0 引言
【研究意义】飞蝗(Locusta migratoria)主要以禾本科植物为食,对农业生产造成极大的损害,是重要的农业害虫[1]。由于长期采用化学防治,不仅对环境造成了严重的污染,而且导致飞蝗对常用杀虫剂马拉硫磷产生抗药性,因此研发新型绿色环保杀虫剂成为重要的社会热点[2-3]。几丁质是昆虫及甲壳动物外骨骼的特有成分,对昆虫正常的生长发育具有不可或缺的重要作用。几丁质脱乙酰基酶(chitin deacetylase,CDA,E.C.3.2.1.41)是几丁质降解通路中的重要酶系,能够催化几丁质中β-1,4糖苷键连接的N-乙酰基葡糖胺的乙酰胺基,使几丁质水解并转化为壳聚糖[4-5]。因此,对飞蝗几丁质脱乙酰基酶(LmCDA)及其酶活力进行研究,对于后续以该酶为靶标研发特异性杀虫剂具有重要的指导意义。【前人研究进展】目前有关昆虫CDA基因的研究大多集中在双翅目、鞘翅目和鳞翅目等昆虫中。ARAKI等[6]在研究真菌细胞壁中几丁质代谢时首次分离纯化出CDA蛋白。后续研究发现各物种之间CDAs生物学功能具有差异性,在拟尺蠖(Trichoplusia ni)中CDA参与调控中肠围食膜的形成[7]。在黑腹果蝇(Drosophila melanogaster)中存在2个CDAs基因序列,分别是serpentine(serp)和vermiform(verm),二者均对胚胎期气管的形成和延伸具有重要作用[8]。随着研究的不断深入,DIXIT等[9]利用生物信息学方法在4种昆虫中做了系统的研究,把CDAs分为5种类型,其中GROUP-I和GROUP-II的CDAs包含几丁质结合域、A型低密度脂蛋白受体结构域和脱乙酰基酶催化结构域;GROUP-III和GROUP-IV中的CDAs包含几丁质结合域和脱乙酰基酶催化结构域;而GROUP-V中的CDAs仅含有脱乙酰基酶催化结构域。此外,该研究还指出不同昆虫的CDAs在功能域结构和mRNA表达特异性方面均存在差异[10-13]。ARAKANE等[14]在赤拟谷盗(Tribolium castaneum)中鉴定出9个CDAs,发现它们在不同组织部位和不同时期的表达模式具有很大的差异,并且敲除TcCDA1后幼虫因不能正常蜕去旧表皮而死亡。笔者课题组前期工作利用RNAi技术研究发现,沉默中华稻蝗(Oxya chinensis)OcCDA1、OcCDA2和OcCDA2a基因后可导致中华稻蝗因蜕皮困难而死亡,而干扰OcCDA2b对中华稻蝗的生长发育没有影响[15-16];此外,在飞蝗中使LmCDA2a的表达下调后出现与中华稻蝗相似的表型[17]。目前关于几丁质脱乙酰基酶的研究主要集中在其生物学功能的探讨上,而有关昆虫CDA酶活力体外测定的报道较少。2014年,ZHONG等[18]在毕赤酵母中成功表达了具有活性的家蚕(Bombyx mori)围食膜几丁质去乙酰基酶。【本研究切入点】根据课题组前期研究,笔者推测LmCDA2a和LmCDA2b基因沉默后导致飞蝗出现不同表型可能是由于这两个基因的催化功能不同所引起。因此,本研究将LmCDA1、LmCDA2a和LmCDA2b体外表达并纯化后对其酶活力进行测定。【拟解决的关键问题】成功构建pFastBac-LmCDAs重组表达质粒,并在昆虫Sf9细胞中实现异源表达。采用Western blot方法对表达的目的蛋白进行验证后,探索适合LmCDA重组酶的分离纯化条件及体系,并采用分光光度法以对硝基乙酰苯胺为底物对LmCDA重组酶催化活力进行检测,为研发昆虫特异性的绿色环保杀虫剂提供理论依据。1 材料与方法
试验于2014—2015年在山西大学应用生物学研究所完成。1.1 材料与试剂
材料:LmCDA1、LmCDA2a和LmCDA2b(GenBank登录号KR537804和KR537805)全长质粒为笔者实验室保存。pFastBacTMDual真核表达载体购于Invitrogen公司。草地贪夜蛾卵巢Sf9细胞系购于昆明动物所。大肠杆菌DH10Bac感受态细胞为笔者实验室保存。试剂:ExTaq酶、T4 DNA连接酶购买于Promega公司,DNA Marker、Xba I和Hind Ⅲ限制性内切酶购于NEB公司,IPTG、X-Gal购于TaKaRa公司,Trizol试剂购于Invitrogen;无内毒素质粒提取试剂盒、质粒提取试剂盒、DNA凝胶回收试剂盒均购于OMEGA公司;Ni-NTA亲和层析柱和Q-Sepharose阴离子交换层析柱材料购于GE公司。
