喻召武1, 宋立华2, 童丽艳1, 王远1, 郑雯1, 宫兴文1


1. 浙江工商大学食品与生物工程学院, 浙江 杭州 310018;
2. 上海交通大学农业与生物学院, 上海 200240
收稿日期: 2015-05-18; 修回日期: 2015-07-13; 网络出版日期: 2015-08-19
基金项目: 浙江省自然科学基金(LY12C19011);食品科学与工程浙江省重中之重一级学科资助项目(JYTSP20141052)
通信作者: E-mail: gongxingwen@163.com
摘要: [目的] 开发出与铜绿假单胞菌粘肽合成酶PBP3有高亲和力,具有全新结构的先导化合物。[方法] 以铜绿假单胞菌粘肽合成酶PBP3为靶点,通过分子对接软件DOCK6.5,对含有104万个小分子化合物的数据库进行了大规模虚拟筛选,选取结构相对简单的中选化合物进行合成,得到先导化合物,并验证其抑菌活性。[结果] 通过grid score进行第一轮初筛,筛选出grid score分值小于-30 kcal/mol的6万个化合物,接着以amber score进行第二轮筛选,筛出amber score分值小于-20 kcal/mol的化合物约200个。最终,经过观察分析,从中挑选出4种打分高并且结构新颖的小分子化合物作为先导化合物。合成出的先导化合物及其衍生物对铜绿假单胞菌等常见微生物的最小抑菌浓度(MIC)在175-275 μg/mL之间,对革兰氏阴性菌和阳性菌均有效,MIC值比作为阳性对照的磺胺嘧啶更低,说明先导化合物具有较好的抗菌活性。[结论] 这些先导化合物可以进一步开发为新型抗菌药物,用于解决铜绿假单胞菌的耐药性问题。
关键词: 铜绿假单胞菌粘肽合成酶PBP3耐药性虚拟筛选抑菌活性
Virtual screening and antibacterial activity of lead compounds targeting to penicillin-binding protein 3 (PBP3) of Pseudomonas aeruginosa
Zhaowu Yu1, Lihua Song2, Liyan Tong1, Yuan Wang1, Wen Zheng1, Xingwen Gong1
1. School of Food Science and Biotechnology, Zhejiang Gongshang University, Hangzhou 310018, Zhejiang Province, China;
2. School of Agriculture and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China
Received: 18 May 2015; Revised: 13 July 2015; Published online: 19 August 2015
Supported by the Natural Science Foundation of Zhejiang Province of China (LY12C19011) and by the Foundation of FoodScience and Engineering-the Most Important Discipline of Zhejiang Province (JYTSP20141052)
Corresponding author. E-mail: gongxingwen@163.com
Abstract:[Objective] This study was carried out to obtain lead compounds targeting penicillin-binding protein 3 (PBP3) of Pseudomonas aeruginosa by virtual screening. [Methods] UCSF dock 6.5 was used for the virtual screening from a database containing 1.04 million small molecules. Hit compounds with simple structures were synthesized and then evaluated for their antibacterial activities. [Results] Grid score was used for the first round of screening, and 60000 small molecules whose scores lower than -30 kcal/mol were screened out from the database. These molecules were subjected to the second round of screening using amber score. Approximately 200 hit compounds with scores lower than -20 kcal/mol were analyzed and 4 of them were selected as lead compounds and then synthesized. The minimal inhibition concentrations (MICs) of the lead compounds were between 175-275 μg/mL, which were lower than that of Sulfadiazine (500 μg/mL) significantly. Meanwhile, these compounds were effective for both Gram-negative and Gram-positive bacteria. [Conclusion] The lead compounds had potential to become new antibacterial agents for conquering the drug resistance of P. aeruginosa.
