
 , 秦玉芳1, 于晓庆2
, 秦玉芳1, 于晓庆2 1. 上海海洋大学 信息学院,上海 201306;
2. 上海应用技术大学 理学院,上海 201418
收稿日期:2020-07-23;接收日期:2020-11-23;网络出版时间:2021-01-05
基金项目:上海市科技创新计划(No. 20dz1203800),国家自然科学基金(Nos. 61702325, 11701379),国家重点研发计划(No. 2018YFD0701003),上海市科技创新行动计划(No. 16391902900) 资助
摘要:不同细胞在特定化合物作用下具有不同的扰动信号,基于这些扰动信号预测细胞的活性和挖掘隐藏在表型之下的药物敏感性非常重要。文中开发了一种基于LINCS-L1000扰动信号的SAE-XGBoost细胞活性预测算法。通过对LINCS-L1000、Achilles和CTRP三大数据集匹配和筛选,采用堆栈式深度自动编码器对基因信息进行特征提取,结合RW-XGBoost算法预测药物诱导下的细胞活性,进而在NCI60和CCLE数据集上完成药物敏感性推断。与其他方法相比,该模型取得了良好效果,皮尔逊相关系数为0.85,并进行独立集验证,对应皮尔逊相关系数为0.68。结果表明,所提出的方法有助于发现新型有效的抗癌药物,为精准医疗提供帮助。
关键词:扰动信号细胞活性药物敏感性堆栈式深度自动编码器RW-XGBoost
Prediction of drug-induced cell viability by SAE-XGBoost algorithm based on LINCS-L1000 perturbation signal
Jiaxing Lu1, Ming Chen1
, Yufang Qin1, Xiaoqing Yu2 1. College of Information Technology, Shanghai Ocean University, Shanghai 201306, China;
2. School of Sciences, Shanghai Institute of Technology, Shanghai 201418, China
Received: July 23, 2020; Accepted: November 23, 2020; Published: January 5, 2021
Supported by: Science and Technology Innovation Plan Project (No. 20dz1203800), National Natural Science Foundation of China (Nos. 61702325, 11701379), National Key Research and Development Program of China (No. 2018YFD0701003), Shanghai Science and Technology Innovation Action Plan, China (No. 16391902900)
Corresponding author: Ming Chen. Tel: +86-21-61900296; Fax: +86-21-61900000; E-mail: mchen@shou.edu.cn;
Xiaoqing Yu. Tel: +86-21-60873530; E-mail: xqyu@sit.edu.cn.
Abstract: Different cell lines have different perturbation signals in response to specific compounds, and it is important to predict cell viability based on these perturbation signals and to uncover the drug sensitivity hidden underneath the phenotype. We developed an SAE-XGBoost cell viability prediction algorithm based on the LINCS-L1000 perturbation signal. By matching and screening three major dataset, LINCS-L1000, CTRP and Achilles, a stacked autoencoder deep neural network was used to extract the gene information. These information were combined with the RW-XGBoost algorithm to predict the cell viability under drug induction, and then to complete drug sensitivity inference on the NCI60 and CCLE datasets. The model achieved good results compared to other methods with a Pearson correlation coefficient of 0.85. It was further validated on an independent dataset, corresponding to a Pearson correlation coefficient of 0.68. The results indicate that the proposed method can help discover novel and effective anti-cancer drugs for precision medicine.
Keywords: perturbation signalscell viabilitydrug sensitivitystacked autoencoder deep neural networkRW-XGBoost algorithm
