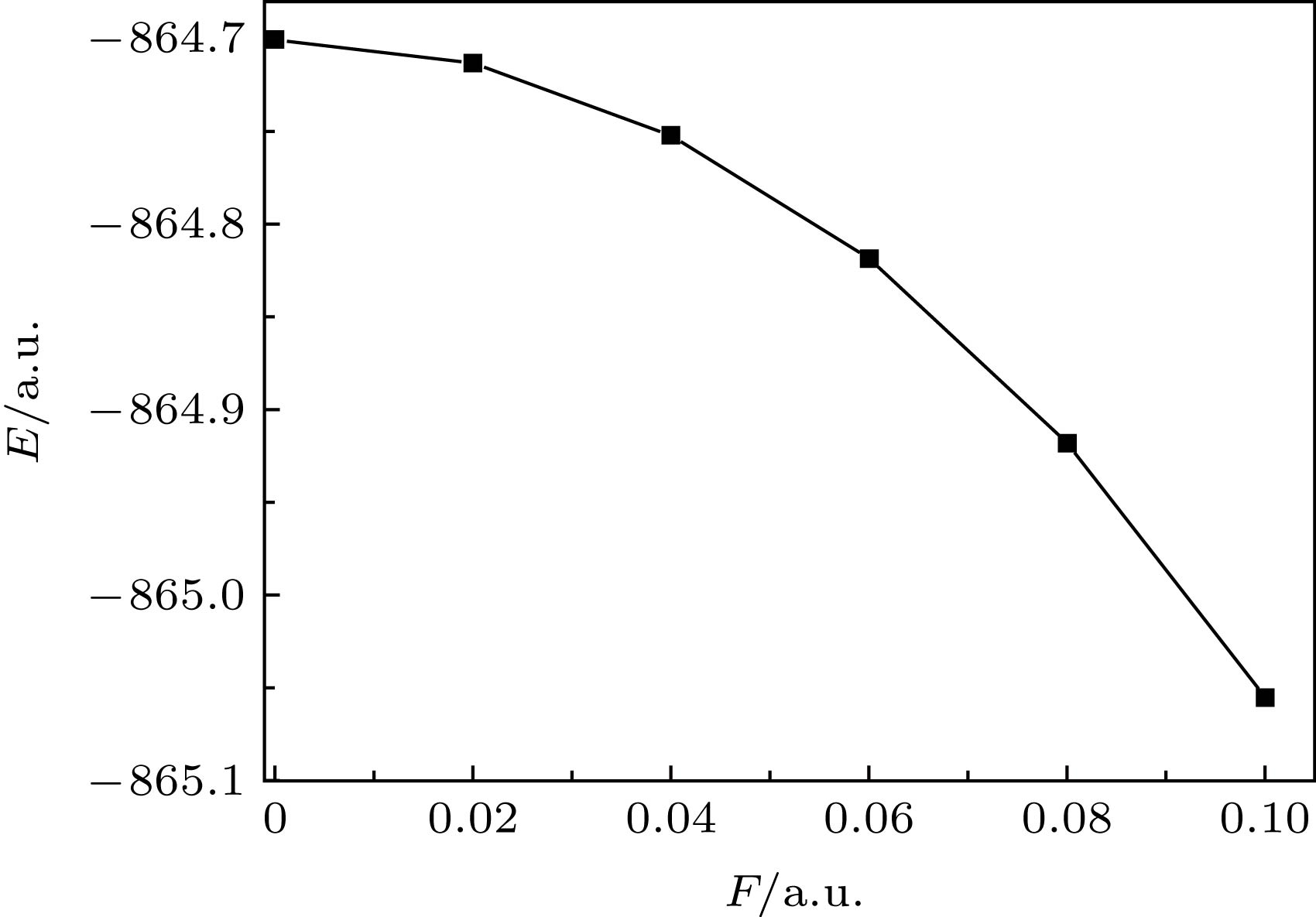 图 4 外电场下分子总能量E的变化
图 4 外电场下分子总能量E的变化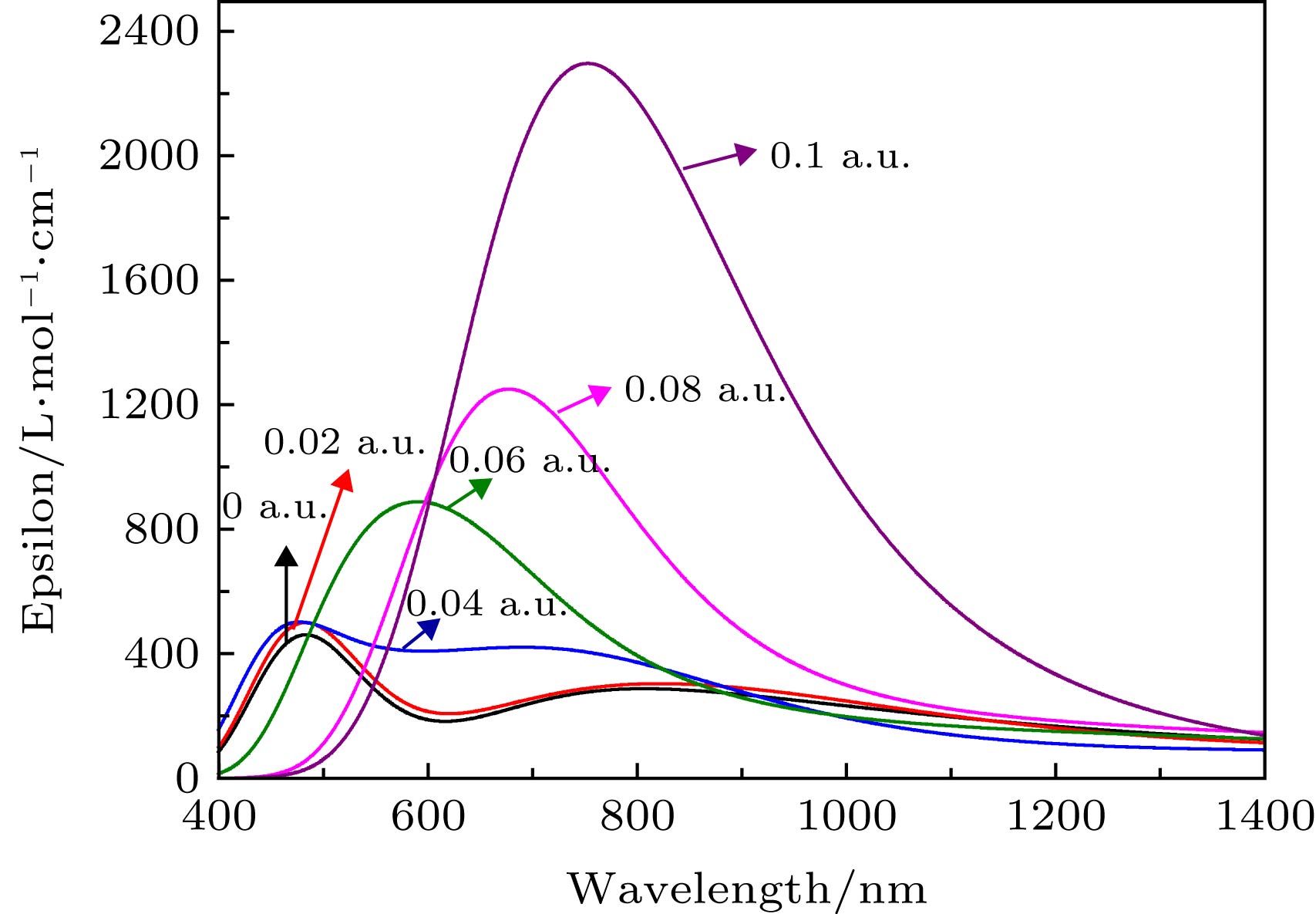 图 5 不同外电场下的MoS2分子UV-Vis吸收光谱(Epsilon: 摩尔吸收系数)