Key words: Pseudomonas aeruginosapenicillin-binding protein 3drug resistancevirtual screeningantibacterial activity
铜绿假单胞菌(Pseudomonas aerμginosa),即绿脓杆菌,是一种革兰氏阴性菌,喜潮湿,广泛分布于自然界和健康人体的皮肤、呼吸道和消化道等部位,是医院感染重要的病原菌。随着抗菌药物的广泛与不合理应用,细菌的耐药性变得越来越严重[1, 2]。铜绿假单胞菌能够通过不同的机制产生药物耐受性,特别是多重耐药和对临床上应用最广的β-内酰胺类抗生素的耐药,导致并发症和死亡率逐渐上升[3]。
β-内酰胺酶的产生是铜绿假单胞菌对β-内酰胺类抗生素耐药最重要的机制之一。β-内酰胺酶通过水解和非水解方式破坏β-内酰胺环,使抗菌素失活,其产生的耐药性可由染色体或质粒介导[4]。此酶包括超广谱β-内酰胺酶(ESBLs)、金属β-内酰胺酶(MBLs)和头孢菌素酶(AmpC酶)等[5]。
青霉素结合蛋白(Penicillin binding proteins,PBPs)是广泛存在于细菌表面的一种膜蛋白,是细菌细胞壁主要结构成分肽聚糖合成过程中所必需的转肽酶,对细菌生长发挥重要的作用。与李斯特菌一样[6],铜绿假单胞菌的粘肽合成酶PBP3是负责细胞壁肽聚糖合成的最后步骤的一种必需酶,对细胞壁合成和细菌的存活起着至关重要的作用,同时也是所有β-内酰胺类抗生素的关键治疗靶位,各种β-内酰胺类抗生素通过与PBP3共价结合从而抑制PBP3的酶活性,阻碍细胞壁肽聚糖的合成,使细菌细胞壁缺损,导致菌体膨胀崩解[7, 8, 9, 10]。
从已有的化合物,包括合成化合物和天然产物中寻找药物或者先导化合物(Lead compounds),是药物发现的一条重要途径[11]。随着计算机技术的更新以及大数据技术的发展,应用虚拟筛选策略发现先导化合物逐渐成为主流。这种策略通过各种算法对大量化合物库进行搜索来获得有功能的化合物分子,其中应用分子对接方法进行药物或功能化合物发现是一项有效的筛选技术,该技术通过将已知结构的小分子配体放入靶标分子的结合位点,按照几何互补、能量互补和化学环境互补等多种原则来寻找配基与受体的最佳匹配模式,从大量的化合物库中筛选出与靶蛋白有相互作用的小分子,从而发现先导化合物[12]。与高通量筛选相比,虚拟筛选方法可以富集活性化合物,降低筛选成本,提高药物筛选的可行性。因此,应用虚拟筛选技术进行药物开发,已成为新药发现的重要方法[13]。
本研究根据β-内酰胺类抗生素与PBPs结合的原理,选择铜绿假单胞菌的粘肽合成酶PBP3作为虚拟筛选的靶点,对含有104万种小分子化合物的数据库进行筛选,希望筛选出能够与PBP3特异性结合,并且有高亲和力的先导化合物,用于竞争性抑制PBP3与底物的结合,从而阻断铜绿假单胞菌的细胞壁合成。目前,在抗菌药物的开发方面,PBPs仍然是一个有吸引力的靶点,因为它们负责催化肽聚糖生物合成的最后一步反应,这一步对细菌的生存至关重要,而且,该反应是发生在细胞膜之外的,这样就可以避免细菌通过降低外膜渗透性等方式产生的其他耐药机制[14]。此外,通过计算机进行大规模虚拟筛选,有利于得到具有全新结构的先导化合物,它们的结构不同于已有的β-内酰胺类抗生素(如青霉素),从而避免被耐药细菌所产生的β-内酰胺酶降解,克服现有的β-内酰胺类抗生素容易被β-内酰胺酶降解所致的细菌耐药性问题。同时,这些先导化合物可以与β-内酰胺酶相结合,抑制其活性,可以用作β-内酰胺类抗生素的增效剂。
1 材料和方法 1.1 材料 1.1.1 DOCK程序、Chimera程序、ZINC数据库: DOCK程序由美国加利福尼亚大学Kuntz等开发,选用版本为DOCK6.5。Chimera程序从加州大学旧金山分校计算机图形实验室下载(http://www.cgl.ucsf.edu/chimera)。ZINC是市售化合物中免费用于虚拟筛选的数据库(http://zinc.docking.org)。本研究的目的是开发新型抗菌药物,因此选用的是ZINC数据库中的Drug-like子数据库。
1.1.2 菌种与培养基: 大肠杆菌标准株CMCC44102和铜绿假单胞菌标准株CMCC10104来自中国医学微生物菌种保藏中心,金黄色葡萄球菌、李斯特菌、枯草芽孢杆菌等菌种由本实验室所保存。MH肉汤培养基、LB肉汤培养基等生物试剂购自杭州百思生物技术有限公司。
1.1.3 主要试剂和仪器: 对乙酰氨基苯磺酰氯、4-氯-7-硝基苯并-2-氧杂-1,3-二唑(NBD-CL)、磺胺嘧啶等常用有机试剂均为分析纯或者纯度大于99%,购自杭州双峰试剂等公司。旋转蒸发仪IKA RV10购自德国艾卡公司,离心机Eppendorf 5804 R购自德国艾本德公司,电泳仪为Bio-Rad公司生产,全自动凝胶成像分析仪JS-680B购自上海培清科技有限公司,平板式扫描仪UMAX Power Look2100XL-USB购自上海力广电子科技有限公司。