1.2 引物设计
本研究所用引物采用Primer Express 5.0软件设计,由生物工程(上海)股份有限公司合成,具体见表1。Table 1
表1
表1飞蝗几丁质脱乙酰基酶基因(LmCDAs)的表达引物
Table 1Primers used for PCR amplification of chitin deacetylase gene (LmCDAs) in L. migratoria
| 引物名称 Primer name | 引物序列 Sequence of primer (5′-3′) |
|---|---|
| LmCDA1 Xba I FW | TGCTCTAGAATGGCGCGGATTGTGTACAGC |
| LmCDA1 Hind ΙΙΙ RV | CCCAAGCTTCTAGTGATGGTGATGGTGATGGAAGAAGCCGTCGCCGGT |
| LmCDA2ab Xba I FW | TGCTCTAGAATGGCCAGGACTACGACTTCGTG |
| LmCDA2ab Hind ΙΙΙ RV | CCCAAGCTTTTAGTGATGGTGATGGTGATGTTTCACAGAGAAGCCATCACCTGTG |
新窗口打开
1.3 几丁质脱乙酰基酶基因的真核表达
1.3.1 LmCDA1、LmCDA2a和LmCDA2b克隆和纯化 以LmCDA1、LmCDA2a和LmCDA2b全长序列为模板,利用PCR扩增技术获得带有双酶切位点(Xba I、Hind III)和编码尾部6×His标签的LmCDA1、LmCDA2a和LmCDA2b。1%的琼脂糖凝胶对PCR产物进行电泳检测,并将目的基因全长序列纯化回收,置于4℃保存备用。1.3.2 pFastBac-LmCDA重组载体的构建 用Xba I和Hind III对pFastBacTMDual质粒和上述3种PCR回收产物分别进行双酶切后,1%琼脂糖凝胶电泳检测酶切产物后完成目的片段的胶回收;双酶切后的pFastBacTMDual质粒片段和目的基因按3﹕1的摩尔比例混合,T4 DNA连接酶16℃连接过夜,连接产物转化至Trans-T1感受态细胞后,在含有100 μg·mL-1的氨苄和100 μg·mL-1庆大霉素的LB琼脂平板上筛选阳性克隆。LB培养基扩大培养后,提取质粒,随后进行酶切及测序验证。
1.3.3 Bacmid重组载体的构建与鉴定 将构建成功的pFastBac-LmCDA1、pFastBac-LmCDA2a和pFastBac- LmCDA2b重组质粒分别转化到DH10 Bac感受态细胞中。重组中间载体通过同源重组整合入DH10 Bac感受态细胞携带的Bacmid质粒中,37℃培养20 h,进行蓝白斑筛选。由于重组Bacmid 基因序列大于135 kb,采用普通酶切鉴定阳性克隆的方法可能导致杂带过多,因此采用转座位点mini-attTn7两侧的M13F/M13R作为引物进行PCR扩增鉴定。扩增产物用1%琼脂糖凝胶电泳检测后,采用OMEGA无内毒素质粒提取试剂盒提取Bacmid重组质粒,用于后续转染试验。
1.3.4 Bacmid重组质粒的转染及表达 将长势良好的昆虫Sf9细胞常规消化后接种到24孔培养板内,以无抗生素的细胞培养液进行培养,次日,待细胞密度接近80%时按照LipofectamineTM3000转染试剂盒说明完成Bacmid重组质粒对Sf9细胞的转染。转染共分为3组:空白对照组、空载体对照组(pFastBacTMDual)和试验组(pFastBac-LmCDAs)。分别转染12、24和72 h,在显微镜下观察Bacmid重组质粒转染Sf9细胞后的形态,并在转染72 h时收集细胞进行检测。
1.3.5 表达产物的Western blot检测与纯化 离心收集的细胞中加入1 mL 50 mmol·L-1的磷酸盐缓冲液(pH 7.2)和10 μL 100 mmol·L-1的PMSF,冰上匀浆,使细胞充分破碎。2 000 r/min,4℃离心5 min,收集上清,得到粗酶液。粗酶液在12%的SDS-PAGE胶上电泳后,100 V恒压转印100 min至 PVDF膜上,随后用5% w/v牛血清蛋白TBST缓冲液于4℃封闭过夜;TBST洗膜3次;按1﹕1 000比例稀释抗His标签鼠单克隆抗体,室温孵育2 h,TBST洗膜3次;按1﹕5 000比例稀释二抗(羊抗鼠 IgG),室温孵育2 h,TBST洗膜3次后,在双色红外激光成像系统中进行避光显色。