近年来,细胞凋亡与肿瘤的关系成为精准医疗研究的热点之一[1],有研究证明,细胞凋亡功能的抑制将导致肿瘤的发生及免疫功能的异常,肿瘤细胞凋亡抑制因子活性的增强,能够抵御细胞凋亡信号通路的激活,其比例变化和异常增殖行为通常与化合物的浓度、时间相关,是肿瘤细胞发生和发展的关键因素之一[2]。由于药物作用的个体差异性[3],对于大多数刚进入临床试验阶段的化合物来说仍然缺乏最佳的应用治疗方案[4],随着大规模药物诱导下的基因组数据被得以公开,应用机器学习方法从分子水平上构建细胞活性预测模型,分析潜在的化学治疗反应,为临床治疗设计最佳策略,依然是目前面临的挑战[5]。
由于细胞扰动信号与细胞生存能力紧密相关,在药物敏感性和抗癌药物反应预测的研究中,通过从不同高覆盖分子数据预测细胞表型,采用化合物控制细胞凋亡通路中的关键蛋白或酶类的表达或功能,来诱导异常细胞凋亡。一种常见的方法是考虑使用实验测量的核糖核酸(Ribonucleic acid,RNA) 表达、蛋白质表达、甲基化、单核苷酸多态性(Single nucleotide polymorphisms,SNP)等基因组特征和对不同药物与细胞系之间的反应作为训练集,并针对每种单独药物设计一个或多个监督预测模型。例如,G?nen和Margolin[6]在训练时采用药物关联方法,依赖于内核的降维和多任务学习的贝叶斯(Kernelized bayesian multitask learning,KBMTL) 方法表现出显著的药物反应预测性能,使其备受青睐。同样,Menden等[7]根据每种细胞的基因组背景,训练了一个神经网络模型,预测其在整个细胞系中的IC50分布,发现癌细胞对药物分子的敏感性是由细胞和药物的特征共同驱动的。在DREAM细胞毒性反应预测挑战赛中,针对884种淋巴细胞系中156个化合物的细胞毒性,随机森林模型以整体性能最佳在两个子任务中均获优胜[8]。Goswami等[9]针对药物反应基因表达测量的最佳药物暴露时间和核心基因集选择,使用新颖的药物相互作用评分算法,预测药物对弥漫性大B细胞淋巴瘤(Diffuse large B cell lymphoma,DLBCL) 癌细胞的相互作用。Tan等[10]通过将相似的细胞系和药物分组,对数据子集训练大量模型,通过整合多个数据库提取细胞系敏感性和药物活性特征,采用模型堆叠概括的方法,结果令人鼓舞。在最近的研究中,Szalai等[11]使用线性回归方法预测差异表达信号对药物敏感性的影响,发现药物引起的细胞凋亡与剂量和时间有关,但其由于只提取了差异表达基因与细胞活性之间的线性特征,在预测过程中存在预测精度较低和拟合较差的问题。
在这项研究中,我们使用差异表达基因与细胞表型信息,通过机器学习算法,预测药物诱导下的细胞活性。通过对扰动转录组学信号LINCS- L1000、癌症治疗反应门户(Cancer treatment response portal,CTRP) 和癌症依赖性图谱Achilles三大数据集的筛选与匹配,划分为9个数据子集。其次,提出基于堆栈式深度自动编码器(Stacked autoencoder,SAE) 算法进行关键基因提取,有效提取差异表达基因与细胞活性之间的非线性特征,使用极端梯度提升(Extreme gradient boosting,XGBoost) 算法对细胞活性进行预测,分析在药物毒性和基因沉默作用下的细胞凋亡反应,同时将随机游走(Random walk,RW) 算法引入XGBoost学习算法中,解决了繁琐的调参问题。为了衡量我们方法的可行性,完成了在不同扰动时间下的化合物与shRNA之间的跨数据集验证,最后在癌细胞系百科全书(Cancer cell line encyclopedia,CCLE) 和NCI60上进行了药物敏感性推断。
1 材料与方法1.1 数据集获取扰动转录组学信号,包括LINCS-L1000- PhaseⅠ和LINCS-L1000-PhaseⅡ扰动谱数据,选自LINCS-L1000数据集,可从GEO数据库(https://www.ncbi.nlm.nih.gov/geo/) 下载获得,对应的基因芯片识别号分别为GSE92742和GSE70138。L1000技术的首次大规模应用使得运用LINCS大数据作为参考库进行化合物分析成为可能,我们选择LINCS项目中Level5水平对应的差异表达信号所含有的978个标记基因组成训练数据集[12-13]。癌症治疗反应门户CTRP涵盖了70 000个癌细胞系化合物敏感性和遗传或谱系特征之间的联系,我们选用后验控制质量的细胞活性数值作为建模的目标,可从https://ocg.cancer.gov/programs/ctd2/data-portal下载获得[14]。为了探究单基因敲降或敲除对癌细胞增殖的影响并预测癌症依赖性,我们从癌症依赖性图谱数据库Achilles (https://portals.broadinstitute.org/achilles) 下载数据,选用shRNA治疗前后的效应改变量大小用于模型分析[15]。NCI60数据集可从https://dtp.cancer.gov/discovery_development/nci-60下载,包括细胞系在不同药物作用下的GI50、LC50和TGI等药物敏感性值,选用GI50值作为药物敏感性的评价标准[16]。CCLE数据集可从https://portals.broadinstitute.org/ccle下载,包括不同细胞系在24种药物的8个浓度点上的EC50、IC50和药物活性面积,选用药物活性面积作为药物敏感性的评价标准[17]。
1.2 数据集预处理由于LINCS-L1000中具有大量的扰动转录组学信号,而CTRP数据集中包含药物诱导下的细胞活性数据,Achilles数据集中包含shRNA治疗前后的效应改变量数据,这3个数据集有大量数据是重叠的,在最新的研究中也有类似的方法,将LINCS-L1000数据集中的扰动转录组学信号分别与CTRP数据集中的细胞生存能力数据和Achilles的shRNA丰度数据进行关联。因此,在本研究中,我们将GEO数据库中获取的两阶段LINCS-L1000扰动谱数据进行合并,获得多种扰动状态下全基因组的基因表达情况,为了进一步研究基于化合物扰动下不同细胞系的细胞活性,将CTRP中药物治疗后的细胞活性数据作为关联,同时基于相同的细胞系和Broad研究所提供的药物识别号进行了数据实例的匹配,并参照公式1匹配浓度相近的若干样本,针对在不同实验批次下,相同浓度对应的细胞活性值,取其细胞活性平均值。