1.2 虚拟筛选 1.2.1 准备受体和配体文件: 从Protein data bank 网站(http://www.pdb.org/pdb/home/home.do)下载铜绿假单胞菌粘肽合成酶PBP3与头孢他啶的复合物晶体结构的受体文件(3pbo.pdb)。在Chimera中选择并删除配体头孢他啶,然后进行受体PBP3的准备工作,包括去除溶剂、修正错误命名的氨基酸残基、加氢和加电荷等内容,保存相应的受体文件。之后,在Chimera中重新打开复合物文件,删除复合物中除了配体以外的所有结构,仅保留配体头孢他啶,然后给配体加氢和合适的电荷,以获得配体文件。
1.2.2 生成球集: 球集是在受体分子表面的内陷位置所产生的负像(或阴影),用于代表配体分子可能结合的位点。为保证对接的精确性,以头孢他啶的结合位点为参照,选择距离该结合位点10 Å 范围内的球集,并对该球集进行编辑,去除位置不佳的球体,最终得到的球集文件用作虚拟筛选时候的活性位点。然后,在活性位点外面生成一个盒子,盒子距离活性位点5 Å,用于对筛选时小分子化合物的结构和对接时的位点变化情况进行约束,以便节省虚拟筛选的运算时间。
1.2.3 生成Grid文件: 本实验用的是基于网格(Grid)的能量打分功能,DOCK的能量打分是一种力场评分。除了需要给受体文件、盒子文件和力场文件指明所在的路径以外,其余参数均用默认值。程序会分别产生以.cnt、.nrg及.bmp为扩展名的接触、能量以及碰撞等用于计算的文件。在本实验中,网格文件命名时以“grid”为前缀,分别为grid.nrg和grid.bmp。
1.2.4 第一轮筛选: 第一轮筛选采用柔性对接,在柔性对接时,配体是柔性的,配体的构象是不断改变的,以便与受体更好的结合,最终找到配体分子在受体上的最佳结合位点和构象,并采用Grid score的默认参数来评价配体分子与受体的亲和力大小。计算结束后,生成的结果文件按打分情况由高到低进行排序,每个配体分子都会给出最优的构象和结合位点信息,用Chimera等视图软件对配体在受体结合位点的结合情况进行观察分析,挑选出分值小于-30 kcal/mol的中选化合物,用于第二轮的筛选。
1.2.5 第二轮筛选: 采用Amber score评价配体分子与受体间的亲和力。Amber score可以在削减计算成本的同时仍然保持精度,其主要优点是在执行中,无论是配体还是受体的活性部位都可以是柔性的,允许小的构象改变,重现所谓的“诱导契合”,因此,计算精度比Grid score更高,但计算量更大,所以通常需要经过Grid score进行初筛,找到配体分子在受体上的最佳结合位点和构象,以节约计算时间。在本实验中,由于能量优化和分子动力学(MD)模拟很耗费时间,所以采用了默认的100次能量优化和3000步MD。同时,设定距离配体5 Å的受体氨基酸残基是柔性的,构象可以变化,以便与配体更好的契合,而其余的参数采用默认值。计算结束后,生成的结果文件按打分情况由高到低进行排序,从中挑选出分值小于-20 kcal/mol的中选化合物进行分析,选择结构新颖并易于合成的化合物进行有机合成。
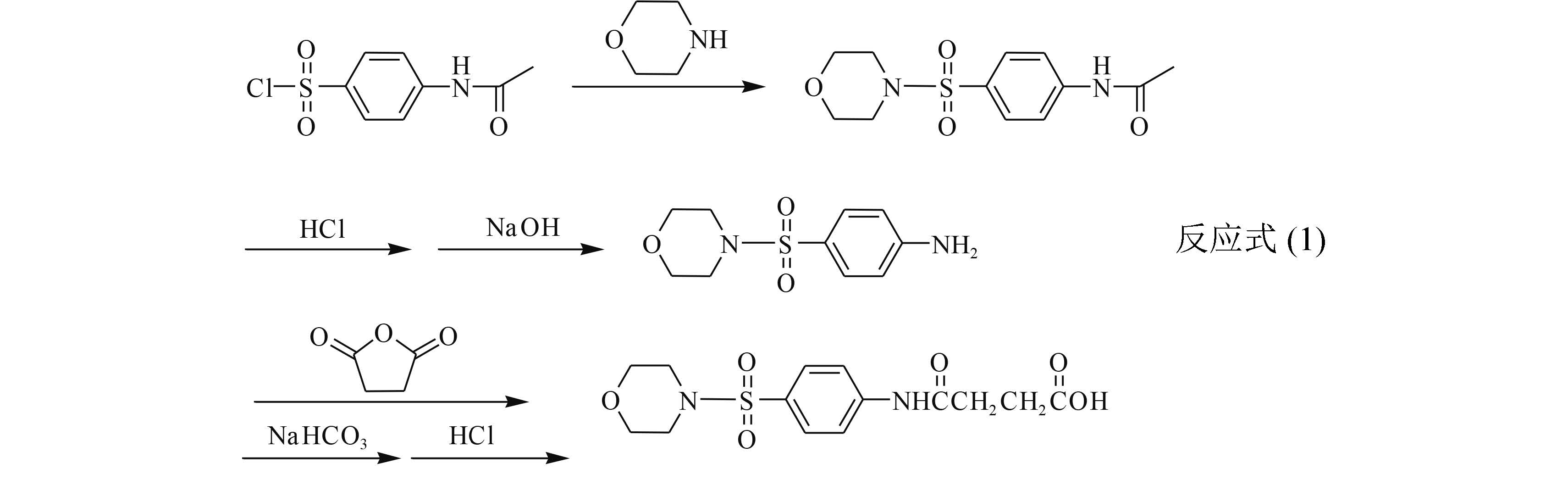
1.3 先导化合物(Lead compounds) A1 (氮取代-苯磺酰吗啡啉-琥珀酰胺酸)的合成[15] 1.3.1 对乙酰氨基苯磺酰吗啡啉的合成: 取对乙酰胺基苯磺酰氯23.3 g (0.1 mol)溶解于30 mL乙醚中,逐滴加入吗啉18.27 g (0.21 mol)。混合物在室温下搅拌1 h,然后倒入冰水中。过滤,取沉淀,得到对乙酰氨基苯磺酰吗啡啉,将产物烘干,备用。
1.3.2 对氨基苯磺酰吗啡啉的合成: 取22.72 g (0.08 mol)对乙酰氨基苯磺酰吗啡啉加入到100mL水中,同时加入25 mL浓盐酸,将混合物加热1h左右,直到溶液变的澄清为止,继续将溶液加热10 min,冷却到室温,加入10% NaOH将溶液调至中性,加入饱和NaHCO3会有絮状沉淀析出,过滤,沉淀用冷水冲洗3次,干燥即可得到对氨基苯磺酰吗啡啉。
1.3.3 氮取代-苯磺酰吗啡啉-琥珀酰胺酸的合成: 取对氨基苯磺酰吗啡啉1.84 g (7.6 mmol)溶于20mL纯丙酮,搅拌,然后加入0.8 g (8 mmol)琥珀酰酐,回流加热2 h,反应结束后,用旋转蒸发器去掉丙酮,残留物用10 mL饱和NaHCO3碱化,搅拌5 min,过滤,取滤液,用浓盐酸调节pH到2.0左右,有沉淀析出,抽滤,沉淀物用10 mL蒸馏水洗涤2次。干燥,得到先导化合物 A1(氮取代-苯磺酰吗啡啉-琥珀酰胺酸)。反应终产物经过硅胶柱纯化后通过核磁分析验证其结构式。
反应流程如反应式(1)所示。
1.4 先导化合物A1系列衍生物的合成 在不改变先导化合物 A1 基本骨架的情况下,通过选择性的改变其胺类和酸酐组成,就可以合成它的一系列衍生物。在本实验中我们选择吗啉、环己胺、苄胺这3种胺类物质和丁二酸酐、戊二酸酐这两种酸酐为组合,合成了5种不同的衍生物。
1.5 抑菌实验 参照美国临床和实验室标准化研究所(CLSI) 的微生物检验标准文件M07-A9进行[16]。
1.5.1 接种物制备: 从18-24 h培养的琼脂平皿中挑选相应的菌落直接在MH肉汤中制成菌悬液,将菌种在37 °C、200 r/min摇床中培养一段时间(大约6 h),直到菌悬浊度达到与0.5麦氏单位相同为止,备用。为准确操作此步骤,在充足的光线下,将接种的试管与0.5麦氏单位标准管同置于白色背景下画有黑色线条的卡片上,用肉眼进行比较。
