 ,, 丛佩华,中国农业科学院果树研究所/农业部园艺作物种质资源利用重点实验室/国家苹果育种中心,辽宁兴城 125100
,, 丛佩华,中国农业科学院果树研究所/农业部园艺作物种质资源利用重点实验室/国家苹果育种中心,辽宁兴城 125100Cloning and Functional Analysis of U6 Promoter in Apple
BIAN ShuXun, HAN XiaoLei, YUAN GaoPeng, ZHANG LiYi, TIAN Yi, ZHANG CaiXia,, CONG PeiHua,Institute of Pomology, Chinese Academy of Agricultural Sciences/Key Laboratory of Fruit Germplasm Resources Utilization, Ministry of Agriculture/National Apple Breeding Center, Xingcheng 125100, Liaoning通讯作者:
责任编辑: 赵伶俐
收稿日期:2019-07-31接受日期:2019-09-19网络出版日期:2019-12-01
| 基金资助: |
Received:2019-07-31Accepted:2019-09-19Online:2019-12-01
作者简介 About authors
卞书迅,E-mail:bb18236767941@163.com
韩晓蕾,E-mail:hanxiaolei@caas.cn

摘要
关键词:
Abstract
Keywords:
PDF (2594KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
卞书迅, 韩晓蕾, 袁高鹏, 张利义, 田义, 张彩霞, 丛佩华. 苹果U6启动子的克隆及功能分析[J]. 中国农业科学, 2019, 52(23): 4364-4373 doi:10.3864/j.issn.0578-1752.2019.23.016
BIAN ShuXun, HAN XiaoLei, YUAN GaoPeng, ZHANG LiYi, TIAN Yi, ZHANG CaiXia, CONG PeiHua.
0 引言
【研究意义】人工优化后的II型CRISPR/Cas9基因编辑体系[1,2],因操作简单、效率高,以及脱靶效应低等优点受到全世界的广泛关注。目前,该系统已应用于多种植物[3,4],在植物分子育种中展现出巨大的前景。具有双链断裂能力的Cas9蛋白和一个人工融合的小向导RNA(single guide RNA)构成了常规的CRISPR/Cas9基因编辑体系[5],其中sgRNA起到靶向结合目的位点的功能,因此,筛选合适的启动子来驱动sgRNA的转录对CRISPR/Cas9基因编辑体系发挥功能有至关重要的意义。【前人研究进展】U6 snRNA(Small nuclear RNA)是一种小的非编码RNA,全长102 bp且序列非常保守[6],受RNA聚合酶III的识别和转录,广泛参与细胞核中mRNA前体(pre-mRNA)的剪接成熟[7,8,9]。驱动U6 snRNA转录的U6启动子常作为CRISPR/Cas9基因编辑载体中驱动sgRNA转录的重要元件。U6启动子在真核生物中发挥作用需具备两个重要的序列元件USE(Upstream sequence element)和TATA-Like box[6,10],均处于转录起始位点之前[11,12]。此外,U6启动子具有明确的转录起始位点‘G’碱基,可以精确转录sgRNA[13],但精确的起始转录要求转录起始位点周围序列保持稳定[14],前人[15,16]对携带5′端不同长度snRNA(+1 bp,+19—+27 bp)的U6启动子进行了转录活性比较,发现携带27 bp snRNA序列的U6启动子转录活性较高。目前,已有多种植物的U6启动子应用于CRISPR/Cas9基因编辑体系[17,18,19,20,21],LI等[3]和FENG等[22]利用拟南芥中已克隆的AtU6启动子驱动sgRNA的转录,在拟南芥中成功建立CRISPR/Cas9基因组编辑体系,并成功敲除拟南芥中AtBRI1、AtJAZ1和AtGAI。JIANG等[17]克隆水稻U6启动子驱动sgRNA的转录,并在水稻原生质体中敲除OsSWEET14和OsSWEET11。JACOBS等[23]克隆了大豆U6启动子,构建靶向敲除绿色荧光蛋白(GFP)基因的Cas9/sgRNA载体,并成功修饰9个大豆内源基因。NISHTANI等[24]利用拟南芥U6启动子驱动sgRNA的转录,成功突变了苹果叶绿素合成相关基因MdPDS。【本研究切入点】虽然多种植物的U6启动子在CRISPR/Cas9基因编辑体系中得到应用,但U6启动子在亲缘关系较远的物种中并不适用[6],LI等[18]发现仅拟南芥U6启动子具备保守的序列元件‘CAT框’,推测其可能是拟南芥U6启动子的物种特异性因子。并且,不同序列长度、不同组织类型对U6启动子的转录活性也存在一定影响[18,25-26]。目前,还未有苹果内源U6启动子介导的CRISPR/Cas9基因编辑体系。【拟解决的关键问题】对苹果U6启动子的转录活性比较,筛选到长度合适且转录活性高的苹果U6启动子,以期为进一步发展苹果CRISPR/ Cas9基因编辑技术奠定基础。1 材料与方法
试验于2018—2019年在中国农业科学院果树研究所/农业部园艺作物种质资源利用重点实验室/苹果育种中心实验室进行。1.1 试验材料
试验所用‘金冠’苹果幼嫩叶片和本氏烟草种子于中国农业科学院果树研究所种质资源圃及实验室中保存;pGreenII-0800-LUC质粒由新西兰皇家植物和食品研究所姚家龙博士惠赠;pYLsgRNA-AtU6-1(Accession No. KR029101.1)质粒由华南农业大学刘耀光院士惠赠;pTOPO-Blunt Clone kit购于北京艾德莱生物科技有限公司;Prime STAR MAX高保真DNA聚合酶购于TaKaRa公司;DH5α和农杆菌GV3101感受态购于庄盟生物科技有限公司;荧光素钠盐购于上海生工生物工程公司;测序及引物合成由天津金唯智生物公司完成。1.2 苹果U6启动子克隆及表达载体构建
用拟南芥保守的102 bp U6 snRNA序列在蔷薇科植物基因组数据库GDR(Table 1
表1
表1本研究使用的引物序列
Table 1
| 引物名称 Prime name | 上游引物 Forward sequence (5′-3′) | 下游引物 Reverse sequence (5′-3′) |
|---|---|---|
| MdU6-6P | F:ggtaccTTTGGAGTTGAAGGATTT | R;aagcttAATTTTATCGGATGTCCC |
| MdU6-7P | F:ggtaccCCCCCGTTTGGATGACCCA | R:aagcttAATTTTATCGGATGTCCC |