 | (1) |
为了使研究更深入,我们尝试使用癌症依赖性图谱数据库Achilles中的表型信息,将训练模型在跨越其他数据集进行独立集测试,探究在shRNA作用下单基因敲降或敲除对癌细胞凋亡或者增殖产生的影响。由于在药物治疗或者shRNA治疗后的细胞存活数量与CTRP或者Achilles中的评价指标成比例,我们为了简便起见,将以上两大数据集中的细胞表型信息统称为细胞活性。
数据关联过程包含扰动信号与细胞表型信息两个部分。(1) 合并LINCS-L1000-PhaseⅠ与LINCS-L1000-PhaseⅡ数据,命名为LINCS- L1000。(2) 将LINCS-L1000数据集中的化合物扰动信号与shRNA扰动信号,分别与CTRP、Achilles数据集按照相关条件进行关联,命名为CTRP-L1000数据集与Achilles-L1000数据集。(3) 将数据集按照不同的扰动时间划分为CTRP- L1000-3 h、CTRP-L1000-6 h、CTRP-L1000-24 h和Achilles-L1000-96 h、Achilles-L1000-120 h、Achilles-L1000-144 h数据集。(4) 将CTRP-L1000-3 h、CTRP-L1000-6 h、CTRP-L1000-24 h按照不含浓度(S1) 和包含浓度(S2) 因素划分为6个子数据集,以上具体过程如图 1所示。匹配完成的最终各数据集大小如表 1所示。
 |
| 图 1 LINCS-L1000分别与CTRP、Achilles数据关联图 Fig. 1 Data association diagram of LINCS-L1000 with CTRP and Achilles respectively. |
| 图选项 |
表 1 各数据集的详细信息Table 1 The detailed description of each dataset
| Screen name | Data points | Cell line | Compounds | shRNAs |
| CTRP-L1000-3h (S1) | 1 130 | 5 | 43 | 0 |
| CTRP-L1000-6h (S1) | 9 099 | 47 | 288 | 0 |
| CTRP-L1000-24h (S1) | 13 363 | 18 | 327 | 0 |
| CTRP-L1000-3h (S2) | 1 328 | 5 | 43 | 0 |
| CTRP-L1000-6h (S2) | 10 063 | 47 | 288 | 0 |
| CTRP-L1000-24h (S2) | 15 610 | 18 | 327 | 0 |
| Achilles-L1000-96h | 69 941 | 10 | 0 | 11 771 |
| Achilles-L1000-120h | 14 563 | 2 | 0 | 12 018 |
| Achilles-L1000-144h | 10 484 | 3 | 0 | 4 837 |
表选项
1.3 模型建立本研究的过程包括:基于扰动转录组学信号LINCS-L1000与癌症治疗反应门户CTRP数据集完成差异表达基因的提取与细胞活性的预测分析,并进行模型结果的评估;其次,在癌症依赖性图谱数据库Achilles上进行独立集测试,获得模型在跨数据集上的性能。(Achilles-L1000系列模型在跨数据集CTRP-L1000上的测试过程,也亦然),同时,根据预测的细胞活性在CCLE与NCI60数据集上完成药物敏感性预测。
为了客观地验证我们所提出方法的预测性能,我们进一步将扰动谱与细胞活性数据空间PertDT随机划分为70%的训练集PertDT-S用于模型的学习、15%的验证集PertDT-V用于模型的调参与优化和15%的测试集PertDT-T用于评估模型在实际使用时的泛化能力。
1.3.1 基于堆栈式深度自动编码器的特征提取堆栈式自动编码器作为一种深度学习模型,使用自动编码器作为堆叠模块逐层构建深度神经网络,以分层和自底向上的方式进行训练[18]。在首次降维完成后,将提取低维表达特征用以训练下一个自动编码器,以获取输入向量的更低维表示,原始数据通过逐层转换到顶层的方式实现了特征的逐层提取。同时模型使用Relu型激活函数获取特征的非线性表达,通过最小化损失函数Θ(X, Z)进行反向传播完成参数的学习:
 | (2) |
基于以上过程,隐藏层最大程度地学习了原始输入的特征信息。在完成无监督的特征训练之后,整个神经网络可以使用标记的数据来微调训练参数。堆栈式自动编码器最高一级的隐藏层具有提取原始数据信息的功能,并且可以进一步应用于回归器。在本研究中,我们建立了堆栈式多层自动编码器,并将XGBoost用作最终的预测器。
1.3.2 基于XGBoost和随机游走的细胞活性预测XGBoost作为机器学习中竞争力最强的预测算法之一,对梯度提升算法的集成方式做了一定改进,在解决分类和回归问题上都具有较高的性能[19]。我们在采用XGBoost算法对细胞活性进行预测时,根据每个输入样本中的基因差异表达,在每棵决策树的叶子节点上都能获得一个预测值,通过多次迭代逐一构建多个弱评估器,细胞活性预测结果定义为所有树的预测分数之和:
 | (3) |
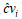
 | (4) |
 | (5) |
1.4 模型评估为了有效地量化不同模型对药物诱导下的细胞活性的预测能力,模型的评价函数选用皮尔逊相关系数(Pearson correlation coefficient)、决定系数(Coefficient of determination) 和均方误差(Mean squared error)。皮尔逊相关系数的取值范围为[?1, 1],绝对值越大,说明真实值与预测值相关性越强;R2用于评估真实值与预测值之间的拟合优度,取值越接近于1,说明拟合程度越高;MSE用于衡量预测值与真实值之间的偏差,其值越小,说明模型具有更好的精确度。其计算公式分别为:
 | (6) |
 | (7) |
 | (8) |