1.5.2 不同浓度药物配制: 将磺胺嘧啶、先导化合物 A1 及其衍生物用二甲基亚砜(DMSO)溶解(个别溶解情况不好的加适量吐温80助溶,但要保证终溶度不超过2‰),配制成浓度为4 mg/mL的母液。使用时,以MH肉汤稀释至所需浓度(DMSO 终浓度不超过10%)。氨苄青霉素以双蒸水溶解,配成4 mg/mL的母液。使用时,以MH肉汤稀释至所需浓度。为检验DMSO对实验的影响,设置DMSO对照组,DMSO终浓度不超过10%。每支试管加入菌悬液100 μL,再加入稀释后的药物溶液9.9 mL,使得每管中最终菌浓度约为5×105 CFU/mL。同时,设置空白对照组,加10 mL MH 肉汤,不加菌悬液。每组设3个平行。
1.5.3 培养与实验结果观察: 将试管置35 &dedeg;C培养箱中培养12 h。实验结果要保证每组第一管长菌而空白对照组不长菌。
1.6 先导化合物A1与PBP3的结合 铜绿假单胞菌菌株的培养及PBP3蛋白的提取参照安艳冬等[17]所采用的方法。
为了方便显示先导化合物A1与PBP3蛋白的结合情况,对化合物A1进行了荧光标记。先将化合物A1的末端羧基转换成酰胺基[18],然后,将转化产物与荧光试剂NBD-CL在室温下搅拌过夜,生成的产物即为化合物A1-NBD复合物。
取50 μL细胞破碎液的上清(PBP3蛋白主要在细胞破碎液上清中),加入到100 μL化合物A1-NBD复合物之中,将混合物在37 °C摇床中振荡孵育2 h,让复合物与蛋白充分接触。之后,加入50μL上样缓冲液,煮沸5 min,SDS-PAGE电泳,在紫外光下观察荧光条带。
2 结果和分析 2.1 获得受体和配体文件 从Protein data bank网站下载铜绿假单胞菌粘肽合成酶PBP3与头孢他啶的复合物晶体结构文件3pbo.pdb,可视化后得图 1-A。对受体蛋白PBP3进行去除溶剂、修正错误命名的氨基酸残基、加氢和加电荷等处理,得图 1-B。再对配体头孢他啶进行加氢和加电荷处理,以获得配体文件,得图 1-C。
 |
| 图 1. 生成的受体、配体和球集结构 Figure 1. Profiles of receptor, ligand and sphere structures. |
| 图选项 |
2.2 生成球集 运行Sphgen程序,产生球集,选择距离头孢他啶10 Å的球集,然后,观察选择出来的球集,去除位置不佳的球体,最终得到由14个球体所组成的球集,用作虚拟筛选时候的活性位点,见图 1-D。围绕活性位点的盒子的距离设为5 Å,以保障盒子的大小足够容纳小分子配体而且不影响计算速度(图 1-D)。
2.3 第一轮虚拟筛选:Grid score 我们对含104万个化合物的ZINC数据库进行了虚拟筛选,因为是进行新药的开发,所以选用的是ZINC数据库的Drug-like子数据库,该数据库内化合物的特性包括:分子量在150-500之间,可旋转键≤7,脂水分配系数≤5,氢键供体≤5,氢键受体≤10。在第一轮筛选中,采用了Grid score来评价化合物与受体的亲和力大小,最终,得到了打分在-30 kcal/mol以上的中选化合物约6万个,进入第二轮筛选。
2.4 第二轮筛选:Amber score 第二轮筛选用Amber score,这种打分方法可以在削减计算成本的同时仍然保持精度,主要优点是在执行中,无论是配体还是受体的活性部位都可以是柔性的,允许小的结构重组,重现所谓的“诱导契合”,因而,比Grid score打分更加精确。经过运算,最终筛出打分高于-20 kcal/mol的化合物约200个,通过查询美国化学文摘(CA)数据库,未发现这些化合物在抗菌方面的相关研究报道,说明是新的化合物。选取4种打分较理想的中选化合物进行了分析(表 1)。
表 1. 筛选出的中选化合物的打分情况 Table 1. Scores of hit compounds
| ZINC serial number | The first round (Grid score) | The second round (Amber score) |
| ZINC00310847 | -51.391228 | -80.280861 |
| ZINC06727143 | -54.174118 | -73.471054 |
| ZINC03912961 | -52.192467 | -70.920242 |
| ZINC12734013 | -54.875553 | -50.909260 |
表选项
从表 1中可以看到,在第一轮筛选中Grid score很高的化合物,其Amber score却未必是最高的;而在第二轮筛选中,Amber score高的化合物,其Grid score也未必高。这是由于第二轮的Amber score才是精筛,因为在Amber score筛选时,受体和配体的相互作用部分都是柔性的,这样才能得到更精确的结果。而第一轮的Grid score只是初筛,主要是为了得到配体在受体上的最佳结合位点和构象,以便减少第二轮Amber score的运算时间。