Western blot鉴定成功后对目的蛋白进行纯化。由于重组蛋白尾部携带6×His标签序列,利用Ni-NTA亲和层析柱与6×His蛋白标签上的组氨酸咪唑基团特异性结合的原理,使用咪唑线性梯度的方法将目标蛋白洗脱下来,完成重组蛋白的初步纯化。保留部分粗酶液作为柱前对照后,将剩余粗酶液与镍柱材料在4℃条件下温和颠倒振荡孵育2 h后,4℃离心收集穿透液。用含20 mmol·L-1咪唑的磷酸缓冲液(50 mmol·L-1,pH 8.0)洗去杂蛋白,再用含有20—300 mmol·L-1咪唑的磷酸缓冲液(50 mmol·L-1,pH 8.0)以2.0 mL·min-1的流速洗脱目的蛋白,最后用12%的SDS-PAGE胶电泳对纯化后的各组分纯度进行检测。
采用阴离子(Q-Sepharose)交换层析柱对上述纯化的蛋白进行进一步纯化。首先将含有目的蛋白的洗脱液直接上样于磷酸钠缓冲液(50 mmol·L-1,pH 8.0)预平衡过的Q-Sepharose层析柱中,之后用含有0—1 mol·L-1氯化钠的磷酸缓冲液(50 mmol·L-1,pH 8.0)洗脱目的蛋白,流速为2.0 mL·min-1,最后用12%的SDS-PAGE蛋白胶对纯化后的各组分进行纯度检测。
1.3.6 目的蛋白CDA酶活力测定 (1)对硝基乙酰苯胺和对硝基苯胺特征吸收波长的确定:分别配制0.07 mmol·L-1对硝基乙酰苯胺、对硝基苯胺的水溶液,采用酶标仪在310—480 nm的波长范围内进行扫描,以去离子水作为参比,测定对硝基乙酰苯胺和对硝基苯胺的特征吸收波长[19]。
(2)对硝基苯胺标准曲线的绘制:称取1 mg对硝基苯胺溶解于10 mL水中,加入微量丙酮助溶,混匀后用蒸馏水进行梯度稀释制备1—10 mg·L-1不同浓度梯度的标准品。随后,以蒸馏水作为参比,测定各浓度标准品在400 nm处的吸光值;以标准品浓度为横坐标,以400 nm处吸光值为纵坐标,绘制对硝基苯胺标准曲线,并通过计算标准曲线斜率确定线性系数K。
(3)酶解反应体系及条件:配制200 mg·L-1的对硝基乙酰苯胺底物溶液作为母液,微量丙酮助溶。向含有100 μL的底物溶液中加入300 μL提前预热的磷酸盐缓冲液后,迅速加入100 μL酶液,于50℃反应15 min后,100℃煮沸终止反应。再加入超纯水定容至1 000 μL,混匀,845×g离心10 min,取200 μL上清于酶标板中,用酶标仪检测其400 nm处吸光值(A400)。空白对照体系:100 μL底物溶液加300 μL磷酸盐缓冲液以及100 μL相应高温预灭活的酶液后,加入超纯水定容至1 000 μL,然后吸取200 μL检测其吸光值(A0)。上述每个反应均包含3个生物学重复。
酶活力单位定义:在上述酶催化反应条件下,每小时产生1 μg对硝基苯胺所需要的酶量定义为1个酶活力单位(U·μL-1)。计算公式:酶活力=(A400-A0)×酶液稀释倍数/KT。式中,A400为待测酶液样品的吸光值;A0为预灭活酶液样品的吸光值;T为酶促反应时间(h);K为线性系数。
2 结果
2.1 LmCDA1、LmCDA2a和LmCDA2b的结构域预测
使用BLASTP和SMART软件,在线预测LmCDA1、LmCD2a和LmCDA2b的结构域。LmCDA1、LmCD2a和LmCDA2b均含有4个结构域: N-端信号肽(signal peptide)、几丁质结合域(chitin binding peritrophin-A,ChBD)、A型低密度脂蛋白受体结构域(low-density lipoprotein receptor class A,LDLa)和脱乙酰基酶催化结构域(catalytic domain,CDA)(图1)。3个基因的几丁质结合域中均包含6个保守的半胱氨酸。LmCDA2a和LmCDA2b两个剪切子除在其第3个半胱氨酸和第4个半胱氨酸之间(67—84 aa)的氨基酸数目和组成及在第4和第6个半胱氨酸(84—106 aa)之间的序列存在差异外,其余部分完全一致。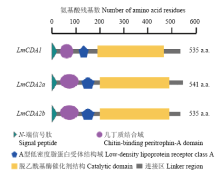 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图1LmCDA1、LmCDA2a和LmCDA2b的结构域预测示意图