为了评估模型在药物敏感性的预测效果,我们将模型的细胞活性值与药物敏感性相关联。在CTRP-L1000数据集中设定细胞活性小于0.8 (认为具有毒性) 的药物为有效药物,在Achilles-L1000数据集中设定细胞活性小于?2 (认为具有毒性) 的药物为有效药物,反之则视为无效药物。我们选择ROC曲线与PR曲线来评价算法的预测性能,并使用混淆矩阵对结果进行度量,计算其对应的查准率和查全率:
 | (9) |
 | (10) |
2 结果与分析2.1 对特征提取的分析在使用堆叠式自动编码器对差异表达基因进行特征提取时,由于网络参数的设置会对训练过程中的堆栈式自动编码器产生一定影响,为了获得较优的准确率,分别设置网络输入层节点数为978,权重衰减成本为0.002,Dropout率为0.2,学习率为0.001,网络层数、各层的节点数以及最后一层用于特征提取的神经元数量因不同数据集而异。以LINCS-L1000-CTRP-24 h数据集为例,本研究所使用的算法与现有的其他方法,主成分分析(Principal component analysis,PCA)、局部线性嵌入(Locally linear embedding,LLE)、核主成分分析(Kernel principal component analysis,KPCA) 和独立成分分析(Independent component analysis,ICA) 进行比较分析,皮尔逊(Pearson) 相关系数均优于其他算法,如图 2A所示。我们还分别使用不同的学习率在LINCS-L1000-CTRP-24 h数据集上进行特征提取,当堆栈自编码器学习率较低时,更有利于模型的学习,如图 2B所示。而在图 2C–D中,通过多次实验,与传统的堆栈式自动编码器对比分析,发现SAE-XGBoost算法拥有较低均方误差,结果相对更稳定。
 |
| 图 2 特征提取算法的对比与分析 Fig. 2 Comparison and analysis of feature extraction algorithms. |
| 图选项 |
2.2 药物诱导下的细胞活性预测分析结合随机游走算法对XGBoost各项参数组合进行更新与调优,在CTRP-L1000与Achilles- L1000系列模型的优化过程中,设定初始行走步长为0.5,迭代次数为10次,输出精度为0.000 01,变量数目为8个,同时分别设定各自的属性步长。以真实值与预测值之间的皮尔逊相关系数为模型评价指标,并使用其作为模型的优化目标,随机游走算法根据预先设定的初始点坐标和初始行走步长开始随机游走,当迭代到满足预置的精度要求时,以较大概率找到最优解或者近似最优解,该算法对XGBoost模型进行参数优化实验的结果展示如表 2所示。
表 2 随机游走算法在XGBoost算法中的迭代结果Table 2 Iterative results of random walk in XGBoost
| The screen used | Pearson correlation | R2 | Mean squared error |
| CTRP-L1000-3 h (S2) | 0.849 0 | 0.702 5 | 0.020 4 |
| CTRP-L1000-6 h (S2) | 0.703 6 | 0.485 3 | 0.051 6 |
| CTRP-L1000-24 h (S2) | 0.892 6 | 0.794 5 | 0.016 9 |
| CTRP-L1000-3 h (S1) | 0.773 4 | 0.594 4 | 0.027 7 |
| CTRP-L1000-6 h (S1) | 0.635 4 | 0.387 6 | 0.063 6 |
| CTRP-L1000-24 h (S1) | 0.846 0 | 0.710 8 | 0.023 8 |
| Achilles-L1000-96 h | 0.599 5 | 0.339 6 | 1.493 1 |
| Achilles-L1000-120 h | 0.487 5 | 0.185 9 | 1.568 6 |
| Achilles-L1000-144 h | 0.511 0 | 0.212 8 | 1.438 2 |
表选项
从以下实验结果可以明显看出,CTRP的细胞活性的测量需要较长的扰动时间,随着扰动时间的增长,预测的可靠性也在不断上升,其中24 h扰动时间预测结果较为可靠;而在CTRP数据集预测中,当考虑浓度因素时,模型的准确性有所提高,可知细胞活性在一定程度上与药物浓度相关。在LINCS-L1000扰动谱与癌症依赖性图谱数据库Achilles项目中,以96 h的扰动时间所产生的模型预测效果最为显著。
2.3 CTRP-L1000与Achilles-L1000独立数据集检验为了验证模型预测的可靠性,我们采用独立数据集来验证模型的预测能力,实现了在CTRP- L1000系列模型与Achilles-L1000系列模型中进行交互性测试,结果如图 3所示。在CTRP-L1000数据集中,以24 h扰动时间的模型最佳,Pearson相关系数为0.846 0,优于3 h与6 h的扰动时间,Achilles-L1000数据集中,以96 h的扰动时间模型最佳,Pearson相关系数为0.599 5,优于120 h与144 h的扰动时间模型。同样在独立集验证方面,CTRP-L1000-24 h数据集在CTRP-L1000-6 h模型、CTRP-L1000-24 h模型和Achilles-L1000- 96 h模型中,Pearson相关系数分别为0.744 8、0.846 0和0.682 9均优于其他模型,进一步证实了药物在较长的扰动时间后能够取得优良的预测性能。
 |
| 图 3 独立数据集验证 Fig. 3 Independent dataset validation: using the Achilles-L1000 series model to predict cell viability in CTRP-L1000 data and vice versa. |
| 图选项 |
2.4 基于细胞活性在NCI60和CCLE数据集上预测药物敏感性通过分析药物诱导下的细胞活性,我们可进一步在NCI60和CCLE上完成药物的敏感性预测。针对NCI60数据,我们以改进的GI50值(50%的癌细胞生长得到抑制或控制时所需的药物浓度)作为衡量药物敏感性的依据。具体地,当GI50值在最大药物浓度测试范围内,且作用于细胞系有效时,记录此时的GI50值为药物敏感性值;反之,若在最大测试浓度测试范围内,该药物无效,则将最大测试浓度作为药物敏感性值。