对表 1中化合物与受体PBP3结合后的情况进行观察分析,可以看出,中选化合物嵌合在受体的活性位点处,形态匹配较好,周围被受体的氨基酸残基所环绕(图 2)。
 |
| 图 2. 中选化合物与受体PBP3的结合情况 Figure 2. Binding of hit compounds at the active site of PBP3. Panels A, B, C, and D represent the binding profiles of ZINC00310847, ZINC12734013, ZINC03912961 and ZINC06727143, respectively. |
| 图选项 |
对中选化合物与受体之间的相互作用情况进行分析,可以看到中选化合物与周围的氨基酸残基形成了较多的疏水作用和氢键作用(图 3),其中,ZINC00310847与PBP3有3个疏水作用和6个氢键,这可能是中选化合物与受体具有较高亲和力的原因。
 |
| 图 3. 中选化合物与受体之间的相互作用分析 Figure 3. Interactions between hit compounds and receptor. Panels A, B, C, and D represent the interactions of ZINC00310847, ZINC12734013, ZINC03912961 and ZINC06727143 with PBP3, respectively. |
| 图选项 |
四种中选化合物的化学结构如图 4所示,这些化合物的结构与已知的β-内酰胺类抗生素不同,因此,可以避免被耐药细菌所产生的β-内酰胺酶降解,克服现有的β-内酰胺类抗生素(如青霉素)容易被β-内酰胺酶降解所致的细菌耐药性问题。而且,这些中选化合物还可以用于抑制β-内酰胺酶的水解作用,用作β-内酰胺类抗生素的增效剂。
 |
| 图 4. 中选化合物的化学结构 Figure 4. Chemical structures of hit compounds. Panels A, B, C, and D represent the structures of ZINC00310847, ZINC12734013, ZINC03912961 and ZINC06727143, respectively. |
| 图选项 |
2.5 先导化合物的合成 通过对图 4中化合物的化学结构进行观察分析,发现ZINC00310847结构比较简单,容易合成,而其他化合物则结构复杂,不易合成或者所需的试剂过于昂贵。同时,从表 1和图 3中可以看出,ZINC00310847的Amber 得分比较高,和受体的相互作用也比较多,因而可能具有比较好的活性。所以,采用有机化学方法合成了该化合物,并命名为A1,同时,合成了它的五种衍生物,依次命名为A2到A6,具体结构见图 5。
 |
| 图 5. 先导化合物A1及其衍生物的化学结构 Figure 5. Chemical structures of lead compound A1 and its derivatives. |
| 图选项 |
将先导化合物A1溶于氘代DMSO中,进行核磁分析,其核磁氢谱如图 6所示。
 |
| 图 6. 先导化合物A1的核磁氢谱 Figure 6. 1H NMR of lead compound A1. 1H NMR (500 MHz,DMSO) d 10.51 (s,1H),7.85 (d,2H,J=8.5Hz),7.66 (d,2H,J=8.9Hz),3.61 (m,4H),2.82 (m,4H),2.61 (t,2H,J=6.45),2.53 (t,2H,J=6.45)。 |
| 图选项 |
2.6 抑菌实验 将先导化合物A1及其衍生物配成不同的浓度,测定它们对不同菌株的最小抑菌浓度(MIC)。选取铜绿假单胞菌(革兰氏阴性菌)、大肠杆菌(革兰氏阴性菌)、李斯特菌(革兰氏阳性菌)、枯草芽孢杆菌(革兰氏阳性菌)、金黄色葡萄球菌(革兰氏阳性菌)5种菌作为实验菌株,抑菌结果见表 2。实验表明,这些化合物对革兰氏阴性菌和阳性菌都有效,但对革兰氏阴性菌效果比阳性菌效果略好,最小抑菌浓度在175-275 μg/mL之间。先导化合物A1对各菌的抑制效果均较好,从侧面验证了虚拟筛选的正确性,表明该化合物是个有潜力的先导化合物。
表 2. 先导化合物A1及其衍生物的最小抑菌浓度 Table 2. Minimal inhibitory concentrations of six compounds (MIC, μg/mL)
| Compound name | E. coli | L. monocytogenes | B. subtilis | S. aureus | P. aeruginosa |
| A1 (Lead compound) | 175 | 200 | 200 | 225 | 175 |
| A2 (Derivative) | 175 | 225 | 225 | 250 | 200 |
| A3 (Derivative) | 200 | 200 | 225 | 225 | 200 |
| A4 (Derivative) | 225 | 225 | 250 | 250 | 225 |