| MdU6-9P | F:aagcttATGCTTCTCCCATGGAAAT | R:ggatccAATTTTATCGGATGTCCC |
| MdU6-10P | F:ggtaccTGGTGACATTGAGGTTCT | R:aagcttAATTTTATCGGATGTCCC |
| MdU6-15P | F:ggtaccTTTGGCGTTGCATTAG | R:ggatccAATTTTATCGGATGTCCC |
| MdU6-17P | F:ggtaccCTCCCCGGAAATGAC | R:ggatccAATTTTATCGGATGTCCC |
| MdU6-10-2P | F:ggtaccTGATATGTGGTGTTTCTAGG | R:aagcttAATTTTATCGGATGTCCC |
| MdU6-10-3P | F:ggtaccGGCGAAAGGTTTATGTTC | R:aagcttAATTTTATCGGATGTCCC |
| MdU6-10-4P | F:ggtaccCTCCTGACTAGTAAAGAAGG | R:aagcttAATTTTATCGGATGTCCC |
| AtU6-1 | F;ggtaccAGAAATCTCAAAATTCCGGCAG | R:aagcttCAATCACTACTTCGTCTCTAACCATAT |
新窗口打开|下载CSV
图1
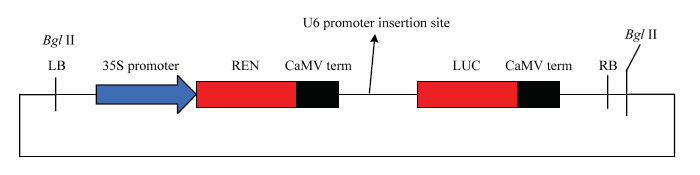 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1U6启动子驱动LUC报告基因的融合表达载体构建
Fig. 1Construction of LUC reporter gene fusion expression vector driven by U6 promoters
1.3 MdU6启动子序列元件分析
利用DNAMAN软件对BLAST检索到的6条苹果U6启动子进行序列比对,分析其保守的序列元件。利用PLACE和Plant CARE启动子在线分析网站预测苹果U6启动子序列元件。1.4 本氏烟草种植
本氏烟草种子于湿润环境中避光发芽后移入土中(蛭石﹕腐殖土﹕田园土=1.5﹕1﹕1)。温度23℃,光照强度12 000 lx,湿度70%,16 h光照/8 h黑暗条件下培养4—6周,选取第4片之后的功能叶进行注射。1.5 本氏烟草叶片中瞬时表达分析
将鉴定正确的表达载体转化至农杆菌GV3101感受态。于液体YEB培养基中摇至OD600值为0.8—1.0,离心收集菌体,重悬浮于注射缓冲液(10 mmol?L-1 MgCl2+200 μmol?L-1 AS)中。将重悬液于28℃静置3 h,注射本氏烟草叶片,避光过夜后于23℃光照培养箱进行16 h光照/8 h黑暗培养96 h。每片叶为一次重复,3次重复。1.6 苹果愈伤组织中瞬时表达分析
方法同1.5,待目的菌液摇至OD600值为0.6—0.8,离心收集菌体,重悬浮于注射缓冲液中,28℃静置活化3 h,取状态良好的苹果‘王林’愈伤组织放入其中,于28℃震荡侵染15 min,无菌水冲洗后用滤纸吸干,置于愈伤培养基中28℃避光共培养96 h,再次冲洗并吸水后放于无抗性愈伤培养基上,重复3次。1.7 生物发光检测
对瞬时转化96 h后的植物组织的荧光信号进行检测。将植物材料与100 mmol?L-1荧光素钠盐进行反应,烟草叶片浸泡3—5 min,苹果愈伤组织浸泡10 min。通过高分辨力制冷CCD检测系统进行数据采集(Tanon 5200Multi,中国),运用成像软件对采集的数据进行分析,将数据转换成彩色图像。1.8 数据分析
试验数据采用Excel 2016进行分析, 并采用SPSS 18.0进行单因素方差分析(P<0.05)。2 结果
2.1 MdU6启动子序列比对分析
基于‘金冠’苹果基因组中的BLAST结果,对E-value<3e-40的6条U6 snRNA进行分析,发现其分别位于苹果第6、7、9、10、15和17号染色体上(表2),全长均为102 bp且序列保守,另外,其转录起始位点均为‘G’碱基。参照PAUL等[15]的结论,本试验选取各U6 snRNA的5′端27 bp及其上游 1 500 bp作为苹果U6启动子候选序列,分别命名为MdU6-6P、MdU6-7P、MdU6-9P、MdU6-10P、MdU6- 15P和MdU6-17P。选取转录起始位点前后150 bp序列进行比对,发现其均存在-30位点的TATA-like box和-60位点的USE(5′-GTCCCACATCG-3′),且两个元件之间的距离较为保守(图2),其中TATA- like box与RNA聚合酶III的识别和结合相关。Table 2
表2
表2苹果U6 snRNA染色体定位
Table 2
| 基因名称 Gene names | 基因ID Gene ID | 染色体定位 Chromosome:Location |
|---|---|---|
| MdU6-06 | MD06G1040500 | Chr06: 5202556..5202455 |
| MdU6-07 | MD07G1079000 | Chr07: 7617645..7617544 |
| MdU6-09 | MD09G1120900 | Chr09: 9346234..9346335 |
| MdU6-10 | MD10G1255200 | Ch10: 34716072..34715971 |
| MdU6-15 | MD15G1130400 | Chr15: 9427466..9427365 |
| MdU6-17 | MD17G1111900 | Chr17: 9588121..9588222 |
新窗口打开|下载CSV
图2
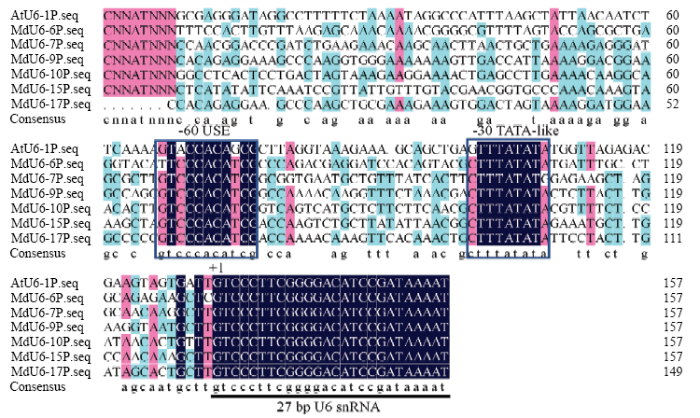 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2MdU6启动子序列分析
Fig. 2Sequence analysis of MdU6 promoters
2.2 苹果U6启动子的克隆及鉴定
从‘金冠’基因组DNA中成功克隆出MdU6-6P、MdU6-7P、MdU6-9P、MdU6-10P、MdU6-15P和MdU6-17P,测序结果显示序列正确。将各启动子片段分别连入pGreenII-0800-LUC载体,并命名为MdU6-6P:: LUC、MdU6-7P::LUC、MdU6-9P::LUC、MdU6-10P::LUC、MdU6-15P::LUC和MdU6-17P::LUC(图3)。图3
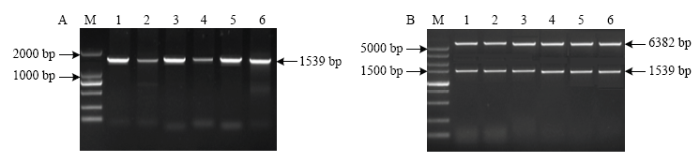 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3苹果U6启动子PCR扩增及鉴定