我们将模型预测的细胞活性值与药物敏感性相关联,根据模型预测的细胞活性在NCI60数据集上进行药物敏感性预测。将GI50值作为药物敏感性评价指标,进行二值化处理,得到有效药物(正例) 和无效药物(反例),同时在模型预测的细胞活性上使用设定的决策阈值判定药物的有效性。我们通过ROC曲线与PR曲线来衡量算法在评判药物有效性方面所作的贡献。在如图 4A所示的ROC曲线中,Achilles-L1000-96 h模型所作的预测最为准确,AUC面积达到0.78,其95%置信区间范围为(0.76, 0.81),且显著性水平小于0.000 1,其余两个模型均表现出不错的性能,其中CTRP-L1000-24 h和CTRP-L1000-6 h模型在ROC曲线下的AUC面积均达到了0.70,其95%置信区间范围都为(0.67, 0.73)。在如图 4B所示的准确率-召回率评估曲线中,Achilles-L1000-96 h模型仍以曲线下的面积AUC=0.94超越其他模型,Achilles-L1000-96 h模型和CTRP-L1000-24 h模型预测的细胞活性在指定分类阈值下,混淆矩阵如图 5所示。通过以上分析,进一步证实了Achilles-L1000-96 h模型在LINCS-L1000-NCI60- 24 h数据集上预测药物敏感性是有效的,可用于后续其他药物的有效性检测。
 |
| 图 4 模型在LINCS-L1000-NCI60-24 h上验证的ROC曲线(A) 与PR曲线(B) Fig. 4 ROC curve (A) and PR curve (B) of the model evaluation on LINCS-L1000-NCI60-24 h dataset. |
| 图选项 |
 |
| 图 5 模型Achilles-L1000-96 h (A) 与模型CTRP-L1000-24 h (B) 在LINCS-L1000-NCI60-24 h上的混淆矩阵 Fig. 5 ROC curve (A) and PR curve (B) of the model evaluation on LINCS-L1000-NCI60-24 h dataset. |
| 图选项 |
除此以外,我们还将LINCS-L1000、CTRP和NCI60数据再次进行关联,采用ROC曲线与PR曲线对结果进行讨论与分析。如图 6所示,在Achilles-L1000-96 h、CTRP-L1000-24 h和CTRP- L1000-6 h模型中,ROC曲线下面积AUC值分别为0.79、0.77和0.64,95%置信区间分别为(0.72, 0.86)、(0.70, 0.84) 和(0.56, 0.72),PR曲线下面积AUC值分别为0.98、0.97和0.96。Achilles-L1000-96 h和CTRP-L1000-24 h模型预测的细胞活性在指定分类阈值下,混淆矩阵如图 7所示。以上结果表明Achilles-L1000-96 h与CTRP- L1000-24 h模型具有卓越的预测性能。
 |
| 图 7 模型Achilles-L1000-96 h (A) 与模型CTRP-L1000-24 h (B) 在LINCS-L1000-CTRP-NCI60-24 h上的混淆矩阵 Fig. 7 ROC curve (A) and PR curve (B) of the model evaluation on LINCS-L1000-NCI60-24 h dataset. |
| 图选项 |
图 6中的CTRP-L1000-AUC为CTRP-L1000数据集中,药物浓度-时间曲线下所围面积AUC的取值,用以比较真实值CTRP-L1000-AUC与模型所预测的细胞活性在推断药物敏感性上是否存在显著差异。依据Achilles-L1000-96 h模型预测的细胞活性值推断药物的有效性方法,与使用真实CTRP-L1000-AUC值推断药物的有效性方法进行对比分析,可以发现两种方法在ROC曲线上所围面积AUC的显著性差异水平为0.958 3,该值大于0.05,表示使用这两种方法在推断药物有效性方面不存在显著差异。而在使用CTRP-L1000- AUC值推断药物有效性方法中,ROC曲线下的面积AUC值为0.79,其95%置信区间为(0.72, 0.85),显著性水平小于0.000 1,PR曲线下的面积AUC值为0.98。
 |
| 图 6 模型在LINCS-L1000-CTRP-NCI60-24 h上验证的ROC曲线(A) 与PR曲线(B) Fig. 6 ROC curve (A) and PR curve (B) of the model evaluation on LINCS-L1000-CTRP-NCI60-24 h dataset. |
| 图选项 |
在此,我们已经完成了通过细胞活性预测值对药物有效性的推断,为了进一步检验有效药物与无效药物的细胞活性值之间是否存在显著差异,采用正态性检验方式,由于检验结果存在一定的偏态,使用非参数类Mann Whitney test检验方法对细胞活性预测结果进行分析(图 8)。不同的模型分别在LINCS-L1000-NCI60-24 h与LINCS- L1000-CTRP-NCI60-24 h数据集上进行预测,结果发现使用Achilles-L1000-96 h模型在区分有效药物与无效药物之间在均值上存在显著差异,显著性水平分别为P < 0.000 1和P=0.000 3;同样地,在使用CTRP-L1000-24 h模型在推断药物有效性上也得到了类似的结果,显著性水平分别为P < 0.000 1和P=0.000 8。
 |
| 图 8 有效药物组与无效药物组的比较 Fig. 8 Comparison of the effective and the ineffective drug group. |
| 图选项 |