| A5 (Derivative) | 225 | 225 | 225 | 250 | 200 |
| A6 (Derivative) | 250 | 250 | 250 | 275 | 225 |
| Sulfadiazine | 500 | 500 | 500 | 500 | 500 |
| Ampicillin sodium | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 |
表选项
以10%的DMSO作为空白对照,结果表明DMSO在浓度小于10%时,对实验结果无干扰作用。选取磺胺嘧啶这种传统抑菌药物为阳性对照,其对五种试验菌株的MIC均为500 μg/mL,这可能与磺胺类药物只有抑菌作用而无杀菌作用有关。当然,这些化合物相对于氨苄青霉素(最小抑菌浓度为12.5 μg/mL)等高效抗菌药来说,抗菌活性还不是很强大,在后续的研究中,我们将考虑对先导化合物A1的化学结构进行优化与修饰,以进一步增强其抗菌活性。
2.7 先导化合物的抗菌机理 制备铜绿假单胞菌的细胞破碎液,取上清与A1-NBD复合物在37 °C混合2 h,然后进行SDS-PAGE,电泳条带如图 7所示。铜绿假单胞菌PBP3的理论分子量为60 kDa,与图 7的结果相一致,而且可以看出PBP3的表达量很大。泳道2、3、4的条带在紫外光下可以看到三条对应的荧光条带,荧光条带位置与电泳条带一致,即先导化合物A1可以与PBP3蛋白有效地结合,这说明虚拟筛选的结果是正确的,得到的先导化合物的确可以结合在铜绿假单胞菌的PBP3上。同时,这也表明先导化合物的抗菌机理可能是由于结合PBP3,通过干扰细胞壁的合成而发挥抗菌活性。
 |
| 图 7. 铜绿假单胞菌细胞破碎液上清与A1-NBD复合物的电泳条带及荧光条带 Figure 7. Electrophoretic and fluorescent profiles of PBP3-A1-NBD complex. Cell pellets of P. aeruginosa were disrupted for analysis. Lane 1: supernatant of cell disruption solution; lanes 2-4: supernatants supplemented with A1-NBD complex; M: protein marker. |
| 图选项 |
3 讨论 计算机辅助药物设计是新药开发的一个重要组成部分,其中,基于受体结构的分子对接由于适于大规模数据库筛选而被广泛应用[19, 20, 21]。通过计算机进行大规模虚拟筛选,利用计算机强大的计算能力从已建立的大规模化合物的三维数据库(含上百万个化合物)中搜寻与靶标生物大分子活性部位或者结合部位相匹配的化合物,其目的是快速发现有苗头的化合物,集中目标进行攻关。通过计算机进行虚拟筛选,可以节省大量的人力、物力和财力,加强药物研发进度,并且有利于得到具有全新结构的先导化合物[22, 23]。
本研究根据β-内酰胺类抗生素的作用机理,靶向铜绿假单胞菌的粘肽合成酶PBP3,对含有104万种小分子化合物的数据库进行虚拟筛选,目标是筛出能与PBP3特异性结合的高亲和力的先导化合物,通过竞争性抑制PBP3底物的结合从而阻断铜绿假单胞菌的细胞壁合成。同时,全新的化学结构可以使先导化合物避免被耐药细菌所产生的β-内酰胺酶降解,克服现有的β-内酰胺类抗生素(如青霉素)容易被β-内酰胺酶降解所致的细菌耐药性问题,而且,这些先导化合物可以结合到β-内酰胺酶上,抑制其酶活,用作β-内酰胺类抗生素的增效剂。
本研究对含有104万个小分子化合物的数据库进行了虚拟筛选,以Grid score进行第一轮初筛,得到了Grid score分值小于-30 kcal/mol化合物约6万个,接着以Amber score进行了第二轮筛选,筛出Amber score分值小于-20 kcal/mol的化合物约200个。通过进一步的观察分析,最终挑选出4种亲和力高并且结构新颖的中选化合物。选择4种化合物中结构相对简单的一个进行了有机合成,该先导化合物显示出了良好的抗菌潜力,同时,该化合物可以与PBP3蛋白结合,验证了虚拟筛选的科学性,并在一定程度上证明了该化合物的抗菌机理是结合PBP3蛋白,干扰细胞壁的合成。在后续研究中,我们将对该化合物的结构进行改造和优化,争取将其开发为新型抗菌药物,用于解决铜绿假单胞菌的耐药性问题。
参考文献
| [1] | Livermore DM. Has the era of untreatable infections arrived?. The Journal of Antimicrobial Chemotherapy, 2009, 64(Suppl 1): i29-i36. |
| [2] | Rice LB. The clinical consequences of antimicrobial resistance. Current Opinion in Microbiology, 2009, 12(5): 476-481. |