A:M:2 kb DNA标准分子量M: DL2000 DNA marker; 1: MdU6-6P; 2: MdU6-7P; 3: MdU6-9P; 4: MdU6-10P; 5: MdU6-15P; 6: MdU6-17P: B:M:5 kb DNA标准分子量M: DL5000 DNA marker; 1: MdU6-6P::LUC; 2: MdU6-7P::LUC; 3: MdU6-9P::LUC; 4: MdU6-10P::LUC; 5: MdU6-15P::LUC; 6: MdU6-17P::LUC
Fig. 3PCR amplification and identification of the MdU6 promoters
2.3 苹果U6启动子转录活性鉴定
将各MdU6-Ps::LUC载体通过农杆菌介导转化法注射于烟草叶片,并以35S::LUC为阳性对照,pGreenII-0800-LUC空载体为阴性对照。96 h后进行荧光素酶活性检测,发现MdU6-10P荧光信号最强,MdU6-9P仅次于MdU6-10P,而MdU6-6P、MdU6-7P、MdU6-15P以及MdU6-17P的荧光信号均较弱(图4)。图4
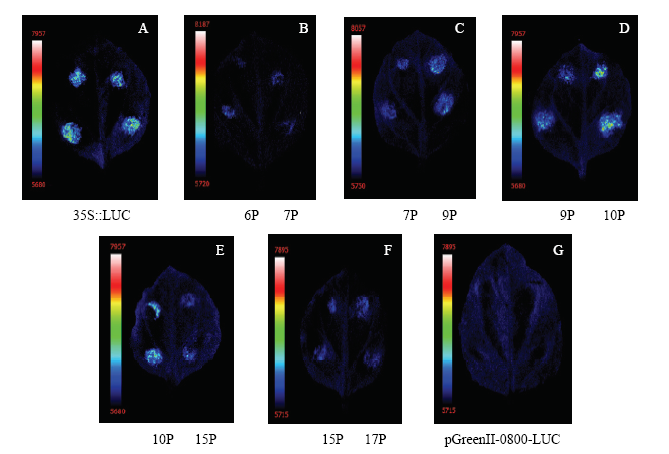 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4MdU6-Ps::LUC本氏烟草叶片中瞬时表达
6P: MdU6-6P::LUC; 7P: MdU6-7P::LUC; 9P: MdU6-9P::LUC; 10P: MdU6-10P::LUC; 15P: MdU6-15P::LUC; 17P: MdU6-17P::LUC
Fig. 4Transient expression of MdU6-Ps::LUC in N. benthamiana leaves
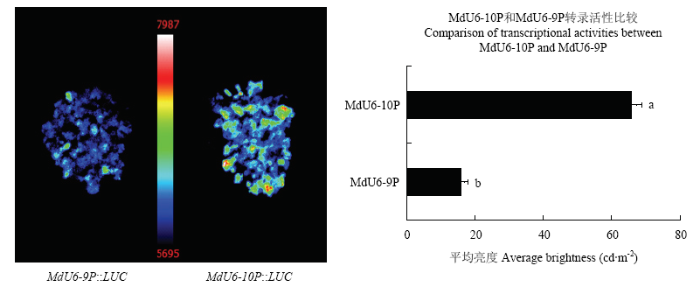
为比较MdU6-9P和MdU6-10P在苹果中的转录活性,进一步将MdU6-9P::LUC和MdU6-10P:: LUC载体瞬时转化苹果愈伤组织,对荧光素酶活性进行检测并将荧光信号数据进行统计分析(图5)。发现MdU6-10P::LUC的荧光信号强度显著高于MdU6-9P::LUC,证明MdU6-10P具有较高的转录活性,因此,选取苹果10号染色体上的U6启动子用于后续试验。
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5MdU6-9P::LUC和MdU6-10P::LUC在苹果愈伤组织中瞬时表达
不同小写字母表示差异显著(P<0.05)。下同
Fig. 5Transient expression of MdU6-9P::LUC and MdU6-10P::LUC in apple callus
Different lowercase letters indicate significant difference (P<0.05). The same as below
2.4 MdU6-10P元件分析
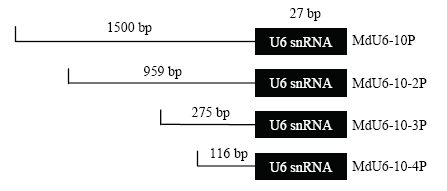
为分析序列长度对MdU6-10P转录活性的影响,使用PLACE和Plant CARE启动子元件在线分析网站对MdU6-10P进行元件分析,以预测的‘CAT框’为依据,对其进行5′端截短(截短长度分别为959、275和116 bp)。截短后的苹果U6启动子分别命名为MdU6-10-2P、MdU6-10-3P和MdU6-10-4P(图6)。图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图65′端截短的苹果U6启动子示意图
Fig. 6Schematic diagram of different MdU6-10-Ps truncated at the 5′ end
2.5 MdU6-10-Ps的克隆及鉴定
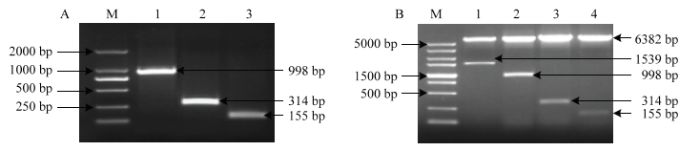
从‘金冠’苹果基因组中成功克隆出MdU6-10-2P、MdU6-10-3P和MdU6-10-4P,将各启动子片段分别连至 pGreenII-0800-LUC载体,将测序正确的各载体分别命名为MdU6-10-2P::LUC、MdU6-10-3P::LUC和MdU6-10-4P::LUC(图7)。图7
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7截短的苹果U6启动子PCR扩增和鉴定
A:M:2 Kb DNA 标准分子量M: DL2000 DNA marker; 1: MdU6-10-2P; 2: MdU6-10-3P; 3: MdU6-10-4P; B:M:5Kb DNA标准分子量M: DL5000 DNA marker; 1: MdU6-10P::LUC; 2: MdU6-10-2P::LUC; 3: MdU6-10-3P::LUC; 4: MdU6-10-4P::LUC
Fig. 7PCR amplification and identification of truncated MdU6 promoters
2.6 MdU6-10-Ps转录活性鉴定
利用农杆菌介导转化法,将MdU6-10P::LUC、MdU6-10-2P::LUC、MdU6-10-3P::LUC和MdU6-10-4P::LUC载体注射烟草叶片。检测荧光素酶活性并进行分析,发现注射后的烟草叶片均具有较强的荧光信号,但并无显著差异(图8)。图8
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8MdU6-10-Ps::LUC在本氏烟草叶片中瞬时表达
10P: MdU6-10P::LUC; 10-2P: MdU6-10-2P::LUC; 10-3P: MdU6-10-3P::LUC; 10-4P: MdU6-10-4P::LUC
Fig. 8Transient expression of MdU6-10-Ps::LUC in N. benthamiana leaves
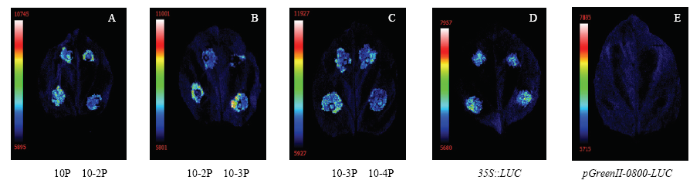
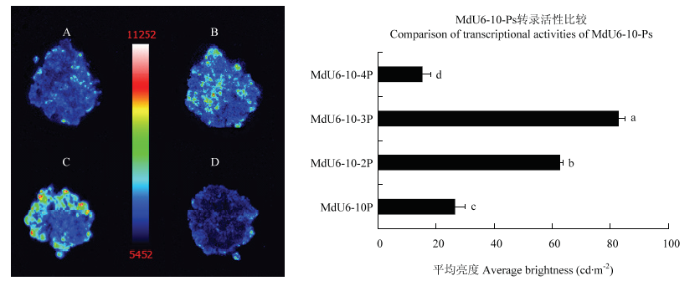
进一步在苹果愈伤组织中验证各长度苹果U6启动子转录活性,荧光素酶活性检测结果与烟草叶片不同,如图9所示。MdU6-10-Ps转染后的愈伤组织荧光信号强度存在显著差异(MdU6-10-3P>MdU6-10-2P>MdU6-10P>MdU6-10-4P),其中,MdU6-10-3P荧光信号最强,而MdU6-10-4P荧光信号强度最弱,但仍可检测到荧光信号。
图9
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图9MdU6-10-Ps::LUC在苹果愈伤组织中瞬时表达