在癌细胞系百科全书CCLE上,采用活性面积作为药物敏感性的评价指标。对此,将CCLE中的活性面积进行零均值标准化处理,高于均值0.8个方差的活性面积定义为有效药物,反之定义为无效药物,完成二值化处理。其次在LINCS- L1000扰动谱数据集中寻找共有细胞系和药物组合对,由于在CCLE数据集中只存在少量的24种药物,借助PubChem数据库寻找同义药物,匹配完成后的数据记为LINCS-L1000-CCLE,同样我们还筛选了扰动时间为24 h对应的药物,且当存在多个药物扰动信号时,选取细胞活性最低值。在实验结果的评价中,使用ROC曲线与PR曲线来对算法的结果进行衡量,发现Achilles-L1000-96 h模型不仅在跨数据集验证上表现出了卓越的性能,且在ROC曲线下的面积AUC值为0.79,95%置信区间范围为(0.67, 0.88),PR曲线下所围面积AUC为0.78。
3 讨论为了评估本文算法的有效性,我们将本研究算法与其他现有方法进行分析比较,分别使用预测值与真实值的Pearson相关系数、决定系数R2和均方误差进行衡量模型的预测性能,如表 3所示。以CTRP-L1000-24 h (S1) 数据集为例,Pearson最大相关系数达到了0.846 0、R2最大决定系数达到了0.710 8、最小的均方误差为0.023 8,均优于其他算法。具体而言,相比于PCA-Lasso、KPCA-RF、LLE-SVR和LLE-KNN算法,本研究算法在Pearson相关系数方面,分别提高了9.02%、10.01%、23.79%和15.16%;在R2方面,分别提高了18.13%、21.65%、57.99%和32.51%;在均方误差方面,分别下降了27.44%、30.61%、47.46%和37.70%。实验结果表明,本研究算法的预测结果得到了进一步的提升。
表 3 本文算法与其他算法的比较Table 3 Comparison of the algorithm in this paper with other algorithms. Taking the CTRP-L1000-24 h (S1) dataset as an example
| The algorithm used | Pearson correlation | R2 | Mean squared error |
| Our model | 0.846 0 | 0.710 8 | 0.023 8 |
| PCA-Lasso | 0.776 0 | 0.601 7 | 0.032 8 |
| KPCA-RF | 0.769 0 | 0.584 3 | 0.034 3 |
| LLE-SVR | 0.683 4 | 0.449 9 | 0.045 3 |
| LLE-KNN | 0.734 6 | 0.536 4 | 0.038 2 |
表选项
除了通过预测模型推断出细胞活性的有效性和可靠性,我们还需将我们的结果与有关细胞活性的文献联系起来进行对比。药物Vorinostat是一种组蛋白脱乙酰化酶(HDAC) 抑制剂,其主要通过转化诱导使细胞的生长停滞,通过观察使用药物Vorinostat在不同细胞系中进行治疗的细胞活性最低值,可以看出Vorinostat在治疗A549、A375、VCAP、PC3、HA1E、HUES3和NPC等细胞系上具有显著效果,文献[21]首次全面鉴定了A549细胞中用于该药治疗的组蛋白赖氨酸乙酰化,阐述了Vorinostat对非小细胞肺癌A549具有临床应用价值(细胞活性:?4.9),文献[22]报道了Vorinostat可恢复源自NPC患者的成纤维细胞中的胆固醇稳态,以达到最终治疗的目的(细胞活性:?5.0)。使用药物Bardoxolone-methyl治疗MCF7细胞系在柱状图中显示出较高的毒性值,据文献[23]报道,该药物具有抗增殖、抗炎和抗纤维化的作用,可抑制MCF7细胞中的迁移和代谢(细胞活性:?4.6)。文献[24]研究了药物Tyrphostin-AG-1478对结直肠癌HT29细胞的生长作用,并探索了其治疗的潜力,表明该药物将抑制细胞生长并诱导其凋亡(细胞活性:?5.0)。我们还分别针对A549、NPC、MCF7、HT29、PC3、A375、HA1E、HELA和YAPC细胞系进行了药物诱导下的细胞活性预测,说明其可能对以上细胞系具有更高的敏感性,结果如表 4所示。
表 4 影响细胞活性的药物Table 4 The drugs that affect the cell viability
| Cell line | A549 | NPC | MCF7 |
| Drug | Mepacrine | Vorinostat | Idasanutlin |
| Teniposide | Sorafenib | Daunorubicin | |
| Gemcitabine | Rigosertib | Selinexor | |
| Topotecan | Fenretinide | Mitoxantrone | |
| Piperlongumine | Pirarubicin | PHA-848125 | |
| Cell line | HT29 | PC3 | A375 |
| Drug | Delanzomib | Bortezomib | Delanzomib |
| Panobinosta | YM-155 | Oprozomib | |
| Ixazomib | MG-132 | PHA-848125 | |
| Trichostatin-a | BGT-226 | Bruceantin | |
| Brotezomid | Bortezomib | Delanzomib | |
| Cell line | HA1E | HELA | YAPC |
| Drug | Perhexiline | AZD-7762 | Ixazomid |
| BVT-948 | Carfilzomib | Bortezomid | |
| Foretinib | MK-1775 | AZD-7762 | |
| Withaferin-a | JNJ-26481585 | Delanzomid | |
| Teniposide | Romidepsin | Brotezomid |
表选项
4 结论基因组数据为开发预测药物敏感性算法提供了宝贵的资源,在本研究中,我们使用3个独立的样本集合,提出了一种新颖的SAE-XGBoost机器学习方法,通过细胞活性与药物基因组学之间的关联,采用堆栈式深度自动编码器完成了差异表达基因的信息提取,将随机游走算法引入到XGBoost学习中,建立模型预测数百种癌细胞中多种药物的反应。这种方法不仅可以使我们的预测模型用于预测新测得的细胞对已经测试的药物的反应,而且可以利用已知的基因组信息预测现有药物对癌细胞的抑制作用,结果表明,我们的模型获得了良好的效果,并在CCLE和NCI60数据集上验证了我们所提出的建模策略作为药物敏感性分析的稳健性和有效性。