图 5 不同外电场下的MoS2分子UV-Vis吸收光谱(Epsilon: 摩尔吸收系数)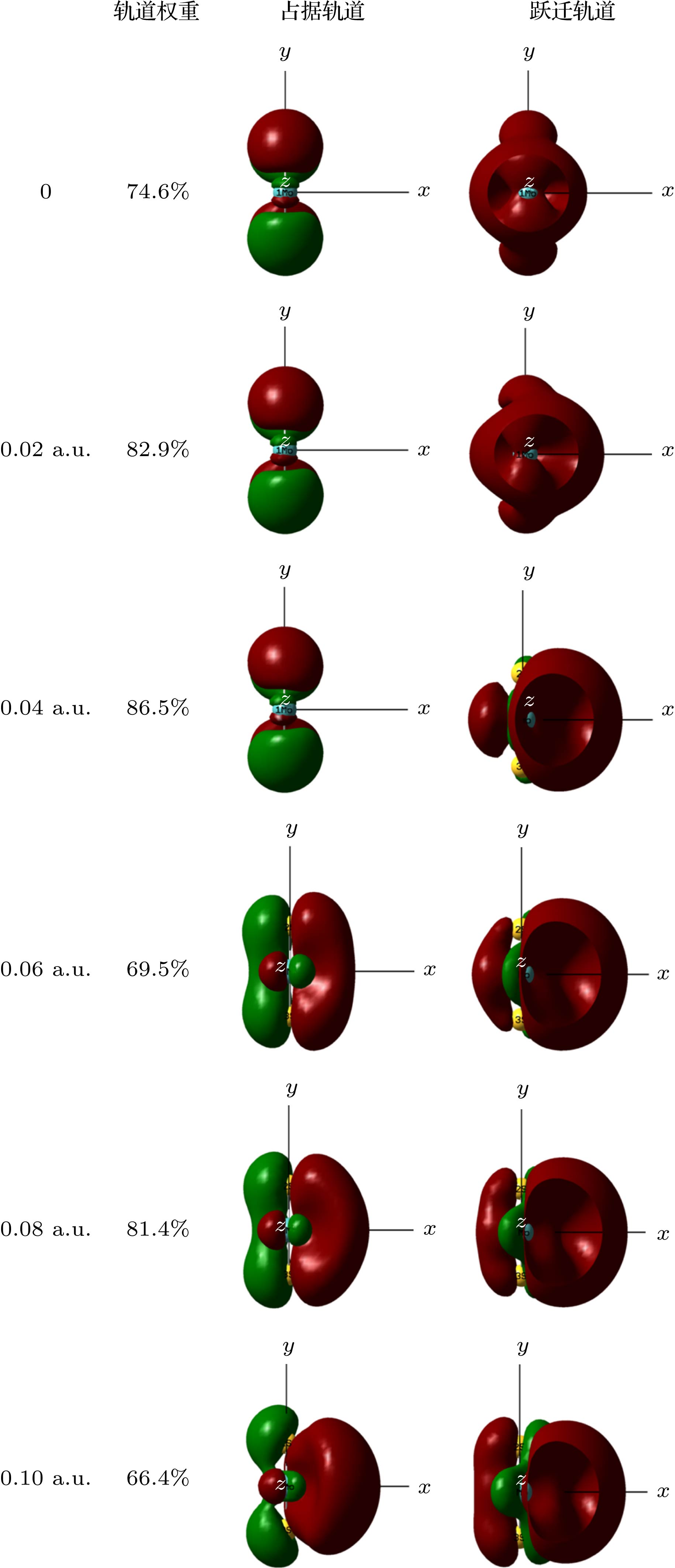 图 6 不同外电场下MoS2激发态的分子轨道(占据轨道, 填充了电子的分子轨道; 跃迁轨道, 分子被激发时, 电子从占据轨道跃迁到的空轨道; 轨道权重, 分子某一激发态, 构成轨道的各个组成部分的贡献, 图中给出的是所占比例最大的部分)
图 6 不同外电场下MoS2激发态的分子轨道(占据轨道, 填充了电子的分子轨道; 跃迁轨道, 分子被激发时, 电子从占据轨道跃迁到的空轨道; 轨道权重, 分子某一激发态, 构成轨道的各个组成部分的贡献, 图中给出的是所占比例最大的部分)