| [3] | Liu R, Luo BR, Li MY. Research progress of the resistant mechanisms of Pseudomonas aeruginosa to β-lactam antibiotics. International Journal of Laboratory Medicine, 2007, 28(8): 716-718. (in Chinese)刘蓉, 罗必蓉, 李明远. 铜绿假单胞菌对β-内酰胺类抗生素耐药机制研究进展. 国际检验医学杂志, 2007, 28(8): 716-718. |
| [4] | Wang LJ, Xu XL, Shi JR, Liu B, Sun HY, Bai YL, Ma CL. Research progress of multi-drug resistant Pseudomonas aeruginosa. International Journal of Laboratory Medicine, 2013, 34(13): 1713-1715. (in Chinese) 王丽娟, 徐修礼, 史皆然, 刘冰, 孙慧英, 白艳玲, 马春丽. 多药耐药铜绿假单胞菌研究进展. 国际检验医学杂志, 2013, 34(13): 1713-1715. |
| [5] | Bush K, Jacoby GA. Updated functional classification of β-lactamases. Antimicrobial Agents and Chemotherapy, 2010, 54(3): 969-976. |
| [6] | Korsak D, Markiewicz Z, Gutkind GO, Ayala JA. Identification of the full set of Listeria monocytogenes penicillin-binding proteins and characterization of PBPD2 (Lmo2812). BMC Microbiology, 2010, 10(1): 239. |
| [7] | Xie XS, Zhao YJ, Zhang DX, Du TT, Zhang B, Li J, Liu MC. Resistant mechanisms of Gram-negative bacteria to β-lactam antibiotics that mediated by PBPs. Chinese Journal of Preventive Veterinary Medicine, 2013, 35(2): 169-172. (in Chinese) 解晓双, 赵玉军, 张德显, 杜婷婷, 张冰, 李杰, 刘明春. PBPs介导革兰氏阴性菌对β-内酰胺类抗生素的耐药机制. 中国预防兽医学报, 2013, 35(2): 169-172. |
| [8] | Lim D, Strynadka NCJ. Structural basis for the β lactam resistance of PBP2a from methicillin-resistant Staphylococcus aureus. Nature Structural & Molecular Biology, 2002, 9(11): 870-876. |
| [9] | Sainsbury S, Bird L, Rao V, Shepherd SM, Stuart DI, Hunter WN, Owens RJ, Ren JS. Crystal structures of penicillin-binding protein 3 from Pseudomonas aeruginosa: comparison of native and antibiotic-bound forms. Journal of Molecular Biology, 2011, 405(1): 173-184. |
| [10] | De León SR, Daniels K, Clarke AJ. Production and purification of the penicillin-binding protein 3 from Pseudomonas aeruginosa. Protein Expression and Purification, 2010, 73(2): 177-183. |
| [11] | Zhang C, Li WZ, Yun LH. Application of the combinatorial chemistry to research of the natural products. Progress in Chemistry, 2003, 15(3): 194-203. (in Chinese) 张城, 李伟章, 恽榴红. 用组合化学建立天然产物类似物库. 化学进展, 2003, 15(3): 194-203. |
| [12] | Kroemer RT. Structure-based drug design: docking and scoring. Current Protein & Peptide Science, 2007, 8(4): 312-328. |