然而,在临床应用中迫切需要确定癌细胞与抗癌疗法之间药物敏感和耐药联系的生物标记,因此,在未来的工作中我们仍然希望设计一种更理想的监督机器学习的算法,揭示癌细胞和药物作用之间的敏感性。由于我们的模型还受到很多因素影响,例如表观遗传和蛋白质水平信息,因此未来可以将其纳入预测模型,从而使模型更稳健,以便获得更高的准确性,为探索癌细胞在细胞水平上的生物学行为提供更多的机会。
参考文献
| [1] | Dull AB, Wilsker D, Hollingshead M, et al. Development of a quantitative pharmacodynamic assay for apoptosis in fixed tumor tissue and its application in distinguishing cytotoxic drug-induced DNA double strand breaks from DNA double strand breaks associated with apoptosis. Oncotarget, 2018, 9(24): 17104-17116. DOI:10.18632/oncotarget.24936 |
| [2] | Mostaghimi H, Mehdizadeh AR, Jahanbakhsh M, et al. Quantitative determination of tumor platinum concentration of patients with advanced breast, lung, prostate, or colorectal cancers undergone platinum-based chemotherapy. J Cancer Res Ther, 2017, 13(6): 930-935. |
| [3] | Cubillos-Ruiz JR, Mohamed E, Rodriguez PC. Unfolding anti-tumor immunity: Er stress responses sculpt tolerogenic myeloid cells in cancer. J Immunother Cancer, 2017, 5(1): 5. DOI:10.1186/s40425-016-0203-4 |
| [4] | Desmedt C, Ruíz-García E, André F. Gene expression predictors in breast cancer: current status, limitations and perspectives. Eur J Cancer, 2008, 44(18): 2714-2720. DOI:10.1016/j.ejca.2008.09.011 |
| [5] | Yousefi MR, Datta A, Dougherty ER. Optimal intervention in markovian gene regulatory networks with random-length therapeutic response to antitumor drug. IEEE Trans Biomed Eng, 2013, 60(12): 3542-3552. DOI:10.1109/TBME.2013.2272891 |
| [6] | G?nen M, Margolin AA. Drug susceptibility prediction against a panel of drugs using kernelized bayesian multitask learning. Bioinformatics, 2014, 30(17): i556-i563. DOI:10.1093/bioinformatics/btu464 |
| [7] | Menden MP, Iorio F, Garnett M, et al. Machine learning prediction of cancer cell sensitivity to drugs based on genomic and chemical properties. PLoS ONE, 2013, 8(4): e61318. DOI:10.1371/journal.pone.0061318 |
| [8] | Eduati F, Mangravite LM, Wang T, et al. Prediction of human population responses to toxic compounds by a collaborative competition. Nat Biotechnol, 2015, 33(9): 933-940. DOI:10.1038/nbt.3299 |
| [9] | Goswami CP, Cheng L, Alexander PS, et al. A new drug combinatory effect prediction algorithm on the cancer cell based on gene expression and dose-response curve. CPT Pharmacometr Syst Pharmacol, 2015, 4(2): 80-90. DOI:10.1002/psp4.9 |
| [10] | Tan M, ?zgül OF, Bardak B, et al. Drug response prediction by ensemble learning and drug-induced gene expression signatures. Genomics, 2019, 111(5): 1078-1088. DOI:10.1016/j.ygeno.2018.07.002 |