A: MdU6-10P::LUC; B: MdU6-10-2P::LUC; C: MdU6-10-3P::LUC; D: MdU6-10-4P::LUC
Fig. 9Transient expression of MdU6-10-Ps::LUC in apple callus
2.7 苹果和拟南芥U6启动子转录活性比较
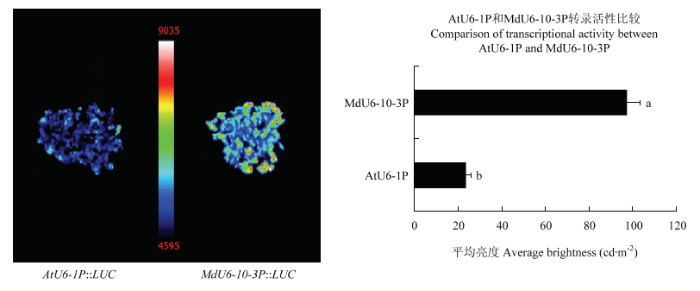
为比较拟南芥和苹果U6启动子在苹果中的转录活性,将AtU6-1P::LUC和MdU6-10-3P::LUC载体分别瞬时转化至苹果愈伤组织,结果如图10所示。MdU6-10-3P::LUC转染的愈伤组织荧光信号强度显著高于AtU6-1P::LUC。说明在苹果愈伤组织中,苹果U6启动子的转录活性高于拟南芥U6启动子。图10
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图10AtU6-1P::LUC和MdU6-10-3P::LUC在苹果愈伤组织中瞬时表达
Fig. 10Transient expression of AtU6-1P::LUC and MdU6-10-3P::LUC in apple callus
3 讨论
RNA介导的第3代人工核酸酶技术CRISPR/Cas9在植物基因功能验证及定向育种中展现出巨大的潜力,但由于Cas9序列较长(大于4 000 bp),且驱动Cas9转录的35S启动子长度也有800 bp,如果构建同时编辑多个靶点的CRISPR/ Cas9基因编辑载体,序列长度将达到15 000 bp,载体过长将会对细菌转化和植物组织转染造成困难。因此,筛选合适的启动子驱动sgRNA转录将为该体系的应用提供便利。U6启动子常被用来驱动sgRNA的转录。U6启动子属于RNA聚合酶III识别的启动子,III型启动子主要是对tRNA、5S rRNA等含发夹结构的小RNA进行转录[8],因此,U6启动子在转录含发夹结构的sgRNA时具有天然优势,并且可以确保转录的sgRNA在细胞核中发挥功能。此外,U6启动子具有精确的转录起始位点‘G’碱基,可以有效降低CRISPR/Cas9基因编辑体系的脱靶效应。本试验将拟南芥U6-1启动子和苹果6条候选U6启动子进行序列比对,发现6条候选的苹果U6启动子的转录起始位点均为‘G’碱基,且均具有RNA聚合酶III发挥作用所需的上游序列元件USE和TATA-like box。由于U6 snRNA序列高度保守,通过荧光定量PCR来比较各U6启动子的转录活性并不可行,因此,本试验通过利用U6启动子驱动报告基因表达的方式对苹果U6启动子的转录活性进行比较。目前,常用于启动子活性检测的报告基因有GUS[27]、eGFP[28,29]和LUC[30],本试验选择LUC为报告基因,通过瞬时转化后检测荧光素酶活性对各启动子的转录活性进行比较,发现所选的6条苹果U6启动子均具有转录活性,但转录活性存在差异,其中MdU6-10P具有较高的转录活性。前人研究表明,U6启动子在具备必要元件时即具有转录活性,反而序列过长可能存在一些抑制因子,使较长的U6启动子的转录活性下降[18]。在番茄[21]上,U6启动子被截取到200 bp左右时仍具有较高的活性,而在棉花[31]上,U6启动子截取到105 bp仍具有较高的活性。KHAOULA等[32]将拟南芥U6启动子截取到79 bp仍可应用于基因编辑体系。本研究对苹果10号染色体上的U6启动子进行5′端截短,并进行转录活性比较,发现在苹果愈伤组织中,长度为275 bp的苹果U6启动子转录活性最高,表明在苹果U6启动子中可能存在一些抑制因子,且存在于5′末端。荧光素酶活性检测结果还显示,116 bp长度的U6启动子活性虽减弱,但并未丧失转录活性,说明苹果U6启动子具有必要元件即有转录活性,而在序列较短时转录活性下降可能是因为缺少了一些增强转录活性的元件。至于在本氏烟草和苹果愈伤组织中瞬时转染展现出不同的结果,或许是因为某些物种特异性因子参与其中,导致U6启动子在亲缘关系较远的物种中转录活性存在一定差异,这与前人得出的结论一致[6]。植物的不同组织类型可能对U6启动子的转录活性也存在一定影响[18],而苹果不同组织类型对U6启动子转录活性的影响程度还有待进一步研究。
4 结论
从苹果品种‘金冠’中克隆获得6条长度为1 500 bp的U6启动子,均具有转录活性,其中10号染色体上的U6启动子转录活性最高,将该启动子从5′端进行截短(长度分别为959、275和116 bp),其中275 bp的启动子转录活性最高。此外,在苹果愈伤组织中,苹果U6启动子的转录活性高于拟南芥U6启动子。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1007/s00299-013-1539-6URLPMID:24277082 [本文引用: 1]

Recent advances in genome engineering indicate that innovative crops developed by targeted genome modification (TGM) using site-specific nucleases (SSNs) have the potential to avoid the regulatory issues raised by genetically modified organisms. These powerful SSNs tools, comprising zinc-finger nucleases, transcription activator-like effector nucleases, and clustered regulatory interspaced short palindromic repeats/CRISPR-associated systems, enable precise genome engineering by introducing DNA double-strand breaks that subsequently trigger DNA repair pathways involving either non-homologous end-joining or homologous recombination. Here, we review developments in genome-editing tools, summarize their applications in crop organisms, and discuss future prospects. We also highlight the ability of these tools to create non-transgenic TGM plants for next-generation crop breeding.