-->Fig. 1Schematic diagram of deduced domains of LmCDA1, LmCDA2a and LmCDA2b
-->
2.2 LmCDA1、LmCDA2a和LmCDA2b克隆和纯化
以LmCDAs全长序列为模板,用所设计的蛋白表达引物进行PCR扩增,扩增产物使用1%琼脂糖凝胶电泳检测。结果如图2所示,以DL5000 DNA Ladder为参照,得到3条与预期大小一致的扩增产物(图中箭头位置所示)。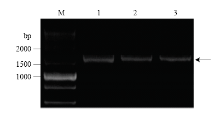 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图2飞蝗几丁质脱乙酰基酶基因的PCR扩增 M:5000 bp DNA分子量标准 5000 bp DNA Ladder;1、2和3分别为LmCDA1、LmCDA2a和LmCDA2b的PCR扩增产物
-->Fig. 2PCR amplification of chitin deacetylase in L. migratoriaPCR products of LmCDA1, LmCDA2a and LmCDA2b, respectively
-->
2.3 pFastBacTM-LmCDA重组载体的构建
使用胶回收试剂盒对PCR扩增产物进行回收纯化。将携带有酶切位点的目的基因序列和中间载体质粒pFastBacTMDual分别经过Hind Ⅲ和Xba I进行双酶切后,回收目的基因序列和载体片段;用T4 DNA连接酶连接目的基因序列和载体片段,获得重组载体质粒。通过双酶切方法对重组载体质粒进行1%琼脂糖电泳检测。泳道2、4和6显示pFastBac-LmCDA1、pFastBac-LmCDA2a、pFastBac-LmCDA2b重组载体经过Hind Ⅲ和Xba I双酶切后得到pFastBacTMDual载体骨架及目的基因序列(图3)。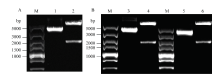 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图3重组质粒的双酶切鉴定 M:5000 bp DNA分子量标准 5000 bp DNA Ladder;1、3、5:pFastBacTM Dual载体 pFastBacTM Dual Vector;2、4、6:分别为重组质粒 pFastBac-LmCDA1、pFastBac-LmCDA2a、pFastBac-LmCDA2b双酶切鉴定结果
-->Fig. 3Identification of recombinant plasmid by Hind Ⅲ and Xba IIdentification of recombinant plasmid pFastBac-LmCDA1, pFastBac-LmCDA2a, and pFastBac-LmCDA2b, respectively, by Hind Ⅲ and Xba I
-->
2.4 Bacmid重组载体的构建及鉴定
由于重组Bacmid 基因序列大于135 kb,采用普通酶切鉴定阳性克隆方法可能导致杂带过多,因此采用转座位点mini-attTn7两侧的M13F/M13R作为引物进行PCR扩增后,1%琼脂糖电泳检测。泳道1为转染空pFastBacDual质粒的Bacmid重组载体的阴性对照,该PCR扩增产物约为2 560 bp;泳道2、3和4分别为Bacmid-LmCDA1、Bacmid-LmCDA2a、Bacmid-LmCDA2b的PCR鉴定结果,其目的片段大小分别为4 198、4 216、4 198 bp。PCR鉴定结果表明上述3种重组Bacmid质粒构建成功(图4)。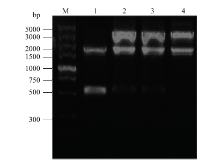 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图4重组Bacmid质粒的PCR鉴定M:5000 bp DNA分子量标准 5000 bp DNA Ladder;1:转染空pFastBacTMDual质粒的重组Bacmid载体(对照) Recombinant Bacmid vector with the pFastBacTMDual transfected;2、3、4:分别为Bacmid- LmCDA1、Bacmid-LmCDA2a和Bacmid-LmCDA2b