| [11] | Szalai B, Subramanian V, Holland CH, et al. Signatures of cell death and proliferation in perturbation transcriptomics data—from confounding factor to effective prediction. Nucleic Acids Res, 2019, 47(19): 10010-10026. DOI:10.1093/nar/gkz805 |
| [12] | Subramanian A, Narayan R, Corsello SM, et al. A next generation connectivity map: L1000 platform and the first 1 000 000 profiles. Cell, 2017, 171(6): 1437-1452.e.17. DOI:10.1016/j.cell.2017.10.049 |
| [13] | Liu CL, Su J, Yang F, et al. Compound signature detection on lincs l1000 big data. Mol Biosyst, 2015, 11(3): 714-722. DOI:10.1039/C4MB00677A |
| [14] | Seashore-Ludlow B, Rees MG, Cheah JH, et al. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov, 2015, 5(11): 1210-1223. DOI:10.1158/2159-8290.CD-15-0235 |
| [15] | Tsherniak A, Vazquez F, Montgomery PG, et al. Defining a cancer dependency map. Cell, 2017, 170(3): 564-576.e16. DOI:10.1016/j.cell.2017.06.010 |
| [16] | Shoemaker RH. The nci60 human tumour cell line anticancer drug screen. Nat Rev Cancer, 2006, 6(10): 813-823. DOI:10.1038/nrc1951 |
| [17] | Ghandi M, Huang FW, Jané-Valbuena J, et al. Next-generation characterization of the cancer cell line encyclopedia. Nature, 2019, 569(7757): 503-508. DOI:10.1038/s41586-019-1186-3 |
| [18] | Vincent P, Larochelle H, Bengio Y, et al. Extracting and composing robust features with denoising autoencoders. In Proceedings of the 25th International Conference on Machine Learning ACM. New York, NY, United States: ACM, 2008: 1096-1103. |
| [19] | Li W, Yin YB, Quan XW, et al. Gene expression value prediction based on XGBoost algorithm. Front Genet, 2019, 10: 1077. DOI:10.3389/fgene.2019.01077 |
| [20] | Lawler GF, Limic V. Random Walk: A Modern Introduction (Cambridge Studies in Advanced Mathematics). Cambridge: Cambridge University Press, 2010. |
| [21] | Wu Q, Xu WQ, Cao LJ, et al. Saha treatment reveals the link between histone lysine acetylation and proteome in nonsmall cell lung cancer A549 cells. J Proteome Res, 2013, 12(9): 4064-4073. DOI:10.1021/pr4004079 |
| [22] | Subramanian K, Rauniyar N, Lavalleé-Adam M, et al. Quantitative analysis of the proteome response to the histone deacetylase inhibitor (hdaci) vorinostat in niemann-pick type c1 disease. Mol Cell Proteom, 2017, 16(11): 1938-1957. DOI:10.1074/mcp.M116.064949 |
| [23] | Refaat A, Pararasa C, Arif M, et al. Bardoxolone-methyl inhibits migration and metabolism in mcf7 cells. Free Rad Res, 2017, 51(2): 211-221. DOI:10.1080/10715762.2017.1295452 |
| [24] | Partik G, Hochegger K, Sch?rkhuber M, et al. Inhibition of epidermal-growth-factor-receptor- dependent signalling by tyrphostins a25 and ag1478 blocks growth and induces apoptosis in colorectal tumor cells in vitro. J Cancer Res Clin Oncol, 1999, 125(7): 379-388. DOI:10.1007/s004320050290 |