DOI:10.1038/nprot.2013.143URL [本文引用: 1]
Targeted nucleases are powerful tools for mediating genome alteration with high precision. The RNA-guided Cas9 nuclease from the microbial clustered regularly interspaced short palindromic repeats (CRISPR) adaptive immune system can be used to facilitate efficient genome engineering in eukaryotic cells by simply specifying a 20-nt targeting sequence within its guide RNA. Here we describe a set of tools for Cas9-mediated genome editing via nonhomologous end joining (NHEJ) or homology-directed repair (HDR) in mammalian cells, as well as generation of modified cell lines for downstream functional studies. To minimize off-target cleavage, we further describe a double-nicking strategy using the Cas9 nickase mutant with paired guide RNAs. This protocol provides experimentally derived guidelines for the selection of target sites, evaluation of cleavage efficiency and analysis of off-target activity. Beginning with target design, gene modifications can be achieved within as little as 1-2 weeks, and modified clonal cell lines can be derived within 2-3 weeks.
DOI:10.1038/nbt.2654URLPMID:23929339 [本文引用: 2]
DOI:10.1111/pbi.13315URLPMID:31821678 [本文引用: 1]
MicroRNAs (miRNAs) are 20-24 nucleotides (nt) small RNAs functioning in eukaryotes. The length and sequence of miRNAs are not only related to the biogenesis of miRNAs, but are also important for downstream physiological processes like ta-siRNA production. To investigate these roles, it is informative to create small mutations within mature miRNA sequences. We used both TALENs (transcription activator-like effector nucleases) and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) to introduce heritable base pair mutations in mature miRNA sequences. For rice, TALEN constructs were built targeting five different mature miRNA sequences and yielding heritable mutations. Among the resulting mutants, mir390 mutant showed a severe defect in the shoot apical meristem (SAM), a shootless phenotype, which could be rescued by the wild type MIR390. Small RNA sequencing showed the two base pair deletion in mir390 substantially interfered with miR390 biogenesis. In Arabidopsis, CRISPR/Cas9 mediated editing of the miR160* strand confirmed that the asymmetric structure of miRNA is not a necessary determinant for secondary siRNA production. CRISPR/Cas9 with double guide RNAs successfully generated mir160a null mutants with fragment deletions, at a higher efficiency than a single guide RNA. The difference between the phenotypic severity of miR160a mutants in Col-0 versus Ler backgrounds highlights a diverged role for miR160a in different ecotypes. Overall, we demonstrated that TALENs and CRISPR/Cas9 are both effective in modifying miRNA precursor structure, disrupting miRNA processing, and generating miRNA null mutant plants.
DOI:10.1186/s12870-014-0327-yURL [本文引用: 1]
URLPMID:3027083 [本文引用: 4]
U6 RNA is an abundant, capped small nuclear RNA (snRNA) associated with hnRNP particles (Reddy, R., and Busch, H. (1983) Prog. Nucleic Acid Res. Mol. Biol. 30, 127-162). Small nuclear ribonucleoprotein particles containing U4 and U6 RNAs are required components for splicing of pre-mRNAs (Berget and Robberson, 1986; Black and Steitz, 1986). In this study the Drosophila U6 RNA genes have been isolated and characterized. The Drosophila genome contains three U6 snRNA genes which are clustered in a 2-kilobase-pairs long DNA fragment. The U6 RNA coding regions are 100% homologous in all three genes, but the flanking sequences diverged significantly from each other. A possible secondary structure model for the Drosophila U4/U6 RNA complex is presented. Consistent with our previous observation that U6 RNA is a RNA polymerase III product (Reddy, R., Henning, D., Das, G., Harless, M., and Wright, D. (1987) J. Biol. Chem. 262, 75-81), all three genes contained a region homologous to the consensus intragenic regulatory region and a cluster of T residues on the 3'-end, characteristic of genes transcribed by RNA polymerase III. A TATA box was found between nucleotides -23 and -31, and a stretch of 28 nucleotides from -43 to -71 was conserved in the 5'-flanking region of all three U6 RNA genes. The Drosophila U6 RNA genes were transcribed in vitro by Drosophila nuclear extracts but were not transcribed by Novikoff hepatoma or HeLa cell extracts. Similarly, a mouse U6 RNA gene was transcribed in Novikoff hepatoma or HeLa cell extracts but not in Drosophila nuclear extracts. These results suggest that species-specific factor(s) are involved in the transcription of U6 snRNA genes.
DOI:10.1055/s-0039-3400233URLPMID:31842235 [本文引用: 1]
Thoracic aortic aneurysm is a typically silent disease characterized by a lethal natural history. Since the discovery of the familial nature of thoracic aortic aneurysm and dissection (TAAD) almost 2 decades ago, our understanding of the genetics of this disorder has undergone a transformative amplification. To date, at least 37 TAAD-causing genes have been identified and an estimated 30% of the patients with familial nonsyndromic TAAD harbor a pathogenic mutation in one of these genes. In this review, we present our yearly update summarizing the genes associated with TAAD and the ensuing clinical implications for surgical intervention. Molecular genetics will continue to bolster this burgeoning catalog of culprit genes, enabling the provision of personalized aortic care.
DOI:10.1016/0092-8674(86)90344-2URLPMID:2427201 [本文引用: 2]
The requirement for individual U RNAs in splicing and polyadenylation was investigated using oligonucleotide-directed cleavage of snRNAs in in vitro processing extracts. Cleavage of U1, U2, or U4 RNA inhibited splicing but not polyadenylation of short precursor RNAs. Thus each snRNA and the snRNP in which it is assembled participates in the splicing reaction. Splicing activity was recovered when extracts containing cleaved U RNAs were mixed in pairwise combinations, indicating that U1, U2, and U4/U6 snRNPs independently interact with the assembling spliceosome. The involvement of multiple snRNPs in the splicing of simple precursor RNAs suggests that the spliceosome is a large complex assembly consisting of multiple snRNPs whose activity is dependent on the structural integrity of the individual U RNAs.