-->Fig. 4Identification of recombinant Bacmid plasmid by PCR Recombinant Bacmid vector of Bacmid-LmCDA1, Bacmid-LmCDA2a and Bacmid-LmCDA2b, respectively
-->
2.5 表达产物的Western blot检测
表达产物经12% SDS-PAGE胶电泳后进行Western blot 检测。如图5所示,只有转染Bacmid重组质粒的试验组(泳道3、4和5)有约为61 kD左右的特异性蛋白条带(黑色箭头所指位置),其蛋白分子量大小均与在线预测的蛋白分子量大小相符;而空载体对照组(泳道2)和空白对照组(泳道1)均无特异性蛋白条带,表明目的蛋白在昆虫Sf9细胞中成功表达,并具有免疫活性,可被抗体特异识别。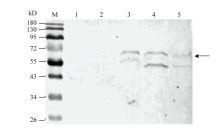 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图5Western blot检测重组质粒在Sf9细胞中表达M:蛋白质分子量标准 Protein molecular weight standards;1:Sf9昆虫细胞超声破碎产物 Sonication product of insect cells of Sf9;2:转染空载体的 Sf9 细胞破碎产物 Sonication product of transfected empty vector of insect cells of Sf9;3:转染重组质粒LmCDA1的Sf9细胞破碎产物 Sonication product of transfected recombinant plasmid LmCDA1 of insect cells of Sf9;4:转染重组质粒LmCDA2a的Sf9细胞破碎产物 Sonication product of transfected recombinant plasmid LmCDA2a of insect cells of Sf9;5:转染重组质粒LmCDA2b的Sf9细胞破碎产物 Sonication product of transfected recombinant plasmid LmCDA2b of insect cells of Sf9
-->Fig. 5Western blot detected the expression of recombinant plasmid in the cells of Sf9
-->
2.6 表达产物的纯化
表达产物与Ni-NTA亲和层析柱结合,采用咪唑线性梯度的方法对目的蛋白进行初步纯化后,用12% SDS-PAGE电泳检测各组分洗脱样品。结果如图6-A—6-C所示,泳道5、7、9、11中均含有明显的目的条带(黑色箭头所示,大小约为61 kD),但是各组分样品仍可见杂蛋白条带,需要通过其他方法进一步分离纯化。 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图6目的蛋白LmCDA1、LmCDA2a和LmCDA2b的纯化 M:蛋白质分子量标准 Protein molecular weight standards;Bc:过柱前组分 Before column fraction;Ft:穿透液 Flow through fraction;图A、B、C中泳道W:含20 mmol·L-1咪唑洗脱液洗脱结合有目的蛋白的Ni-NTA柱洗脱的组分 Lane W in Fig. A, B, C: The eluted fraction of targeted protein binding with Ni-NTA column using 20 mmol·L-1 imidazole eluent;图A、B、C中泳道1—21:不同浓度的咪唑洗脱液对结合有目的蛋白的Ni-NTA柱进行洗脱的组分 Lane 1-21 in Fig. A, B, C: The eluted fraction of targeted protein binding with Ni-NTA column using different concentrations of imidazole