| [13] | Song XR, Li D, Chen J, Zhao Y. Computer aided drug screening platform and its application. Chinese Journal of Bioinformatics, 2014, 12(4): 300-304. (in Chinese) 宋新蕊, 李达, 陈洁, 赵勇. 计算机辅助药物筛选平台及应用. 生物信息学, 2014, 12(4): 300-304. |
| [14] | Zervosen A, Sauvage E, Frère JM, Charlier P, Luxen A. Development of new drugs for an old target: the penicillin binding proteins. Molecules, 2012, 17(11): 12478-12505. |
| [15] | Hadizadeh F, Moradi A, Naghibi G, Vojdani M, Behravan J, Ramezani M. Synthesis and antitumor activity of substituted succinamides using a potato disc tumor induction assay. International Journal of Biomedical Science, 2007, 3(1): 60-64. |
| [16] | CLSI. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard-ninth edition. CLSI document M07-A9. Wayne, PA: Clinical and Laboratory Standards Institute, 2012. |
| [17] | An YD, Du QZ, Tong LY, Yu ZW, Gong XW. Cloning, expression and purification of penicillin-binding protein 3 from Pseudomonas aeruginosa CMCC 10104. Protein Expression and Purification, 2015, 110: 37-42. |
| [18] | 李宇. 三苯基膦与碘温和活化下羧酸的酰胺化反应研究. 西南大学硕士学位论文, 2011. |
| [19] | Zhu W, Chen KJ, Xu XJ. Application of computerized virtual screening technique in traditional Chinese medicine. Chinese Journal of Integrated Traditional and Western Medicine, 2007, 27(3): 263-266. (in Chinese)朱伟, 陈可冀, 徐筱杰. 计算机药物虚拟筛选技术在中医药领域中的应用前景. 中国中西医结合杂志, 2007, 27(3): 263-266. |
| [20] | Xu J. A new approach to finding natural chemical structure classes. Journal of Medicinal Chemistry, 2002, 45(24): 5311-5320. |
| [21] | Koch MA, Waldmann H. Protein structure similarity clustering and natural product structure as guiding principles in drug discovery. Drug Discovery Today, 2005, 10(7): 471-483. |
| [22] | Luo XM, Jiang HL, Shen JH, Chen KX. The progress of drug design. Bulletin of Chinese Academy of Science, 2003, 18(4): 255-259. (in Chinese) 罗小民, 蒋华良, 沈建华, 陈凯先. 药物分子设计研究进展. 中国科学院院刊, 2003, 18(4): 255-259. |
| [23] | Xu WR, Tang LD, Fu HX, Liu BN, Liu P. Molecular simulation and virtual evaluation of new drugs. Chinese Pharmacologist, 2009, 26(2): 76. (in Chinese) 徐为人, 汤立达, 符海霞, 刘冰妮, 刘鹏. 分子模拟与新药虚拟评价. 中国药理通讯, 2009, 26(2): 76. |