DOI:10.1016/0092-8674(86)90345-4URLPMID:2427202 [本文引用: 1]
Selective cleavage of U4 or U6 RNA in a HeLa cell nuclear extract inhibits splicing of pre-mRNAs containing an adenovirus or a simian virus 40 intron. RNAs in the U4/U6 small nuclear ribonucleoprotein (snRNP) were specifically degraded with RNAase H and deoxyoligonucleotides. Two oligomers complementary to U4 RNA and two complementary to U6 RNA cleave their target RNAs and inhibit the appearance of both spliced products and reaction intermediates. Splicing is reconstituted by mixing an extract containing cleaved U4 or U6 RNA with one in which splicing has been inhibited by degrading U2 RNA. All four abundant snRNPs, containing U1, U2, U5, or U4 and U6 RNAs, are now implicated in pre-mRNA splicing. Possible interactions of the U4/U6 snRNP with other components of the splicing complex are discussed.
DOI:10.1093/nar/18.12.3451URLPMID:2362802 [本文引用: 1]
Previously we have demonstrated that the U2 snRNA genes from the higher plant Arabidopsis thaliana contain two upstream elements, the USE with sequence RTCCCACATCG and a -30 'TATA' box, which are essential for transcription by RNA polymerase II, and that the conserved spacing of about four helical DNA turns between these elements is important for optimal promoter function. We have now isolated three genes encoding U6 RNA in Arabidopsis. Transcription of these genes in transfected protoplasts of Nicotiana plumbaginifolia is resistant to alpha-amanitin indicating that they are transcribed by RNA polymerase III. The upstream regions of three Arabidopsis U6 genes contain USE and -30 TATA-like elements similar to those found to be important for transcription of U2 RNA genes but the spacing between the two elements is about 10 bp closer than in the U2 genes. Using synthetic U6 genes we demonstrate that the USE and TATA elements are indispensable for their transcription, the TATA boxes of U2 and U6 genes are interchangeable, and that the intragenic A box-like sequence of U6 gene is not essential. Increasing the distance between the USE and TATA by 10 bp inactivates U6 gene transcription, demonstrating that proper positioning of the elements is also important for transcription by RNA polymerase III. The data indicate that the structure of U-snRNA gene promoters and the determinants of polymerase specificity are completely different between vertebrates and plants.
DOI:10.1016/0092-8674(80)90385-2URLPMID:7357604 [本文引用: 1]
[本文引用: 1]
DOI:10.1126/science.1232033URL [本文引用: 1]
Bacteria and archaea have evolved adaptive immune defenses, termed clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems, that use short RNA to direct degradation of foreign nucleic acids. Here, we engineer the type II bacterial CRISPR system to function with custom guide RNA (gRNA) in human cells. For the endogenous AAVS1 locus, we obtained targeting rates of 10 to 25% in 293T cells, 13 to 8% in K562 cells, and 2 to 4% in induced pluripotent stem cells. We show that this process relies on CRISPR components; is sequence-specific; and, upon simultaneous introduction of multiple gRNAs, can effect multiplex editing of target loci. We also compute a genome-wide resource of similar to 190 K unique gRNAs targeting similar to 40.5% of human exons. Our results establish an RNA-guided editing tool for facile, robust, and multiplexable human genome engineering.
DOI:10.1038/mtna.2014.12URLPMID:24803291 [本文引用: 1]
Pol III promoters such as U6 are commonly used to express small RNAs, including small interfering RNA, short hairpin RNA, and guide RNA, for the clustered regularly interspaced short palindromic repeats genome-editing system. However, whether the small RNAs were precisely expressed as desired has not been studied. Here, using deep sequencing to analyze small RNAs, we show that, for mouse U6 promoter, sequences immediately upstream of the putative initiation site, which is often modified to accommodate the restriction enzyme sites that enable easy cloning of small RNAs, are critical for precise transcription initiation. When the promoter is kept unmodified, transcription starts precisely from the first available A or G within the range of positions -1 to +2. In addition, we show that transcription from another commonly used pol III promoter, H1, starts at multiple sites, which results in variability at the 5' end of the transcripts. Thus, inaccuracy of 5' end of small RNA transcripts might be a common problem when using these promoters to express small RNAs based on currently believed concepts. Our study provides general guidelines for minimizing the variability of initiation, thereby enabling more accurate expression of small RNAs.
DOI:10.1038/nbt0502-505URLPMID:11981566 [本文引用: 2]
In many eukaryotes, expression of nuclear-encoded mRNA can be strongly inhibited by the presence of a double-stranded RNA (dsRNA) corresponding to exon sequences in the mRNA (refs 1,2). The use of this &quot;RNA interference&quot; (RNAi) in mammalian studies had lagged well behind its utility in lower animals because uninterrupted RNA duplexes longer than 30 base pairs trigger generalized cellular responses through activation of dsRNA-dependent protein kinases. Recently it was demonstrated that RNAi can be made to work in cultured human cells by introducing shorter, synthetic duplex RNAs (approximately 20 base pairs) through liposome transfection. We have explored several strategies for expressing similar short interfering RNA (siRNA) duplexes within cells from recombinant DNA constructs, because this might allow long-term target-gene suppression in cells, and potentially in whole organisms. Effective suppression of target gene product levels is achieved by using a human U6 small nuclear RNA (snRNA) promoter to drive nuclear expression of a single RNA transcript. The siRNA-like parts of the transcript consists of a 19 base pair siRNA stem with the two strands joined by a tightly structured loop and a U1-4 3' overhang at the end of the antisense strand. The simplicity of the U6 expression cassette and its widespread transcription in human cell types suggest that this mode of siRNA delivery could be useful for suppressing expression of a wide range of genes.
DOI:10.1254/jphs.93.214URLPMID:14578591 [本文引用: 1]
RNA interference (RNAi), a process of sequence-specific gene suppression, has been known as a natural gene regulatory mechanism in a wide range of organisms. Recently, a small-interference RNA (siRNA) technology has been reported to produce post-transcriptional gene silencing in mammalian cells. In the present study, we constructed a human U6 promoter-driven mammalian expression vector to produce hairpin double-stranded RNA and transfected this into a human cell line. Using this siRNA system, we were able to knock down the gene expression of an enhanced green fluorescence protein. This result indicates that the plasmid vector-based siRNA system is a promising method to downregulate gene expression in human cells.
DOI:10.1093/nar/gkt780URLPMID:23999092 [本文引用: 2]
The type II CRISPR/Cas system from Streptococcus pyogenes and its simplified derivative, the Cas9/single guide RNA (sgRNA) system, have emerged as potent new tools for targeted gene knockout in bacteria, yeast, fruit fly, zebrafish and human cells. Here, we describe adaptations of these systems leading to successful expression of the Cas9/sgRNA system in two dicot plant species, Arabidopsis and tobacco, and two monocot crop species, rice and sorghum. Agrobacterium tumefaciens was used for delivery of genes encoding Cas9, sgRNA and a non-fuctional, mutant green fluorescence protein (GFP) to Arabidopsis and tobacco. The mutant GFP gene contained target sites in its 5' coding regions that were successfully cleaved by a CAS9/sgRNA complex that, along with error-prone DNA repair, resulted in creation of functional GFP genes. DNA sequencing confirmed Cas9/sgRNA-mediated mutagenesis at the target site. Rice protoplast cells transformed with Cas9/sgRNA constructs targeting the promoter region of the bacterial blight susceptibility genes, OsSWEET14 and OsSWEET11, were confirmed by DNA sequencing to contain mutated DNA sequences at the target sites. Successful demonstration of the Cas9/sgRNA system in model plant and crop species bodes well for its near-term use as a facile and powerful means of plant genetic engineering for scientific and agricultural applications.