eluent;图D、E、F中泳道W:含0.05 mmol·L-1磷酸钠缓冲液对结合有目的蛋白的Q-Sepharose柱进行洗脱的组分 Lane W in Fig. D, E, F: The eluted fraction of targeted protein binding with Q-Sepharose column using 0.05 mmol·L-1 sodium phosphate buffer;图D、E、F中泳道1—21:不同浓度的氯化钠洗脱液对结合有目的蛋白的Q-Sepharose柱进行洗脱的组分 Lane 1-21 in Fig. D, E, F: The eluted fraction of targeted protein binding with Q-Sepharose column using different concentrations of sodium chloride eluent
-->Fig. 6The purification of the purpose proteins of LmCDA1, LmCDA2a and LmCDA2b
-->
将经过Ni-NTA亲和层析且含目标条带的蛋白组分混合,然后通过Q-Sepharose交换层析柱进行更精细的纯化。将组分用氯化钠线性梯度洗脱后收集的各馏分进行SDS-PAGE电泳检测,结果如图6-D—6-F中黑色箭头所示。图6-D的泳道1、图6-E的泳道7、图6-F的泳道11均可见在61 kD处目的蛋白条带单一,表明3种重组蛋白纯度符合后续酶活检测要求。
2.7 目的蛋白CDA酶活力测定
2.7.1 对硝基乙酰苯胺和对硝基苯胺特征吸收波长的测定 分别配制0.07 mmol·L-1对硝基乙酰苯胺、对硝基苯胺的水溶液,以去离子水作为参比,用酶标仪在310—480 nm波长范围内进行吸光度扫描,测定其特征吸收波长(图7-A)。对硝基乙酰苯胺的最大吸收峰处于317 nm,而对硝基苯胺的最大吸收峰在380 nm处。由于380 nm处存在对硝基乙酰苯胺的干扰,而在400 nm处对硝基乙酰苯胺干扰较小,吸收仍然强烈,因此选择400 nm作为检测波长。 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图7对硝基乙酰苯胺和对硝基苯胺紫外可见吸收光谱(A)、对硝基苯胺标准曲线(B)及LmCDA2a和LmCDA2b酶活力差异显著性分析(C)
-->Fig. 7Absorption spectrum of p-nitroacetanilide and p-nitroaniline (A), standard curve of p-nitroaniline (B) and significant difference analysis of LmCDA2a and LmCDA2b enzyme activity (C)
-->
2.7.2 对硝基苯胺标准曲线的绘制 以标准品浓度为横坐标,400 nm处吸光值为纵坐标,建立对硝基苯胺标准曲线(图7-B)。对硝基苯胺标准曲线的R2为0.998,表明在OD0—OD1范围内不同浓度的对硝基苯胺溶液与400 nm处的吸光值具有良好的线性关系,符合比尔定律,可以通过分光光度法进行定量测定对硝基苯胺的量。
2.7.3 目的蛋白酶活力检测 LmCDA1、LmCDA2a和LmCDA2b的酶活力分别为0.268、0.354、0.228 U·μL-1(表2),说明以上3种重组蛋白酶体外表达均具有几丁质脱乙酰基酶酶活力。对LmCDA2a和LmCDA2b酶活力进行差异显著性分析发现LmCDA2a和LmCDA2b两者之间的酶活力具有显著差异(P<0.05)(图7-C)。
Table 2
表2
表2体外表达的LmCDAs的酶活力
Table 2Enzyme activity of in vitro expressed LmCDAs
| 酶 Enzyme | 吸光值 A400 | 吸光值 A0 | 酶活力 Enzyme activity (U·μL-1) | 酶活力均值 Average enzyme activity (U·μL-1) |
|---|---|---|---|---|
| LmCDA1 | 0.255 0.264 0.249 | 0.120 0.117 0.113 | 0.260 0.283 0.262 | 0.268±0.013 |