DOI:10.1093/nar/gkg331URLPMID:12711679 [本文引用: 5]
Vertebrate U6 small nuclear RNA (snRNA) gene promoters are among the founding members of those recognized by RNA polymerase III in which all control elements for initiation are located in the 5'-flanking region. Previously, one human U6 gene (U6-1) has been studied extensively. We have identified a total of nine full-length U6 loci in the human genome. Unlike human U1 and U2 snRNA genes, most of the full-length U6 loci are dispersed throughout the genome. Of the nine full-length U6 loci, five are potentially active genes (U6-1, U6-2, U6-7, U6-8 and U6-9) since they are bound by TATA-binding protein and enriched in acetylated histone H4 in cultured human 293 cells. These five all contain OCT, SPH, PSE and TATA elements, although the sequences of these elements are variable. Furthermore, these five genes are transcribed to different extents in vitro or after transient transfection of human 293 cells. Of the nine full-length U6 loci, only U6-7 and U6-8 are closely linked and contain highly conserved 5'-flanking regions. However, due to a modest sequence difference in the proximal sequence elements for U6-7 and U6-8, these genes are transcribed at very different levels in transfected cells.
DOI:10.1038/nbt.2650URLPMID:23929338 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.3864/j.issn.0578-1752.2018.02.011URL [本文引用: 2]
【目的】从番茄中克隆高效转录的SlU6启动子,构建CRISPR/Cas9基因编辑载体,并在番茄中建立CRISPR/Cas9系统,为番茄功能基因组学和分子育种研究提供技术基础。【方法】采用PCR方法从‘中蔬四号’番茄品种中克隆4种SlU6启动子,利用Transfer PCR方法分别对4个启动子进行两种不同长度的截短,分别构建8个截短的SlU6启动子驱动GUS的植物融合表达载体。利用农杆菌瞬时转化法分别转染番茄叶片,通过GUS染色筛选出在番茄叶片中转录活性较高的SlU6-2启动子。采用DNA重组技术构建以SlU6-2为启动子驱动sgRNA,以番茄白粉病相关基因MLO1和EDR1为靶序列的CRISPR/Cas9基因组编辑载体。载体构建成功后,采用PEG法转化番茄原生质体,提取基因组DNA,采用酶切/PCR法分析内源基因突变情况;采用测序法分析内源基因突变的类型。利用突变位点频率分布图来验证番茄内源启动子在番茄CRISPR/Cas9系统中的有效性。【结果】经过两轮PCR,共获得4种8个不同长度的番茄U6启动子,其长度分别是452、202、448、206、433、190、448和218 bp,启动子序列比对分析发现番茄U6启动子与拟南芥U6启动子一样,也含有比较保守的两个元件,USE和TATA框。成功构建了8个SlU6启动子分别驱动GUS的植物融合表达载体。番茄叶片染色结果显示转化后的番茄叶片均被染成蓝色,表明克隆的番茄8个SlU6启动子均具有转录活性。选择SlU6-2P4为启动子驱动sgRNA,成功构建番茄白粉病相关基因MLO1和EDR1为靶序列的CRISPR/Cas9基因组编辑载体,验证结果表明番茄内源启动子SlU6-2P4能有效地驱动sgRNA的转录,并成功实现对番茄内源基因的编辑。内源基因突变的类型都为碱基替换,突变热点仅存在于内源基因靶序列区。【结论】成功克隆了4种在番茄叶片中高效转录的SlU6启动子;基于SlU6-2启动子的CRISPR/Cas9基因组编辑载体,在番茄中成功实现对内源基因的编辑。
DOI:10.3864/j.issn.0578-1752.2018.02.011URL [本文引用: 2]
【目的】从番茄中克隆高效转录的SlU6启动子,构建CRISPR/Cas9基因编辑载体,并在番茄中建立CRISPR/Cas9系统,为番茄功能基因组学和分子育种研究提供技术基础。【方法】采用PCR方法从‘中蔬四号’番茄品种中克隆4种SlU6启动子,利用Transfer PCR方法分别对4个启动子进行两种不同长度的截短,分别构建8个截短的SlU6启动子驱动GUS的植物融合表达载体。利用农杆菌瞬时转化法分别转染番茄叶片,通过GUS染色筛选出在番茄叶片中转录活性较高的SlU6-2启动子。采用DNA重组技术构建以SlU6-2为启动子驱动sgRNA,以番茄白粉病相关基因MLO1和EDR1为靶序列的CRISPR/Cas9基因组编辑载体。载体构建成功后,采用PEG法转化番茄原生质体,提取基因组DNA,采用酶切/PCR法分析内源基因突变情况;采用测序法分析内源基因突变的类型。利用突变位点频率分布图来验证番茄内源启动子在番茄CRISPR/Cas9系统中的有效性。【结果】经过两轮PCR,共获得4种8个不同长度的番茄U6启动子,其长度分别是452、202、448、206、433、190、448和218 bp,启动子序列比对分析发现番茄U6启动子与拟南芥U6启动子一样,也含有比较保守的两个元件,USE和TATA框。成功构建了8个SlU6启动子分别驱动GUS的植物融合表达载体。番茄叶片染色结果显示转化后的番茄叶片均被染成蓝色,表明克隆的番茄8个SlU6启动子均具有转录活性。选择SlU6-2P4为启动子驱动sgRNA,成功构建番茄白粉病相关基因MLO1和EDR1为靶序列的CRISPR/Cas9基因组编辑载体,验证结果表明番茄内源启动子SlU6-2P4能有效地驱动sgRNA的转录,并成功实现对番茄内源基因的编辑。内源基因突变的类型都为碱基替换,突变热点仅存在于内源基因靶序列区。【结论】成功克隆了4种在番茄叶片中高效转录的SlU6启动子;基于SlU6-2启动子的CRISPR/Cas9基因组编辑载体,在番茄中成功实现对内源基因的编辑。
DOI:10.1038/cr.2013.114URLPMID:23958582 [本文引用: 1]
DOI:10.1371/journal.pone.0136064URLPMID:26284791 [本文引用: 1]
As a new technology for gene editing, the CRISPR (clustered regularly interspaced short palindromic repeat)/Cas (CRISPR-associated) system has been rapidly and widely used for genome engineering in various organisms. In the present study, we successfully applied type II CRISPR/Cas9 system to generate and estimate genome editing in the desired target genes in soybean (Glycine max (L.) Merrill.). The single-guide RNA (sgRNA) and Cas9 cassettes were assembled on one vector to improve transformation efficiency, and we designed a sgRNA that targeted a transgene (bar) and six sgRNAs that targeted different sites of two endogenous soybean genes (GmFEI2 and GmSHR). The targeted DNA mutations were detected in soybean hairy roots. The results demonstrated that this customized CRISPR/Cas9 system shared the same efficiency for both endogenous and exogenous genes in soybean hairy roots. We also performed experiments to detect the potential of CRISPR/Cas9 system to simultaneously edit two endogenous soybean genes using only one customized sgRNA. Overall, generating and detecting the CRISPR/Cas9-mediated genome modifications in target genes of soybean hairy roots could rapidly assess the efficiency of each target loci. The target sites with higher efficiencies can be used for regular soybean transformation. Furthermore, this method provides a powerful tool for root-specific functional genomics studies in soybean.