| LmCDA2a | 0.351 0.363 0.341 | 0.181 0.153 0.171 | 0.328 0.405 0.328 | 0.354±0.044 |
| LmCDA2b | 0.293 0.270 0.268 | 0.158 0.159 0.159 | 0.260 0.214 0.210 | 0.228±0.028 |
新窗口打开
3 讨论
体外表达并制备出有活性的蛋白对于更好地研究飞蝗CDA基因的生物学功能至关重要。本研究采用昆虫Bac-to-Bac杆状病毒表达系统构建LmCDAs的重组质粒,通过体外转染昆虫sf9细胞,获得具有生物活性的功能CDA蛋白,并对其酶活力进行检测。本研究采用Western blot技术检测目的蛋白在昆虫Sf9细胞中的表达情况,结果显示pFastBac-LmCDAs重组产物对应的泳道内检测到约61 kD蛋白分子量的特异性目的条带,与在线预测的目的蛋白分子量大小一致,而空白对照组和空载体对照组pFastBac-LmCDAs在61 kD蛋白分子量处均无条带出现,表明标记有组氨酸标签的目标蛋白在昆虫Sf9细胞中成功表达。但是,在pFastBac-LmCDAs重组产物对应的泳道内,除了约61 kD的特异性目标条带外,还分别可以看到约58、55和55 kD较小的蛋白片段。这与经过Ni-NTA亲和层析纯化后得到的蛋白组分一致,推测这3个较小蛋白条带是His标签抗体检测到的非特异性蛋白。
探索并建立了飞蝗几丁质脱乙酰基酶的纯化体系,但采用Ni-NTA亲和层析法得到的纯化蛋白仍然存在少量杂蛋白,推测可能是由于草地贪夜蛾Sf9真核表达体系中除了带有组氨酸标签的目的蛋白外自身存在着其他组氨酸含量较高的蛋白,或者是目的蛋白的随机水解片段,这些蛋白均可被Ni-NTA柱识别并被高浓度咪唑洗脱。2013年,LIU等[20]在Bm- DNAPOL纯化中使用Ni-NTA亲和纯化的基础上,进一步使用Mono Q 阴离子交换层析柱对蛋白进行更彻底的纯化,从而有效的分离目的蛋白。基于此,本研究将经过Ni-NTA亲和层析且含目标条带的蛋白组分混合后,采用Q-Sepharose交换层析柱对其进行进一步的纯化。结果显示经过两步纯化后,LmCDA1、LmCDA2a和LmCDA2b的目的蛋白和杂蛋白均得到了有效的分离。
由于几丁质不溶于水,若以几丁质为底物,需预先处理制成水溶性衍生物,并且几丁质脱乙酰产物检测也缺乏简便方法,造成酶活力检测困难,所以直接用几丁质作底物进行酶活力测定的报道较少。目前CDA酶活力的测定方法主要有3种,即放射标记法、NADH显色法和吲哚/盐酸法比色法[21-26]。ZHONG等[18]在毕赤酵母中成功表达家蚕围食膜的几丁质去乙酰基酶,以对硝基乙酰苯胺为底物测得其酶活力为0.00185 U·μL-1。对硝基乙酰苯胺含有乙酰基,它可以在CDA酶的催化下脱去乙酰基形成对硝基苯胺。该方法具有操作简便、稳定性和重现性好等优点。因此,本研究以对硝基乙酰苯胺为底物对其进行酶活测定,证明了LmCDA1、LmCDA2a和LmCDA2b蛋白在体外具有脱乙酰化的功能。笔者实验室前期对LmCDA2的生物学功能进行研究,RNAi干扰结果表明LmCDA2a的沉默导致飞蝗因蜕皮困难而死亡,对其生长发育起着重要的作用,而LmCDA2b对飞蝗的生长发育没有影响[17]。为了探讨LmCDA2a和LmCDA2b在飞蝗生长发育中功能的差异是否由其催化功能不同所导致,本研究在前期对LmCDA2a、LmCDA2b的体内功能分析的基础上,体外真核表达获得LmCDA2a和LmCDA2b具有酶活性的蛋白,并且发现二者酶活力具有显著差异。这一结果为后续进一步研究飞蝗几丁质去乙酰基酶生理功能提供了基础资料,并为从基因水平上寻找新型杀虫剂靶标、研发新型绿色环保杀虫剂打下了基础。
4 结论
利用Bac-to-Bac真核表达系统成功表达出具有生物活性的LmCDA1、LmCDA2a和LmCDA2b 蛋白,发现LmCDA2a和LmCDA2b酶活力具有显著性差异。从酶学角度为之前提出的推测(LmCDA2a和LmCDA2b基因沉默后导致飞蝗表型不同可能是由于它们的催化功能不同)提供了理论依据。The authors have declared that no competing interests exist.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}