DOI:10.1038/srep31481URLPMID:27530958 [本文引用: 1]
Genome editing is a powerful technique for genome modification in molecular research and crop breeding, and has the great advantage of imparting novel desired traits to genetic resources. However, the genome editing of fruit tree plantlets remains to be established. In this study, we describe induction of a targeted gene mutation in the endogenous apple phytoene desaturase (PDS) gene using the CRISPR/Cas9 system. Four guide RNAs (gRNAs) were designed and stably transformed with Cas9 separately in apple. Clear and partial albino phenotypes were observed in 31.8% of regenerated plantlets for one gRNA, and bi-allelic mutations in apple PDS were confirmed by DNA sequencing. In addition, an 18-bp gRNA also induced a targeted mutation. These CRIPSR/Cas9 induced-mutations in the apple genome suggest activation of the NHEJ pathway, but with some involvement also of the HR pathway. Our results demonstrate that genome editing can be practically applied to modify the apple genome.
[D].
[本文引用: 1]
[D].
[本文引用: 1]
DOI:10.1261/rna.760908URLPMID:18367720 [本文引用: 1]
RNA polymerase III (Pol III) as well as Pol II (35S) promoters are able to drive hairpin RNA (hpRNA) expression and induce target gene silencing in plants. siRNAs of 21 nt are the predominant species in a 35S Pol II line, whereas 24- and/or 22-nucleotide (nt) siRNAs are produced by a Pol III line. The 35S line accumulated the loop of the hpRNA, in contrast to full-length hpRNA in the Pol III line. These suggest that Pol II and Pol III-transcribed hpRNAs are processed by different pathways. One Pol III transgene produced only 24-nt siRNAs but silenced the target gene efficiently, indicating that the 24-nt siRNAs can direct mRNA degradation; specific cleavage was confirmed by 5' rapid amplification of cDNA ends (RACE). Both Pol II- and Pol III-directed hpRNA transgenes induced cytosine methylation in the target DNA. The extent of methylation is not correlated with the level of 21-nt siRNAs, suggesting that they are not effective inducers of DNA methylation. The promoter of a U6 transgene was significantly methylated, whereas the promoter of the endogenous U6 gene was almost free of cytosine methylation, suggesting that endogenous sequences are more resistant to de novo DNA methylation than are transgene constructs.
URLPMID:3327686 [本文引用: 1]
We have used the Escherichia coli beta-glucuronidase gene (GUS) as a gene fusion marker for analysis of gene expression in transformed plants. Higher plants tested lack intrinsic beta-glucuronidase activity, thus enhancing the sensitivity with which measurements can be made. We have constructed gene fusions using the cauliflower mosaic virus (CaMV) 35S promoter or the promoter from a gene encoding the small subunit of ribulose bisphosphate carboxylase (rbcS) to direct the expression of beta-glucuronidase in transformed plants. Expression of GUS can be measured accurately using fluorometric assays of very small amounts of transformed plant tissue. Plants expressing GUS are normal, healthy and fertile. GUS is very stable, and tissue extracts continue to show high levels of GUS activity after prolonged storage. Histochemical analysis has been used to demonstrate the localization of gene activity in cells and tissues of transformed plants.
DOI:10.1016/s0960-9822(02)00483-9URLPMID:8805250 [本文引用: 1]
The green-fluorescent protein (GFP) of the jellyfish Aequorea victoria has recently been used as a universal reporter in a broad range of heterologous living cells and organisms. Although successful in some plant transient expression assays based on strong promoters or high copy number viral vectors, further improvement of expression efficiency and fluorescent intensity are required for GFP to be useful as a marker in intact plants. Here, we report that an extensively modified GFP is a versatile and sensitive reporter in a variety of living plant cells and in transgenic plants.
DOI:10.1006/bbrc.1996.1573URLPMID:8885998 [本文引用: 1]
The green fluorescent protein (GFP) from the jellyfish Aequorea victoria has become an important marker of gene expression. However, the sensitivity of wild-type GFP has been below that of standard reporter proteins, such as beta-galactosidase, which utilize enzymatic amplification. To improve the detection of GFP in transfected mammalian cells, we have constructed a unique GFP variant which contains chromophore mutations that make the protein 35 times brighter than wild-type GFP, and is codon-optimized for higher expression in mammalian cells. These changes in the GFP coding sequence provide an enhanced GFP (EGFP) that greatly increases the sensitivity of the reporter protein. We show that the EGFP expression vector delivered into mammalian cells gives rise to bright fluorescence that is readily detectable following a 16-24 hr transfection interval. Visual detection of transfected cells with EGFP appears to be more sensitive than equivalent measurements with beta-galactosidase catalyzed conversion of the X-gal substrate. We conclude that EGFP allows sensitive and convenient detection of gene transfer in mammalian cells.
DOI:10.1016/s0958-1669(97)80038-9URLPMID:9353237 [本文引用: 1]
The use of reporters such as green fluorescent protein (GFP) and firefly luciferase permit highly sensitive and nondestructive monitoring of gene transfer and expression. Modifications in GFP which increase intensity and thermostability, as well as alter its spectral qualities, have facilitated the use of GFP in a variety of gene transfer methods. Improvements in imaging technologies and their increased application in biological research have allowed the expanded use of luciferase-based reporters in gene transformation, particularly in genetic screens and in monitoring temporal changes in gene expression.
[D].
[本文引用: 1]
[D].
[本文引用: 1]
DOI:10.1186/1746-4811-9-39URLPMID:24112467 [本文引用: 1]
Targeted genome engineering (also known as genome editing) has emerged as an alternative to classical plant breeding and transgenic (GMO) methods to improve crop plants. Until recently, available tools for introducing site-specific double strand DNA breaks were restricted to zinc finger nucleases (ZFNs) and TAL effector nucleases (TALENs). However, these technologies have not been widely adopted by the plant research community due to complicated design and laborious assembly of specific DNA binding proteins for each target gene. Recently, an easier method has emerged based on the bacterial type II CRISPR (clustered regularly interspaced short palindromic repeats)/Cas (CRISPR-associated) immune system. The CRISPR/Cas system allows targeted cleavage of genomic DNA guided by a customizable small noncoding RNA, resulting in gene modifications by both non-homologous end joining (NHEJ) and homology-directed repair (HDR) mechanisms. In this review we summarize and discuss recent applications of the CRISPR/Cas technology in plants.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}